
Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) exert analgesic effects by inhibiting peripheral cyclooxygenases (COXs). It is now clear that these drugs also have central actions that include the modulation of descending control of spinal nociception from the midbrain periaqueductal gray (PAG). Descending control is a powerful determinant of the pain experience and is thus a potential target for analgesic drugs, including COX inhibitors. Noxious information from the periphery is conveyed to the spinal cord in A- and C-fiber nociceptors, which convey different qualities of the pain signal and have different roles in chronic pain. This in vivo study used different rates of skin heating to preferentially activate A- or C-heat nociceptors to further investigate the actions of COX inhibitors and prostaglandins in the PAG on spinal nociceptive processing. The results significantly advance our understanding of the central mechanisms underlying the actions of NSAIDs and prostaglandins by demonstrating that (1) in the PAG, it is COX-1 and not COX-2 that is responsible for acute antinociceptive effects of NSAIDs in vivo; (2) these effects are only evoked from the opioid-sensitive ventrolateral PAG; and (3) prostaglandins in the PAG exert tonic facilitatory control that targets C- rather than A-fiber-mediated spinal nociception. This selectivity of control is of particular significance given the distinct roles of A- and C-nociceptors in acute and chronic pain. Thus, effects of centrally acting prostaglandins are pivotal, we suggest, to both the understanding of nociceptive processing and the development of new analgesic drugs.
- nociception
- periaqueductal gray
- descending control
- prostaglandin
- cyclooxygenase
- C-nociceptor
Introduction
Prostaglandins and their synthetic enzymes, cyclooxygenases (COXs), have well established functions in pain and inflammation, roles clearly shown by the effectiveness of nonsteroidal anti-inflammatory drugs (NSAIDs) as analgesics. The analgesic actions of NSAIDs, nonselective inhibitors of the two well characterized COX isoforms (Vane et al., 1998), cannot, however, be explained solely by their ability to inhibit peripheral prostaglandin production. In this respect, it is now recognized that the COX–prostaglandin pathway plays an important role in spinal nociceptive processing (Vanegas and Schaible, 2001), including the regulation of descending control of spinal nociception from the CNS, which is a powerful determinant of the pain experience (Vanegas and Tortorici, 2002; Heinricher et al., 2004).
The periaqueductal gray (PAG) region of the midbrain is a key site for the coordination of descending control (Lovick and Bandler, 2005). Pain modulatory drugs such as opioids and cannabinoids exert central effects in the PAG (Yaksh et al., 1976; Morgan et al., 1992; Maione et al., 2006), and several lines of evidence now indicate a central role for prostaglandins in this brain region. For instance, COX-1 and COX-2 are expressed in the PAG (Breder et al., 1992, 1995), and it displays [3H]-prostaglandin E2 (PGE2) binding (Matsumura et al., 1992) and prostaglandin EP3 receptor mRNA and immunoreactivity (Ek et al., 2000; Nakamura et al., 2000). Furthermore, nonspecific inhibition of all COX isoforms in the PAG inhibits acute nociceptive processing in vivo (Tortorici and Vanegas, 1995; Vanegas et al., 1997), and microinjection of PGE2 into the PAG produces hindpaw hyperalgesia (Heinricher et al., 2004). Despite mounting evidence that prostaglandins have functional roles in modulation of descending control systems, the relative contributions of the different COX isoforms to nociceptive processing in vivo remains unknown.
Information about peripheral tissue damage is conveyed to the spinal cord in A- and C-fiber nociceptors. The experience of pain evoked by activation of C-nociceptors (slow, burning, poorly localized) is very different from that evoked by A-fiber activation (sharp, well localized) (Schady et al., 1983; Torebjork and Ochoa, 1990; Magerl et al., 2001). Furthermore, these classes of nociceptive neurons have different properties (Lawson, 2002; Meyer et al., 2006) and play distinct roles in chronic pain (Fuchs et al., 2000; Magerl et al., 2001). However, very few studies have investigated systematically the central processing and descending control from the CNS of C- versus A-nociceptive input [but see Koutsikou et al. (2007), McMullan and Lumb (2006a,b), Simpson et al. (2007), and Waters and Lumb (2007)]. To facilitate studies of differential control of A- versus C-fiber-evoked nociception, we have developed a method to preferentially activate either C- or A-heat nociceptors using different rates of skin heating (McMullan et al., 2004). In the present study, we provide additional support for the approach, which enables us to compare the levels of control exerted by the PAG on responses evoked by these functionally distinct populations of nociceptors. The aim of the present study was, therefore, to investigate the role of prostaglandins in the PAG in the spinal processing of C- versus A-nociceptive input.
