
Abstract
Repeated injections of cocaine and morphine in laboratory rats cause a variety of molecular neuroadaptations in the cAMP signaling pathway in nucleus accumbens and ventral tegmental area. Here we report similar neuroadaptations in postmortem tissue from the brains of human smokers and former smokers. Activity levels of two major components of cAMP signaling, cAMP-dependent protein kinase A (PKA) and adenylate cyclase, were abnormally elevated in nucleus accumbens of smokers and in ventral midbrain dopaminergic region of both smokers and former smokers. Protein levels of the catalytic subunit of PKA were correspondingly higher in the ventral midbrain dopaminergic region of both smokers and former smokers. Protein levels of other candidate neuroadaptations, including glutamate receptor subunits, tyrosine hydroxylase, and other protein kinases, were within normal range. These findings extend our understanding of addiction-related neuroadaptations of cAMP signaling to tobacco smoking in human subjects and suggest that smoking-induced brain neuroadaptations can persist for significant periods in former smokers.
- cAMP
- nicotine
- nucleus accumbens
- addiction
- neuroadaptations
- PKA
Introduction
A major goal of addiction research is to characterize the difference between the addicted and the non-addicted brain. In addition to differences in memories associated with drug experiences and drug-seeking habits (Berke and Hyman, 2000; Wise, 2000), repeated drug exposures induce neuroadaptations that appear to alter subsequent responsiveness to the drug. Current addiction theory remains divided between models that posit drug-opposite neuroadaptations producing tolerance or desensitization to the drug (Koob and Bloom, 1988; Kreek and Koob, 1998; Koob et al., 2004) and models that posit neuroadaptations with the opposite effect: “reverse” tolerance or sensitization to the drug (Lett, 1989; Piazza et al., 1990; Robinson and Berridge, 1993; Carlezon and Nestler, 2002; Vezina et al., 2002; Suto et al., 2004). Both models posit drug-induced neuroadaptive changes in the mesolimbic dopamine system that includes dopamine neurons in the ventral tegmental area (VTA) and their synaptic targets in the forebrain and is thought to mediate the reinforcing effects of drugs of abuse.
Most current hypotheses focus on molecular and cellular changes in the VTA and one of its synaptic targets, the nucleus accumbens (Berke and Hyman, 2000; Nestler, 2001; Wise, 2004). Changes in the cAMP signaling pathway and glutamate receptors (GluRs) within these brain regions are thought to mediate altered neurotransmission that produces altered drug-related behaviors. It has been established in animal models that repeated administration of cocaine or morphine causes several cAMP-related neuroadaptations in these brain regions (Nestler, 1992, 2001) and that experimental manipulations of this signal transduction pathway can change behavioral responses to these drugs (Tolliver et al., 1996, 1999; Carlezon et al., 1998; Self et al., 1998; Bibb et al., 2001; Lynch and Taylor, 2005). Glutamate receptor subunit levels are altered by repeated administration of cocaine (Fitzgerald et al., 1996; Churchill et al., 1999; Lu et al., 2003; Scheggi et al., 2004), and experimental manipulations of these receptors can also change behavioral responses to these drugs (Carlezon et al., 1997; Wolf, 1998; Sutton et al., 2003; Kalivas, 2004).
Nicotine is thought to mediate most of the addictive properties of tobacco smoke (Fowler et al., 1996; Gamberino and Gold, 1999; Quattrocki et al., 2000). Nicotine (Corrigall et al., 1992), like cocaine (De Wit and Wise, 1977) and morphine (Bozarth and Wise, 1981; Laviolette et al., 2002), has reinforcing properties that are mediated at least in part by the mesolimbic dopamine system. Several neuroadaptations have been observed in this system in human smokers. Nicotinic receptors levels are altered (Benwell et al., 1988; Breese et al., 1997a,b, 2000; Court et al., 1998; Perry et al., 1999; Buisson and Bertrand, 2002; Mousavi et al., 2003; Staley et al., 2006), as are dopamine metabolites (Court et al., 1998), D1 dopamine receptors (Dagher et al., 2001), but not dopamine transporters (Staley et al., 2001). However, neuroadaptations in the cAMP signaling system or glutamate receptors of human smokers have not yet been reported. The purpose of the present study was to examine these two systems in postmortem tissue from smokers, former smokers, and nonsmokers.
Materials and Methods
Human postmortem tissue.
The brains of 24 human subjects were obtained from the human brain bank of the University of Colorado Health Sciences Center. Human brains were collected at autopsy after family donation. Hospital and autopsy records were reviewed, and family members and physicians were interviewed to learn the age, sex, race, cause of death, and history of mental illnesses, and cigarette, alcohol, and drug use of the donor. Nicotine and cotinine levels were not assessed. Although alcohol abusers were excluded from the study, some subjects had a history of low to moderate alcohol use. In males, this is defined as 1–14 drinks per week; in females, this is defined as one to seven drinks per week. The brain was weighed, examined for gross pathology, and divided into separate hemispheres. One hemisphere was preserved in Formalin for neuropathological analysis. The other hemisphere was sliced into 1-cm-thick coronal slices, from which regions of interest were dissected into 1 g blocks, frozen in powdered dry ice, packaged for storage, and stored at −80°C.
Subjects were selected to be representative of three different groups. The “smokers” group (n = 8) comprised subjects who died without an abstinent period between regular smoking and death. The “former smokers” group (n = 8) comprised subjects who died after several months or years of abstinence from regular smoking. The “nonsmokers” group (n = 8) comprised subjects who died after a lifetime of not smoking. Subject characteristics are detailed in Table 1.
Clinical history of each subject
Tissue preparation for biochemical analysis.
Nucleus accumbens and ventral midbrain tissue were dissected from the frozen coronal slices. A dark melanin-pigmented strip of tissue was further dissected from ventral midbrain. This strip of tissue contains dopaminergic neurons from substantia nigra compacta and ventral tegmental area (Paxinos and Mai, 2004). Tissue from the nucleus accumbens and ventral midbrain dopaminergic region were homogenized in 50 mm TrisCl, pH 7.5, 1 mm EDTA, and Complete Mini peptidase inhibitors from Roche Applied Sciences (Indianapolis, IN) using a Potter-Elvehjem homogenizer with a motor-driven pestle. A 1 ml aliquot from each homogenized sample was immediately centrifuged at 20,800 × g for 30 min at 4°C. Supernatants were frozen at −80°C until assays for protein kinase A (PKA) activity. The pellets were resuspended in 1 ml of adenylate cyclase (AC) homogenization buffer (20 mm TrisCl, pH 7.4, 1 mm EDTA) and kept on ice for AC assays performed the same day. Another 1 ml aliquot from each initial homogenate was mixed with 10% SDS to a final concentration of 1% SDS and sonicated before freezing at −80°C until analysis by Western blotting. The remainder of the initial homogenates were frozen at −80°C. Protein concentrations of all samples were determined using the bicinchoninic acid assay (BCA) assay of Pierce (Rockford, IL).
Adenylate cyclase assays.
Basal and forskolin-stimulated AC activities were assayed using the same protocol described by Lu et al. (2003). Pellet samples were diluted with AC homogenization buffer to a protein concentration of 0.2 μg/μl, and dithiothreitol (DTT) was added to a final concentration of 1 mm. In a total volume of 100 μl, 25 μl of sample protein were mixed with (final concentrations) 50 mm triethanolamine-HCl, pH 7.4, 0.1 mm EGTA, 2 mm MgCl2, 1 mm DTT, 0.2% bovine serum albumin, 50 μm GTP, and 0, 0.5, or 5 μm forskolin (in 0.05% ethanol vehicle). The assay tubes were incubated for 10 min at 30°C before 5 μl of 1 mm ATP was added to start the reactions that continued for 5 min at 30°C. The assays were terminated by the addition of 0.4 ml of 0.125 m HCl and frozen at −30°C. The next day, the amount of cAMP product in each assay tube was determined using the cAMP ELISA kit (DE-0355) from R & D Systems (Minneapolis, MN) according to the instructions for assaying non-acetylated cAMP. Basal and forskolin-stimulated AC activity in our assays increased linearly with increasing amounts of sample protein (0–10 μg) and assay time (0–10 min).