Materials and Methods
Animal preparation
All experiments were performed in accordance with United Kingdom Animals (Scientific Procedures) Act (1986) and associated guidelines. Male adult Wistar rats (280–320 g) were housed in standard conditions and handled daily to minimize stress on the day of the experiment. Anesthesia was induced using 4% halothane in O2, and a branch of the external jugular vein was cannulated for anesthetic maintenance using constant intravenous propofol (∼30 mg · kg−1 · h−1; Rapinovet; Schering Plough Animal Health, South Harefield, UK) or alphadolone/alphaxalone (∼20 mg · kg−1 · h−1; Saffan; Schering Plough Animal Health) infusion. Preliminary experiments showed no differences in heat ramp responses or in drug effects between anesthetics (data not shown). Despite the lack of difference between the two anesthetics, the potential confounding effects of general anesthesia must be borne in mind when interpreting the data. A branch of the carotid artery was exposed and cannulated for recording of blood pressure, and the trachea was cannulated to allow artificial ventilation of the animal if required. Body temperature was maintained within physiological limits by means of a feedback-controlled heating blanket and rectal probe. In some animals (n = 19), a laminectomy was performed between T11 and T13, to record from dorsal horn neurons, and in others (n = 66) a craniotomy was performed, to allow access to the PAG with micropipettes. Animals were then positioned in a stereotaxic frame. In neuronal recording experiments, anesthesia was maintained at a level at which there were no precipitous changes in blood pressure to minor noxious stimuli, and in electromyographic (EMG) recording experiments it was reduced to a level at which animals were moderately responsive to firm pinch of the contralateral forepaw and brushing of the cornea using a cotton swab. Animals were allowed to stabilize at these levels for a minimum of 30 min before recording of neuronal or EMG activity.
Recording of dorsal horn neuronal activity
The vertebral column was clamped at each end of the laminectomy to maximize the stability of the preparation. The dura was removed, a pool was made with skin flaps, and the whole area was filled with agar to further stabilize the preparation. Once set, a small window was cut out of the agar over the desired recording site and filled with warm paraffin oil. A glass-coated tungsten microelectrode (∼5 MΩ) (Merrill and Ainsworth, 1972) was lowered into the cord at the rostrocaudal location at which maximum dorsum potentials had been observed in response to electrical stimulation of the hairy skin of the contralateral hindpaw. Single-unit neuronal activity was amplified (5000×) and filtered (500 Hz to 10 kHz; Neurolog system) before being captured for subsequent analysis via a 1401plus (Cambridge Electronic Design, Cambridge, UK) onto a PC running Spike2 version 5 software (Cambridge Electronic Design).
Recording of electromyographic activity
An intramuscular bipolar electrode, custom-made from two short lengths of Teflon-coated 0.075-mm-diameter stainless steel wire (Advent Research Materials, Eynsham, UK), was inserted into the biceps femoris of the left hind leg to record EMG activity during the withdrawal reflex. The EMG signal was amplified (5000×) and filtered (50 Hz to 5 kHz; Neurolog system), before being captured for subsequent analysis via a 1401plus (Cambridge Electronic Design) onto a PC running Spike2 version 5 software (Cambridge Electronic Design).
Preferential activation of A- or C-nociceptors
A- or C-heat nociceptors were stimulated using a custom-made heating lamp system to deliver fast or slow rates of skin heating respectively to the dorsal surface of the hindpaw, as described previously (McMullan et al., 2004). In brief, heat from a sputter-coated projector bulb was focused onto a blackened copper disk positioned at the focal point. A T-type thermocouple (made in-house: 0.02-mm-diameter copper/constantine) was fixed to the outer surface of the copper plate and therefore measured the surface temperature of the skin when placed in firm, even contact with the hindpaw dorsum. Using a constant bulb voltage, fast rates of heating (7.5 ± 1°C · s−1) were used to preferentially activate A-fiber (myelinated, capsaicin-insensitive) heat nociceptors, whereas slow rates of heating (2.5 ± 1°C · s−1) were used to preferentially activate C-fiber (unmyelinated, capsaicin-sensitive) heat nociceptors. Our previous studies (McMullan et al., 2004) demonstrate that these heating rates measured at the skin surface reliably reproduce the same subcutaneous heating rates as those described by Yeomans and Proudfit (1996) and Yeomans et al. (1996) to preferentially activate A- versus C-nociceptors. The cutoff temperature of the heat lamp was controlled via a Spike2 script to prevent tissue damage. Fast or slow heat ramps were performed at 8 min intervals to preferentially activate A-fiber (myelinated, capsaicin-insensitive) nociceptors or C-fiber (unmyelinated, capsaicin-sensitive) nociceptors.
Experimental protocols and statistical analysis
Dorsal horn neuronal recordings.
Wide dynamic range neurons were located using low-threshold search stimuli (brush, tap, and prod) to the ipsilateral hindpaw followed by high-threshold noxious stimuli (pinch and heat). Once a cell was located, its receptive field was characterized using both low- and high-threshold stimuli. The afferent input to the cell was characterized using percutaneous electrical stimuli (1 ms square pulse) delivered to the center of its receptive field. Thresholds for A- and C-fiber activation were established, and repeated sweeps were made at both 1.5 and 3 times C-fiber threshold voltage (a train of 20 1 ms square pulses delivered at 0.1 Hz). Responses of each cell to both fast and slow heat ramps were recorded.