PKA assays.
PKA activity was assayed using a modification of the protocol described by Lu et al. (2003). All supernatants were diluted to 0.3 μg/μl with the initial homogenization buffer, and DTT was added to a final concentration of 1 mm. Duplicate assays were performed for each sample. In a total volume of 55 μl, 3 μg of sample protein were mixed with PKA assay buffer (final concentrations): 50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 10 mm MgCl2, 0.05% NP-40 (Fluka, Milwaukee, WI), 10 mm DTT, 44 μm kemptide substrate (K-1127; Sigma, St. Louis, MO), 50 μm 32P-labeled ATP (50 μCi/ml; 1 μCi/nmol), and either 1 mm of the PKA activator 8-bromo-cAMP or 0.1 mg/ml of the PKA inhibitor (PKI) (P-0393; Sigma). ATP was added last to begin the kinase assays, which continued for 3 min at 21°C. The assays were terminated by the addition of 100 μl of 100 mm phosphoric acid. One hundred microliters of each reaction sample were transferred to a well plate with P81 phosphocellulose filters (Millipore, Bedford, MA). The samples were drawn through the filters by vacuum, washed three times with 100 mm phosphoric acid, and allowed to dry overnight. Scintillation fluid (200 μl; Biosafe II; Research Products International, Mt. Prospect, IL) was added to each well and counted for 1 min each in a Top Count microplate scintillation counter (PerkinElmer, Boston, MA). To calculate PKA-specific kinase activity in each sample, the value obtained with PKI was subtracted from the value obtained with 8-bromo-cAMP. PKA activity in our assays increased linearly with increasing amounts of sample protein (0–6 μg) and assay time (0–10 min).
Western blotting.
Frozen SDS-treated samples were thawed and further diluted with 1% SDS to equalize the protein concentrations in each sample. Loading buffer (16% glycerol, 20% β-mercaptoethanol, and bromophenol blue) was added to each sample (3:1 sample/loading buffer) before the samples were boiled for 3 min. Samples were allowed to cool and subjected to SDS-PAGE (10% acrylamide/0.27% N,N′-methylenebisacrylamide resolving gel) for 3 h at 150 V. For each electrophoresis run, increasing amounts of protein pooled from the brain region being tested were used to produce a standard curve. Proteins were transferred electrophoretically to Immobilon-P transfer membranes (Millipore) at 0.3 A for 3 h.
Membranes were washed four times for 15 min in blocking buffer composed of 2% nonfat dry milk in PBST (PBS plus 0.05% Tween-20) for tyrosine hydroxylase (TH), cyclin-dependent kinase 5 (cdk5), GluR1, GluR4, NR2A, NR2B, PKA catalytic subunit, extracellular signal-regulated kinase (ERK), calcium/calmodulin-dependent kinase II (CaMKII), and calbindin, with 2% polyvinylpyrollidone (PVP) in PBST for GluR2, NR1, CaMKIV, and cAMP-regulated element binding protein (CREB). Membranes were then incubated overnight at 4°C with primary antibody diluted in their respective blocking buffers plus 0.05% sodium azide. The antibodies used were anti-TH (1:10,000 dilution; Diasorin, Stillwater, MN), anti-cdk5 (1:5000 dilution of sc-173; Santa Cruz Biotechnology, Santa Cruz, CA), anti-GluR1 (1:2000 dilution of catalog #AB1504; Chemicon, Temecula, CA), anti-GluR2 (1:2000 dilution; Chemicon), anti-GluR4 (1:500 dilution; Upstate Biotechnology, Lake Placid, NY), anti-NR1 (1:1000 dilution; BD Biosciences PharMingen, San Diego, CA), anti-NR2A (1:2500 dilution; Chemicon), anti-NR2B (1:2000 dilution; Upstate Biotechnology), anti-PKA catalytic subunit (1:10,000 dilution of catalog #610980; BD Biosciences PharMingen), anti-ERK (1:2000 dilution; Cell Signaling Technology, Beverly, MA), anti-CaMKII (1:10,000 dilution; Upstate Biotechnology), anti-CaMKIV (1:1000 dilution; BD Biosciences PharMingen), anti-CREB (1:2000 dilution of catalog #9192; Cell Signaling Technology), and anti-calbindin (1:20,000 dilution of clone CB-955; Sigma).