Peristimulus time histograms were constructed from repeated electrical sweeps at both 1.5 and 3 times C-fiber threshold voltage, and the response (mean spikes per sweep, corrected for the spontaneous activity of the cell) of each cell at C-fiber latency (90–350 ms after stimulus) was determined. Responses to slow heat ramps were quantified by dividing the total number of spikes by the length of the ramp, giving spikes per ramp per second. This value was then corrected for spontaneous activity of the cell, measured over the 10 s before ramp onset. Correlations of response to slow ramps versus spikes at C-fiber latency were made for both 1.5 and 3 times C-fiber threshold.
EMG recordings.
Once a steady baseline of paw-withdrawal thresholds to fast or slow heat ramps had been achieved, a glass micropipette containing drug or vehicle solution was lowered vertically into the PAG ipsilateral to the stimulated hindpaw between 4.2 and 5.5 mm below the cortical surface. Drug solutions were microinjected using a custom-made paraffin-filled pressure injection system attached to a 1 μl syringe (SGE, Ringwood, Australia) over 1 min (starting at t = 0 min on graphs). Fast or slow heat ramps were resumed at 1 min, and paw-withdrawal thresholds were measured for an additional 60–90 min. In 19 of 66 animals, it was possible to obtain a second complete dataset.
At the end of experiments, animals were killed by overdose of sodium pentobarbital (30 mg, i.v. bolus; Sigma, Poole, UK) and transcardially perfused with 100 ml of mammalian Ringer solution followed by 300 ml of 4% paraformaldehyde in 0.1 m phosphate buffer. After postfixing and cryoprotection in 30% sucrose solution for at least 24 h, brains were sectioned at 60 μm, and PAG injection sites were localized with reference to the rat brain atlas of Paxinos and Watson (2005).
In a separate experimental group (n = 9), after baseline fast or slow heat ramps, peripheral sensitization of A- or C-fiber nociceptors was achieved by topical application of dimethyl sulfoxide (DMSO; 100%; 50 μl; Sigma) or capsaicin (10 mm in 50% ethanol; 50 μl; Sigma) to the hindpaw dorsum using a pipette. Fast or slow heat ramps were resumed 15 min after application, and paw-withdrawal thresholds were measured.
No attempt was made to blind the experimenter to the drugs administered, because all peripheral stimuli were delivered by electronically controlled devices, and EMG activity was captured on-line and analyzed off-line using predetermined protocols.
All EMG data are displayed as mean ± SEM. The effects of COX inhibitors or PGE2 were compared with vehicle using two-way ANOVA followed by Bonferroni's posttest to compare drug effects versus vehicle at each time point. Area under curve (AUC; t = 0–60 or 90 min, as shown in Figs. 3, 5⇓–7) was also calculated, and comparison of fast versus slow AUC was made using unpaired Student's t test. Data from peripheral sensitization experiments were pooled into control and posttreatment groups and compared using Student's t test. All statistical analysis was performed using GraphPad (San Diego, CA) Prism 4.
Drugs
Stock solutions of all drugs for microinjection were made up in DMSO (Sigma) at 10 mm and stored at −20°C until required. On the day of the experiment, an aliquot was thawed and diluted to working concentrations in the appropriate vehicle (70% physiological saline, 30% DMSO for COX inhibitors; PBS for PGE2) containing 5% pontamine sky blue dye to mark injection sites. Final drug doses used were based on effective doses as described by others (de Beaurepaire et al., 1990; Futaki et al., 1994; Masferrer et al., 1994; Cryer and Feldman, 1998; Smith et al., 1998; Heinricher et al., 2004) and were as follows: PGE2, 50 fg in 200 nl; ketoprofen, 10 μg in 300 nl (both from Sigma); SC-560, 5.3 pg in 300 nl (50 nm); NS-398, 0.47 and 9.4 ng in 300 nl (5 and 100 μm, respectively; all from Cayman Chemical, Ann Arbor, MI).
Results
Fast and slow rates of skin heating preferentially activate A- and C-fiber heat nociceptors, respectively
The interpretation of data with respect to A- or C-nociceptor function is critically dependent on the ability of our skin heating protocol to preferentially activate either A- or C-fiber heat nociceptors. There is direct evidence from peripheral nerve recordings (Yeomans and Proudfit, 1996) that the same slow and fast rates of heating as used in the present study preferentially activate primary afferents that conduct in the C- and A-fiber range, respectively. In addition, we have shown previously that response characteristics of dorsal horn neurons to slow versus fast rates of skin heating closely resemble those of peripheral C- and A-nociceptive afferents, respectively (McMullan and Lumb, 2006b). We now provide additional evidence derived from two different lines of investigation to support the use of slow and fast rates of skin heating to preferentially activate C- versus A-fiber nociceptors: (1) differential sensitization of withdrawal reflexes to slow and fast rates of skin heating by capsaicin and DMSO, and (2) a positive correlation between the magnitude of C-fiber-evoked activity of dorsal horn neurons and the magnitude of their responses to slow heat ramp stimulation.