A second set of Western blots used different antibodies against GluR1 and CREB to confirm the identity of bands identified with the first set of antibodies. The antibodies used for the second set of blots were anti-GluR1 (1:1000 dilution of sc-7608; Santa Cruz Biotechnology) and anti-CREB (1:2000 dilution of catalog #06-863; Upstate Biotechnology).
After four 15 min washes in blocking buffer, the blots were incubated for 2 h at room temperature with horseradish peroxidase-conjugated secondary antibodies in blocking buffer. Finally, the blots were washed six times for 10 min each time in PBST and developed for 60 s using the enhanced chemiluminescence (ECL) procedure of Amersham Biosciences. Luminescence from the blots was detected using Amersham Biosciences ECL Hyperfilm followed by digital scanning in transparency mode. Band intensities were quantified using Quantity One software (version 4.0.3) from Bio-Rad (Hercules, CA). Band intensities from each test sample were compared with the band intensities from the standard curve samples. Standard curves run with all Western blots in our assays indicated that the amount of protein loaded for the test samples were within the linear range of detection.
Data analysis.
Data from the PKA activity assays and Western blotting assays were analyzed using one-way ANOVAs. Data from adenylate cyclase assays were analyzed using two-way ANOVAs using smoking group and forskolin concentration (0, 0.5, and 5 μm) as the two factors. In some cases, data were analyzed using ANOVA using alcohol user as the main factor. The values used for statistical analyses were PKA-specific levels of phosphorylated kemptide product for PKA activity assays, levels of cAMP product for adenylate cyclase assays, and optical densities for Western blotting results. Values more than 2 SDs from the mean were considered outliers and removed from analysis. Fisher's PLSD test was used for all post hoc analyses, and significant differences are reported for p < 0.05.
Results
Adenylate cyclase enzyme activity
In nucleus accumbens, adenylate cyclase activity levels were significantly different among smoker, former smoker, and nonsmoker groups (Fig. 1A) (two-way ANOVA, effect of smoking group, F(2,40) = 3.79, p < 0.05; effect of forskolin F(2,40) = 47.3, p < 0.0001; and interaction of smoking group × forskolin, F(4,40) = 2.84, p < 0.05). Post hoc comparisons indicate that forskolin-stimulated activity in smokers was significantly higher than activity in former smokers and nonsmokers. Basal activity levels, assayed with 0 μm forskolin, were not different between groups. Average levels of basal AC activity were ∼40 pmol · min−1 · mg−1 protein for all three smoking groups. The addition of 5 μm forskolin to the assay stimulated AC activity ∼2.2- and 2.8-fold in the nucleus accumbens of nonsmokers and smokers, respectively.

AC enzyme activity was increased in nucleus accumbens tissue from smokers and in ventral midbrain dopaminergic region tissue from smokers and former smokers. Activities were assayed in the presence of 0, 0.5, and 5 mm forskolin. Enzyme activity values are expressed as picomoles of cAMP produced per minute per milligram of protein in the assay (mean ± SEM; n = 8 per group). *p < 0.05, significantly higher levels of enzyme activity compared with levels in tissue obtained from nonsmokers.
In the ventral midbrain dopaminergic region, adenylate cyclase activity levels were significantly different among smoker, former smoker, and nonsmoker groups (Fig. 1B) (two-way ANOVA, effect of smoking group, F(2,38) = 4.72, p < 0.05; effect of forskolin F(2,38) = 21.8, p < 0.0001; and interaction of smoking group × forskolin, F(4,38) = 1.1, p = 0.35). Post hoc comparisons indicate that forskolin-stimulated and basal activity levels in smokers and former smokers were significantly higher than activity levels in nonsmokers. Activity levels in former smokers were approximately the same as activity levels in smokers. Average levels of basal AC activity were ∼25 and 50 pmol · min−1 · mg−1 protein for nonsmoker and smoker groups, respectively. The addition of 5 μm forskolin to the assay stimulated AC activity ∼1.8- and 1.4-fold in the ventral midbrain dopaminergic region of nonsmokers and smokers, respectively.
Adenylate cyclase levels in both brain regions were not different among low to moderate alcohol users and non-users. For both brain regions, differences among groups remained statistically significant even after removal of the one former smoker subject who had a schizophrenic episode. For nucleus accumbens, differences among groups remained statistically significant after removal of the four subjects with a history of depression or suicide. For the ventral midbrain dopaminergic region, removal of these four subjects reduced group size and increased variability so that ANOVA no longer indicated significant differences. Mean activity levels with 0 and 5 μm forskolin in the assay were essentially unaltered; only the mean activity levels with 0.5 μm forskolin appeared significantly altered.
PKA enzyme activity
In nucleus accumbens, PKA activity levels were significantly different among smoker, former smoker, and nonsmoker groups (Fig. 2A) (one-way ANOVA, F(2,20) = 4.19, p < 0.05). Post hoc comparisons indicate that activity levels in smokers were twofold higher than activity levels in nonsmokers. PKA activity levels were ∼80 pmol · min−1 · mg−1 protein in nonsmokers.

cAMP-dependent protein kinase (PKA) enzyme activity was increased in nucleus accumbens tissue from smokers and in ventral midbrain dopaminergic region tissue from smokers and former smokers. Enzyme activity values are expressed as picomoles of phosphate transferred to the kemptide substrate per minute per milligram of protein in the assay (mean ± SEM; n = 8 per group). *p < 0.05, significantly higher levels of enzyme activity compared with levels in tissue obtained from nonsmokers.
In the ventral midbrain dopaminergic region, PKA activity levels were significantly different among smoker, former smoker, and nonsmoker groups (Fig. 2B) (one-way ANOVA, F(2,21) = 3.59, p < 0.05). Post hoc comparisons indicate that activity levels in smokers and former smokers were 1.6-fold higher than activity levels in nonsmokers; activity levels in former smokers were approximately the same as activity levels in smokers. PKA activity levels were ∼220 pmol · min−1 · mg−1 protein in nonsmokers.
In the nucleus accumbens, PKA activity levels were not different among low to moderate alcohol users and non-users. In the ventral midbrain dopaminergic region, low to moderate alcohol users had 1.4-fold higher activity levels than non-users (one-way ANOVA, F(1,22) = 5.14, p < 0.05). For both brain regions, differences among groups remained statistically significant even after removal of the one former smoker subject who had a schizophrenic episode. In nucleus accumbens, differences among groups remained statistically significant after removal of the four subjects with a history of depression or suicide. In the ventral midbrain dopaminergic region, removal of these four subjects reduced group size and increased variability so that ANOVA no longer indicated significant differences among groups; however, mean activity levels appeared essentially unaltered.
PKA catalytic subunit levels
In nucleus accumbens, PKA catalytic subunit levels were not significantly different among groups (Fig. 3A). In the ventral midbrain dopaminergic region, PKA catalytic subunit levels were significantly different among smoker, former smoker, and nonsmoker groups (Fig. 3B) (one-way ANOVA, F(2,21) = 4.33, p < 0.05). Post hoc comparisons indicate that levels in smokers and former smokers were 1.6- and 1.4-fold higher than levels in nonsmokers. Immunoblotting for PKA catalytic subunits produced one strong band with a molecular weight of ∼42 kDa (Fig. 3, inset). Low to moderate alcohol users had 1.3-fold higher activity levels than non-users (one-way ANOVA, F(1,22) = 4.85, p < 0.05).

Western blotting indicates that levels of the catalytic subunit for PKA were increased in nucleus accumbens tissue from smokers and in ventral midbrain dopaminergic region tissue from smokers and former smokers. All values are expressed as a percentage of the mean values from the nonsmokers group for each protein (mean ± SEM; n = 8 per group). *p < 0.05, significantly higher levels of catalytic subunit protein compared with levels in tissue obtained from nonsmokers. Inset shows representative Western blots of PKA catalytic subunits from nucleus accumbens and ventral midbrain dopaminergic regions of nonsmokers, former smokers, and smokers.