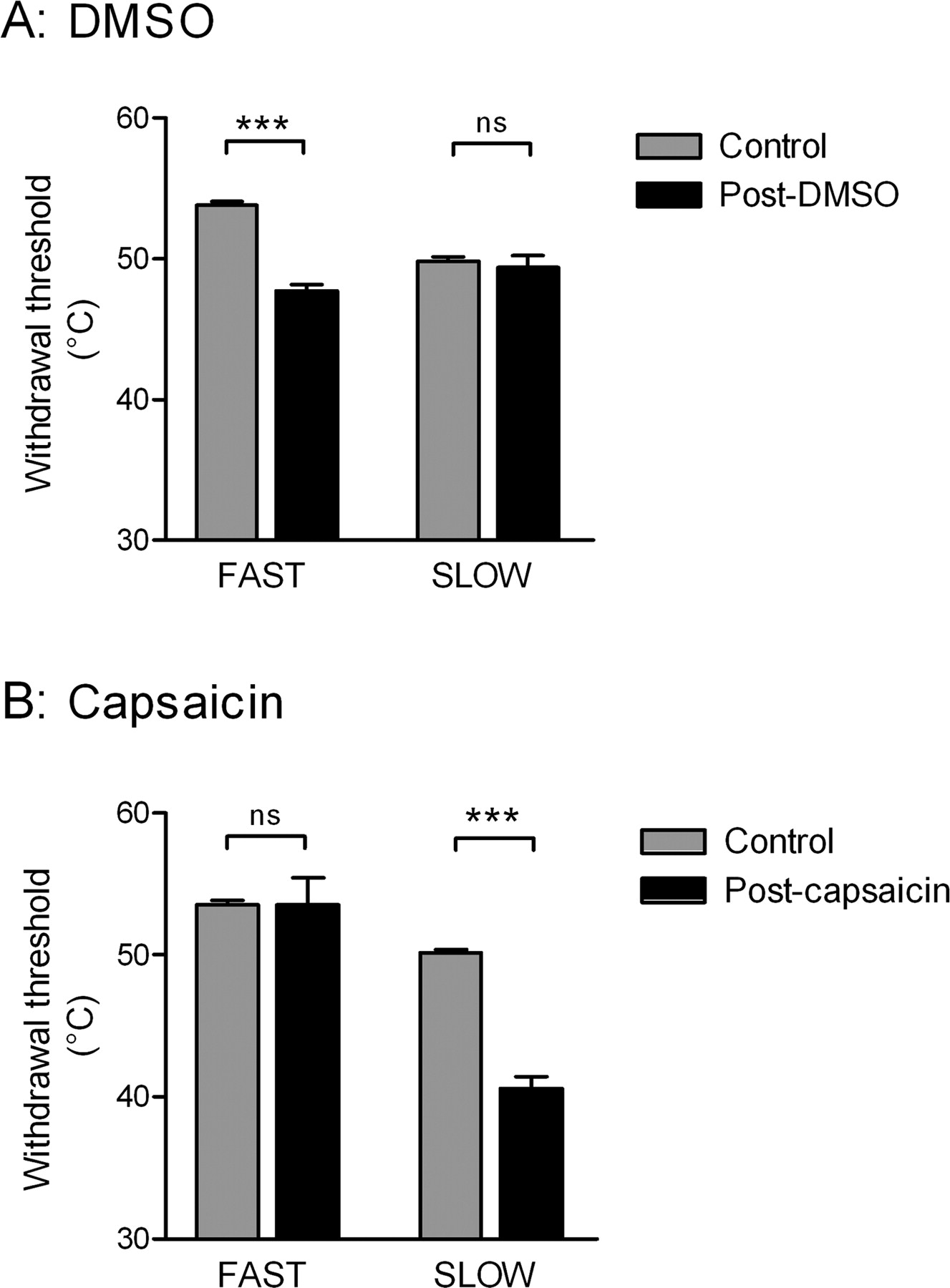
DMSO, which by an unknown mechanism is reported to sensitize A-fiber cutaneous nociceptors (Baumann et al., 1991; Wilson et al., 1999), significantly reduced withdrawal thresholds to fast (p < 0.001; n = 4) but not slow heating ramps (Fig. 1A). Conversely, capsaicin, which when applied to the skin in low concentrations sensitizes TRPV1-expressing primary afferents, the majority of which have C-fibers (Caterina et al., 1997; Kobayashi et al., 2005), significantly lowered withdrawal thresholds to slow (p < 0.001; n = 5) but not fast heating ramps (Fig. 1B) (see also McMullan et al., 2004). Other studies have clearly shown that topical application of capsaicin decreases the latency of withdrawal reflexes and sensitizes peripheral C-fiber nociceptors to the same rates of slow skin heating, while having no effect on responses to fast rates of skin heating (Yeomans and Proudfit, 1996; Yeomans et al., 1996).

Fast and slow heat ramps preferentially activate A- and C-fiber nociceptors, respectively. Effect of peripheral chemical sensitization of nociceptors on withdrawal thresholds to fast and slow heat ramps. A, Topical application of 100% DMSO to the hindpaw dorsum (pooled data of 7 trials from n = 4). B, Topical application of 10 mm capsaicin to the hindpaw dorsum (pooled data of 5 trials from n = 5). Data are expressed as mean ± SEM. Statistical analysis compared control and posttreatment groups by Student's t test. ns, Not significant (p > 0.05); ***p < 0.001.