In the nucleus accumbens, catalytic subunit levels were not different among low to moderate alcohol users and non-users. In the ventral midbrain dopaminergic region, low to moderate alcohol users had 1.3-fold higher activity levels than non-users (one-way ANOVA, F(1,22) = 4.85, p < 0.05). In the ventral midbrain dopaminergic region, differences among groups remained statistically significant even after removal of the one former smoker subject who had a schizophrenic episode. In ventral midbrain dopaminergic region, removal of the four subjects with a history of depression or suicide reduced group size and increased variability so that ANOVA no longer indicated significant differences among groups. However, mean levels of catalytic subunits appeared essentially unaltered, and a t test still indicated a significant difference between nonsmokers and smokers groups.
Protein levels of candidate neuroadaptations
Protein levels of glutamate receptor subunits in either region did not differ among groups (Tables 2, 3). Western blots of AMPA glutamate receptor subunits are shown in Figure 4A. GluR1 Western blots of nucleus accumbens tissue labeled a band of ∼105 kDa, similar to that found in rat brain (Lu et al., 2003; Mattson et al., 2005). This 105 kDa band was weak and only variably present in Western blots of ventral midbrain dopaminergic tissue. The difficulty in detecting GluR1 subunits in the ventral midbrain dopaminergic region may be attributable to posttranslational modifications of GluR1 that make it poorly recognized by the Chemicon antibody against the C-terminus region of GluR1. However, immunoblotting with an antibody from Santa Cruz Biotechnology that recognizes a region near the N terminus of GluR1 again labeled a 105 kDa band in nucleus accumbens without significant labeling in the ventral midbrain dopaminergic region (Fig. 4B), which suggests that GluR1 may instead not be present at significant levels in this brain region. GluR2 