Dorsal horn neurons with strong C-fiber-evoked responses to electrical stimulation in their cutaneous excitatory receptive fields (strong C+ve) showed robust responses to slow rates of skin heating in the same peripheral field (Fig. 2A). In contrast, those with little (weak C+ve) or no (C−ve) C-fiber input gave weak or no response to slow rates of skin heating (Fig. 2A). Both C+ve and C−ve neurons responded to fast rates of skin heating (data not shown). For the population as a whole, there was a strong and highly significant correlation (Spearman's r = 0.90 and r = 0.85; p < 0.001; n = 19 for stimulation at 1.5 and 3 times C-fiber threshold, respectively) between the magnitude of electrically evoked C-fiber activity and the magnitude of slow ramp-evoked activity (Fig. 2B) (i.e., the greater the C-fiber-evoked response, the greater the response to slow ramp stimulation), as would be expected if the afferents activated by slow ramp stimulation conduct in the C-fiber range. Together, these data support the view that fast and slow heating ramps preferentially activate A- versus C-fiber nociceptors, respectively.

Response magnitudes of dorsal horn neurons to slow heat ramps are highly correlated with their degree of C-fiber afferent input. A, Responses of three dorsal horn neurons to slow heat ramps and to electrical stimulation at three times C-fiber threshold. Ratemeter histograms (spikes · s−1) and the corresponding slow heat ramp evoking this response are shown for each cell. After electrical stimulation (at time marked by arrows) of the same peripheral receptive field, all cells show activity at short latency (A-fiber). Both strong and weak C+ve cells show a distinct late volley of action potentials at C-fiber latency, whereas the C−ve cell shows no such response. B, The response magnitude of dorsal horn neurons (n = 19) to slow heat ramps (spikes per ramp per second) is significantly correlated with the magnitude of C-fiber input to the cell evoked by electrical stimulation (mean spikes per sweep) at both 1.5 and 3 times C-fiber threshold (r = 0.90, p < 0.001 and r = 0.85, p < 0.001, respectively; Spearman's nonparametric test for correlation).
Prostaglandin E2 in the ventrolateral PAG facilitates C- but not A-nociceptor-evoked responses
The PAG is organized into functional columns (Bandler and Keay, 1996; Lovick and Bandler, 2005) that exhibit regional sensitivity in the action of analgesic drugs such as μ-opioid agonists (Yaksh et al., 1976). Recent evidence suggests that the antinociceptive actions of systemically administered COX inhibitors depend in part on opioid-sensitive mechanisms in the ventrolateral PAG (VL-PAG) (Tortorici et al., 1996; Vazquez et al., 2005). Therefore, to establish whether effects of prostaglandins on spinal nociception are restricted to this region, we made stereotaxic microinjections of PGE2 within the dorsolateral/lateral (DL/L) or ventrolateral columns of the PAG and compared their effects on paw-withdrawal thresholds (as indicated by biceps femoris EMG activity) in response to slow or fast rates of heating applied to the hindpaw dorsum.
Microinjection of PGE2 into the VL-PAG produced a significant decrease in the withdrawal threshold to C-nociceptor activation compared with vehicle, with no apparent effect of PGE2 on A-nociceptor-evoked responses (Fig. 3; supplemental Table 1, available at www.jneurosci.org as supplemental material). Comparison of the overall drug effect, by measurement of AUC, showed that the pronociceptive effect of PGE2 on C-fiber-evoked reflexes was significantly greater than any effect on A-fiber-evoked reflexes (Fig. 4). In contrast to this facilitatory effect on C-nociceptive input evoked from the VL-PAG, PGE2 had no significant effect on the withdrawal thresholds to either C- or A-nociceptor activation when injected into DL/L-PAG (Fig. 3).

Prostaglandin E2 in the ventrolateral PAG selectively facilitates C-fiber-mediated spinal nociception. A–D, Effect of PGE2 (50 fg in 200 nl) microinjections into the dorsolateral (A, B; n = 3 per group) and ventrolateral (C, D; n = 4–6 per group) PAG on withdrawal thresholds to slow (A, C) and fast (B, D) heat ramps. Data are expressed as mean ± SEM. Statistical analysis compared PGE2 (•, slow; ▾, fast) versus vehicle (○, slow; ▿, fast) using two-way ANOVA followed by Bonferroni's posttest; *p < 0.05; **p < 0.01; ***p < 0.001. E, Injection sites; coordinates are relative to bregma. DM, Dorsomedial; DL, dorsolateral; L, lateral; VL, ventrolateral.

The COX–prostaglandin pathway within the ventrolateral PAG preferentially modulates C-fiber-mediated spinal nociception. Histograms of AUC measurements to illustrate the effects of injection of drugs and their vehicles into the VL-PAG on reflex responses to fast and slow rates of skin heating. In each case, AUC was calculated between t = 0 (injection of drug or vehicle) and the endpoint shown in Figures 3 and 5⇓–7. Data are expressed as mean ± SEM. Statistical analysis compared fast and slow ramp responses for each drug using Student's unpaired t test. ns, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
Nonselective COX inhibition preferentially inhibits C- versus A-nociceptor-evoked responses
Given our finding that prostaglandins preferentially facilitate C-nociceptive input, we hypothesized that NSAID administration into the PAG should produce antinociception that shows at least some selectivity for C-nociceptor-evoked responses.