Western blots labeled a doublet of ∼100 and 110 kDa. GluR4 Western blots labeled a single band of ∼130 kDa. Western blots of NMDA glutamate receptor subunits are shown in Figure 4C. NR1 Western blots labeled major and minor bands of ∼140 and 120 kDa. Band intensities in NR2A Western blots were too weak to quantify. NR2B Western blots labeled a doublet of ∼170 and 180 kDa.
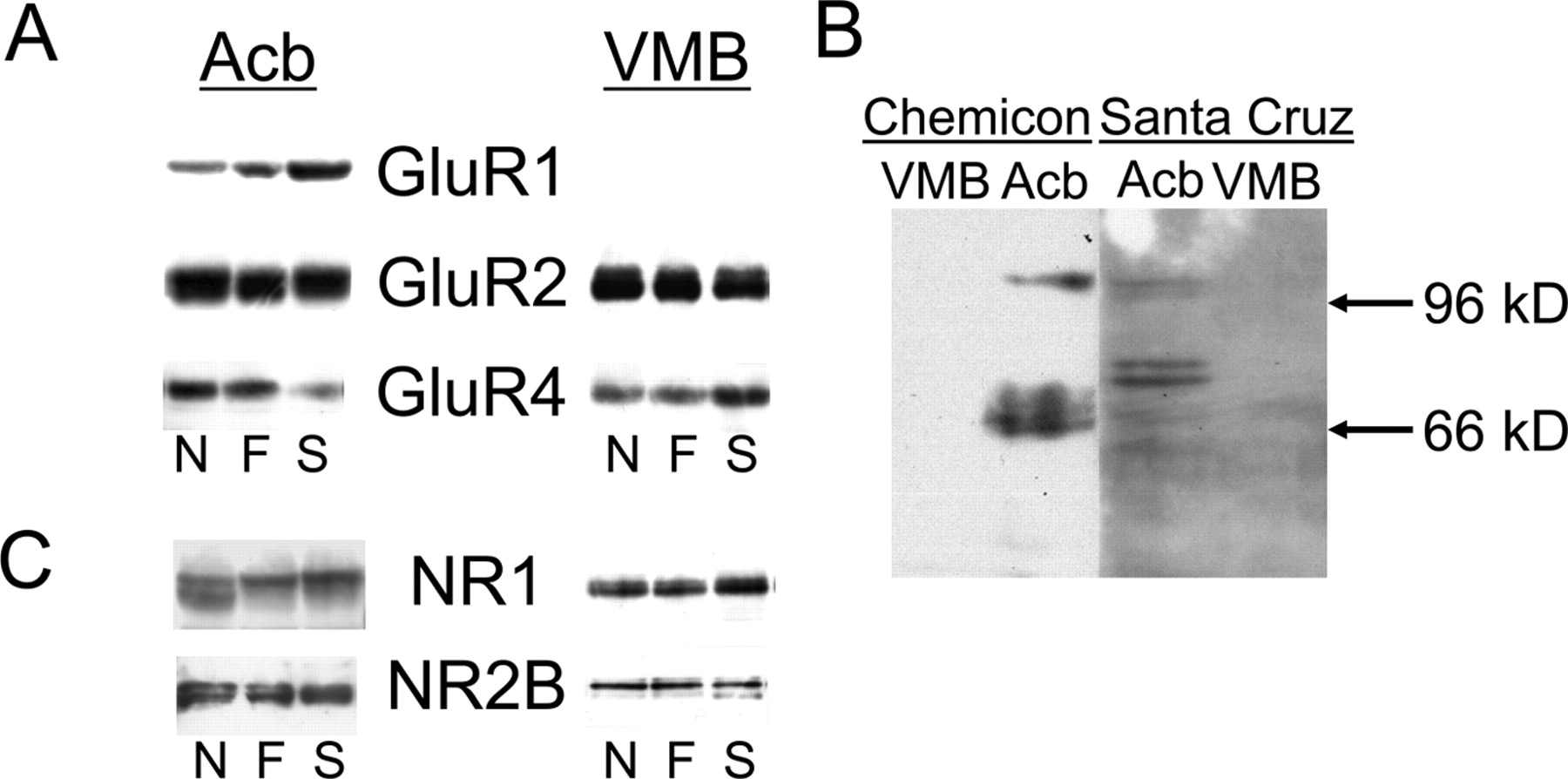
Representative Western blots of glutamate receptors in tissue from nucleus accumbens (Acb) and ventral midbrain dopaminergic region (VMB) of nonsmokers (N), former smokers (F), and smokers (S). A, Western blots of AMPA glutamate receptor subunits (GluR1, GluR2, GluR4). B, Comparison of GluR1 Western blots using antibodies from Chemicon and Santa Cruz Biotechnology. C, Western blots of NMDA glutamate receptor subunits (NR1, NR2A). Labeling of NMDA receptor subunit NR2B was too weak to quantify. Each lane represents a sample from an individual subject.
Results of Western blot assays of tissue from nucleus accumbens
Results of Western blot assays of tissue from ventral midbrain dopaminergic region
Protein levels of calcium/calmodulin-dependent kinases (CaMKII and CaMKIV), ERK (and mitogen-activated protein kinase), and cdk5 were not significantly different among groups (Tables 2, 3). Western blots of CaMKII-, CaMKIV-, and cdk5-labeled single bands of ∼50, 60, and 32 kDa (Fig. 5), similar to the molecular weights we observed in rat brain (Hope et al., 2005; Mattson et al., 2005). Western blots of ERK labeled two bands of ∼42 and 44 kDa (Fig. 5), similar to the molecular weights we found in rat brain (Lu et al., 2003; Mattson et al., 2005).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative Western blots of candidate neuroadaptations in tissue from nucleus accumbens (Acb) and ventral midbrain dopaminergic region (VMB) of nonsmokers (N), former smokers (F), and smokers (S). Each lane represents a sample from an individual subject.