The nonspecific COX inhibitor ketoprofen significantly increased withdrawal thresholds to both C- and A-nociceptor activation compared with vehicle when injected into the VL-PAG (Fig. 5; supplemental Table 1, available at www.jneurosci.org as supplemental material). However, this antinociceptive effect of ketoprofen in the VL-PAG was of far greater duration on C-nociceptor than on A-nociceptor-evoked responses. Comparison of AUC showed that the overall drug effect was significantly greater on C-nociceptor-evoked responses than A-fiber-evoked responses (Fig. 4). In contrast, ketoprofen had no significant effect on the withdrawal thresholds to either C- or A-nociceptor activation when injected into DL/L-PAG (Fig. 5). Together this implies that COX isoforms are tonically active in the VL-PAG and their products exert a facilitatory effect on acute spinal nociceptive processing, which preferentially targets C-nociceptor-evoked spinal events.

Nonselective COX inhibition in the ventrolateral PAG preferentially inhibits C-fiber-mediated spinal nociception. A–D, Effect of the nonselective COX inhibitor ketoprofen (10 μg in 300 nl) or vehicle microinjections into the dorsolateral (A, B; n = 3–4 per group) and ventrolateral (C, D; n = 4 per group) PAG on withdrawal thresholds to slow (A, C) and fast (B, D) heat ramps (n = 4 in each group). Data are expressed as mean ± SEM. Statistical analysis compared ketoprofen (•, slow; ▾, fast) versus vehicle (○, slow; ▿, fast) using two-way ANOVA followed by Bonferroni's posttest. *p < 0.05; **p < 0.01; ***p < 0.001. E, Injection sites; coordinates are relative to bregma. DM, Dorsomedial; DL, dorsolateral; L, lateral; VL, ventrolateral.
COX-1 mediates the acute antinociception produced by NSAIDs in the PAG
Having identified the VL- rather than the DL-PAG as being sensitive to nonselective COX inhibition, we then went on to determine the particular COX isoform responsible for generation of pronociceptive prostaglandins within the PAG. The COX-1 inhibitor SC-560 (50 nm) mimicked the effects of ketoprofen, producing a significant antinociception on both C-nociceptor and A-nociceptor-evoked responses (Fig. 6). As with ketoprofen, SC-560 exerted a greater antinociceptive effect on C-fiber-evoked than A-fiber-evoked reflexes (Fig. 4; supplemental Table 1, available at www.jneurosci.org as supplemental material). At this concentration, SC-560 is a highly specific COX-1 inhibitor (Smith et al., 1998); therefore, these observations suggest that inhibition of COX-1 in the PAG mediates the antinociceptive effects of NSAIDs in this region. However, both COX isoforms are reported to be expressed in the PAG, and inhibition of either COX-1 or COX-2 alters excitability of PAG neurons in vitro. Therefore, we also tested the effects of VL-PAG microinjection of the COX-2 inhibitor NS-398. At a concentration of 5 μm, which is specific for COX-2 (Futaki et al., 1994; Masferrer et al., 1994), NS-398 had no significant effect on withdrawal thresholds to either C- or A-fiber-evoked reflexes (Fig. 7). However, at a higher concentration, no longer selective for COX-2 (100 μm) (Cryer and Feldman, 1998), NS-398 significantly increased withdrawal thresholds to both C- and A-fiber-evoked reflexes, presumably by inhibiting COX-1. Consistent with this, comparison of the overall effect of NS-398 at the higher dose showed that the antinociceptive effect on C-fiber-evoked reflexes was significantly greater than its effect on A-fiber-evoked reflexes (Fig. 4; supplemental Table 1, available at www.jneurosci.org as supplemental material). Thus inhibition of COX-1 and not COX-2 mimics the antinociceptive effect of NSAIDs in the PAG, which indicates a role for prostaglandins synthesized by this enzyme in the tonic control of acute spinal nociceptive processing.

COX-1 inhibition mimics the antinociception produced by NSAIDs in the ventrolateral PAG. A, B, Effect of SC-560 (50 nm) microinjection into the ventrolateral PAG on withdrawal thresholds to slow (A; n = 4 per group) and fast (B; n = 4 per group) heat ramps. Data are expressed as mean ± SEM. Statistical analysis compared SC-560 (•, slow; ▾, fast) versus vehicle (○, slow; ▿, fast) using two-way ANOVA followed by Bonferroni's posttest. *p < 0.05; **p < 0.01; ***p < 0.001. C, Injection sites; coordinates are relative to bregma. DM, Dorsomedial; DL, dorsolateral; L, lateral; VL, ventrolateral.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
COX-2 inhibition in the ventrolateral PAG does not alter nociceptive thresholds in vivo. A, B, Effect of NS-398 (5 or 100 μm) microinjection into the ventrolateral PAG on withdrawal thresholds to slow (A; n = 4 per group) and fast (B; n = 4 per group) heat ramps. Data are expressed as mean ± SEM. Statistical analysis compared NS-398 (5 μm, •, slow; ▾, fast; 100 μm, ■, slow; ▴, fast) versus vehicle (○, slow; ▿, fast) using two-way ANOVA followed by Bonferroni's posttest. *p < 0.05; **p < 0.01; ***p < 0.001. C, Injection sites; coordinates are relative to bregma. DM, Dorsomedial; DL, dorsolateral; L, lateral; VL, ventrolateral.
Discussion
The importance of peripheral COX and prostaglandins in the generation and maintenance of pain and inflammation is beyond doubt and is manifested by the fact that NSAIDs are the most widely used drugs in the world for pain relief (Vane et al., 1998; Simmons et al., 2004). However, the contributions of COX and prostaglandins to nociceptive processing in the periphery do not fully explain their analgesic actions, and recent studies have demonstrated that they have additional central roles (Ferreira et al., 1978; Bjorkman et al., 1992; Vanegas and Schaible, 2001; Vanegas and Tortorici, 2002; Heinricher et al., 2004). The current study has significantly advanced our understanding of the central mechanisms underlying the actions of NSAIDs and prostaglandins by demonstrating that (1) in the PAG, it is COX-1 and not COX-2 that is responsible for acute antinociceptive effects of NSAIDs in vivo, (2) these effects are only evoked from the opioid-sensitive VL-PAG, and (3) prostaglandins in the PAG exert tonic facilitatory control that targets C- rather than A-fiber-mediated spinal nociception.
COX-1 products in the PAG modulate descending control of spinal nociception
The current study confirms previous reports (Heinricher et al., 2004) that administration of PGE2 into the VL-PAG has pronociceptive effects on spinal reflexes and that a synthetic prostaglandin analog in the PAG alters behavioral responses in the formalin test (Oliva et al., 2006). Prostaglandins, including PGE2, are products of COX activity, and nonselective inhibition of all COX isoforms in the PAG produces antinociception (Carlsson and Jurna, 1987; de Beaurepaire et al., 1990; Tortorici and Vanegas, 1995). Both COX-1 and COX-2 isoforms are expressed in the PAG (Breder et al., 1992, 1995), although there is little information on the columnar distribution of the different isoforms, and controversy exists over which of the isoforms might mediate the antinociceptive effects of NSAIDs. In vitro studies indicate that either isoform may have a role in the modulation of neuronal activity in this region. Differences in the experimental methods used [mechanically dissociated PAG neurons (Shin et al., 2003) vs brain slices (Vaughan, 1998)] most likely account for these contradictory findings. Our study shows in vivo, for the first time, that it is specifically COX-1 that is responsible for the antinociceptive effects of NSAIDs within the PAG. Furthermore, the effects of inhibition of COX-1 activity indicate that prostaglandins exert tonic facilitatory control in normal animals and, as a consequence, have the capacity to set the gain of spinal nociception and to contribute to acute pain behaviors.
Effects of COX-1 and prostaglandins on spinal nociception are localized to the opioid-sensitive ventrolateral region of the PAG
The midbrain PAG is a key region in the central control of nociceptive processing and is organized into functionally distinct columns (Bandler and Keay, 1996; Lovick and Bandler, 2005) that exhibit regional sensitivity in the action of analgesic drugs. Here we have shown that the modulation of spinal nociception by the COX–prostaglandin pathway in the PAG is restricted to the VL sector, which is also the target of μ-opioid analgesics in the PAG (Yaksh et al., 1976). This anatomical overlap is consistent with recent reports that the antinociceptive actions of systemically administered COX inhibitors depend in part on μ-opioid-sensitive mechanisms in the VL-PAG (Tortorici et al., 1996; Vazquez et al., 2005). Indeed, there is clinical evidence of synergistic actions of NSAIDs and opioids (Beaver, 1984) and experimental evidence (Maves et al., 1994) that COX inhibitors and μ-opioid agonists share common neural substrates, including modulation of neuronal excitability in GABAergic interneurons in the PAG (Vaughan et al., 1997).
It is generally accepted that, to exert effects at the spinal level, alterations in GABAergic tone modulate output neurons in the VL-PAG that predominantly target the rostral ventromedial medulla (RVM) (Vanegas et al., 1984; Cheng et al., 1986; Fields et al., 1988; Morgan et al., 1992). Not surprisingly, therefore, the behavioral antinociception produced by nonselective COX inhibition and the pronociceptive effect of PGE2 administration into the PAG is associated with altered neuronal activity within pain modulatory circuitry of the RVM (Tortorici and Vanegas, 1994, 1995; Heinricher et al., 2004).
Differential control of C- versus A-fiber-evoked spinal nociception
A major finding of the current study is that tonic facilitatory control exerted by PGE2 and/or other COX-1 products in the PAG targets C- rather than A-fiber-evoked spinal nociception. Our previous studies of nociceptive reflexes demonstrated that differential control of C- versus A-fiber-evoked events was also produced by nonselective neuronal activation in the PAG with an excitatory amino acid (McMullan and Lumb, 2006a). Subsequent single-unit spinal cord recordings revealed that descending control from the PAG targets dorsal horn neurons with C-nociceptive inputs, indicating that differential control is at least partly mediated on the sensory limb of nociceptive reflexes (Waters and Lumb, 2007) and is likely to include postsynaptic mechanisms (McMullan and Lumb, 2006b).
Differential effects on A- versus C-spinal nociception have also been reported after activation of pain control centers in the medulla (Lu et al., 2004). Furthermore, the targeting of C-fiber-evoked activity could explain the thermal behavioral hyperalgesia after infusion of PGE2 into VL-PAG observed by Heinricher et al. (2004), in which the parameters of heat stimulation would, almost certainly, have preferentially activated C-nociceptive afferents.
There are several factors that might account for the observed differences in the selectivity and the time course of effects of PGE2 and COX-1 inhibitors on C- versus A-fiber-evoked responses. These include the stability of PGE2 compared with COX inhibitors, the more robust response of primary afferents and dorsal horn neurons to fast versus slow rates of skin heating (Yeomans et al., 1996; McMullan and Lumb, 2006b), the possibility of additional effects of COX-1 products other than PGE2, and additional effects of NSAIDs on non-COX targets such as acid-sensing ion channels, which have been described recently (Voilley et al., 2001).
Functional significance
Human studies have provided good evidence that myelinated, rapidly conducting, A-nociceptors mediate well localized sharp, pricking sensations of “first” pain, whereas unmyelinated, slowly conducting C-nociceptors mediate “second” pain, which is often described as dull, burning, and diffuse (Schady et al., 1983; Torebjork and Ochoa, 1990; Magerl et al., 2001) (see also Lawson, 2002). However, despite the fact that A- and C-fiber nociceptors convey different sensory information in humans, very few studies have compared their central processing, which is of considerable behavioral significance, because it is likely that the balance of inputs from A- and C-nociceptors determines many aspects of the autonomic, motor, and emotional responses to noxious stimuli.
In normal animals, the VL-PAG has protective functions in that neurons in this region modulate spinal nociception as part of coordinated passive coping strategies that, it is proposed, become active in response to inescapable stressors (e.g., deep and visceral pain), after intense exercise, during recuperation, and possibly as part of the sickness response (Keay and Bandler, 2001; Lumb, 2002; Lovick and Bandler, 2005). Preferential control of C-fiber input by a COX-dependent mechanism in the VL-PAG, as described here, is therefore clinically and behaviorally important and could contribute to the protective function of descending control from this midbrain area, because C-fiber-mediated pain is difficult to control and difficult to tolerate.
It is now apparent that alterations in descending controls from brainstem structures (Urban and Gebhart, 1999; Pertovaara, 2000; Porreca et al., 2002; Ren and Dubner, 2002; Vanegas and Schaible, 2004; Ossipov et al., 2005), including the PAG (Pertovaara et al., 1996, 1997; Monhemius et al., 2001; Pertovaara and Wei, 2003), have pivotal roles in chronic pain and that A- and C-nociceptors have different roles in animal models of hyperalgesia. One school of thought is that increased activity in C-nociceptors arising from inflamed tissue leads to the central sensitization of A-nociceptive inputs that underlies mechanical secondary hyperalgesia (Fuchs et al., 2000; Magerl et al., 2001). As such, alteration of endogenous COX-1 pathways in the VL-PAG that selectively facilitate C-fiber-evoked spinal nociception could potentially contribute to altered spinal excitability and provide an effective target for centrally acting NSAIDs during the transition from acute to chronic pain.
The current findings do not support a role for COX-2 in the PAG in acute nociception. However, COX-2 is induced in higher centers in animal models of inflammatory pain (Samad et al., 2001), and similar induction may also play a role in the PAG, as suggested by the report that nimesulide (a COX-2-selective inhibitor) inhibits neuronal activation in the VL-PAG in a model of nitroglycerin-induced hyperalgesia (Tassorelli et al., 2003).
In conclusion, the current findings suggest that, at the level of the PAG, prostaglandins exert tonic facilitatory control over spinal nociception and thus have the capacity to set the gain of spinal nociception. The finding that COX–prostaglandin-regulated descending control from the PAG preferentially targets C-nociceptor-evoked activity is an observation that will underpin future studies of the roles of C- versus A-nociceptors in chronic pain. We suggest that this tonically active descending control system may have a dynamic role in setting the gain of acute spinal nociceptive processing and that the preferential targeting of C-fiber-evoked nociception will have important consequences in terms of development of more efficacious analgesics.
Footnotes
- Received January 18, 2007.
- Revision received August 23, 2007.
- Accepted August 28, 2007.
-
This work was supported by the Biotechnology and Biological Sciences Research Council United Kingdom [Collaborative Awards in Science and Engineering Studentship with GlaxoSmithKline (J.L.L.)] and the Wellcome Trust.
- Correspondence should be addressed to Dr. Lucy F. Donaldson, Department of Physiology, School of Medical Sciences, University of Bristol, University Walk, Bristol BS8 1TD, UK. lucy.donaldson{at}bristol.ac.uk
- Copyright © 2007 Society for Neuroscience 0270-6474/07/2711296-10$15.00/0