Protein levels of TH, CREB, and calbindin were not significantly different among groups (Tables 2, 3). Western blots of TH labeled a strong doublet of ∼58 kDa as well as minor bands at 54 and 62 kDa (Fig. 5), which likely correspond to the four different isoforms of TH previously observed in human brain (Lewis et al., 1993). Immunoblotting for CREB with the Upstate Biotechnologies antibody that recognizes amino acids 5–24 in the N-terminal region of CREB labeled a band of ∼44 kDa (Fig. 5), similar to the molecular weight observed previously in human brain (Yamamoto et al., 2001b). Immunoblotting with the Cell Signaling Technology antibody that recognizes the middle region surrounding Ser-133 labeled a 44 band with the same molecular weight as that shown with the Upstate Biotechnologies antibody (data not shown), but the Cell Signaling Technology antibody also labeled another strong band of ∼46 kDa. Western blots of calbindin labeled major and minor bands of ∼28 and 30 kDa (Fig. 5), similar to the molecular weights observed previously in rat and human brains (Parmentier et al., 1987).
Discussion
The present study confirms that drug-induced neuroadaptations observed in animal models can also be observed in humans. It also extends to tobacco smoking, the observation that repeated treatment with addictive drugs causes neuroadaptations in the mesolimbic dopamine system and its target neurons in nucleus accumbens. Because three of our smokers had not smoked for 25 years or more, these data suggest that such neuroadaptations are very long lasting.
The cAMP signal transduction pathway is elevated in the ventral midbrain dopaminergic region of smokers and former smokers years after cessation of smoking. Levels for two different components of this pathway, adenylate cyclase and PKA, are higher in this brain region in both smokers and former smokers. Levels of adenylate cyclase and PKA are also higher in nucleus accumbens of smokers. Thus, different assays for components of the cAMP signal transduction pathway all indicate that smoking increases levels of cAMP signal transduction components in the mesolimbic dopamine system. Because we did not observe alterations in the levels of glutamate receptor subunits or other candidate neuroadaptations, including components of other signal transduction pathways, it appears that smoking induces alterations specific to cAMP signal transduction.
It is possible, however, that different levels of adenylate cyclase and PKA between groups in our study were not attributable to smoking but were attributable instead to preexisting genetic differences or differences in life experiences such as stress (Gordon, 2002) that may be more common in people who smoke. Animal studies indicate that differences in adenylate cyclase and PKA levels can be present before drug exposure and correlate with differences in drug-associated behaviors (Nestler, 2001). However, although levels of adenylate cyclase and PKA activity in nucleus accumbens of smokers were higher than nonsmokers, levels in nucleus accumbens of former smokers were not different from that in nonsmokers, which suggests that higher levels of these molecules in smokers return to normal levels over time rather than being constitutively higher in people who smoke.
None of the subjects in this study were alcohol abusers; however, there were more low to moderate alcohol users in the smokers group than in the former smokers or nonsmokers groups. These alcohol users had higher levels of PKA activity and catalytic subunit protein in the ventral midbrain dopaminergic region. Thus, at first it appears that low to moderate alcohol use could have contributed to higher levels of these molecules in the ventral midbrain dopaminergic region of the smokers group. However, this seems unlikely because higher levels of these molecules were also observed in the former smokers group, which had approximately the same number of alcohol users as the nonsmokers group. Nevertheless, it would be interesting to test for an interaction between low-level alcohol use and smoking effects on adenylate cyclase and PKA activity levels.
Four of our subjects in the former smoker and smoker groups had a history of depression or suicide. It is highly unlikely that these subjects contributed to our “increased” adenylate cyclase activity levels because all studies have consistently observed “decreased” activity levels in brains from depressed and suicide subjects (Abou-Saleh et al., 1994; Cowburn et al., 1994; Lowther et al., 1996; Dowlatshahi et al., 1999; Reiach et al., 1999; Stewart et al., 2001; Valdizan et al., 2003). As well, PKA activity or protein levels were decreased or not altered in cortex and striatum from depressed and suicide subjects (Odagaki et al., 2001; Dwivedi et al., 2002, 2004; Hsiung et al., 2003; Pandey et al., 2005) and are thus highly unlikely to have contributed to increased activity levels in our study. Most importantly adenylate cyclase and PKA activity levels in nucleus accumbens were not significantly altered by removal of these four subjects from analyses. Even in the ventral midbrain dopaminergic region, mean activity levels were essentially unaltered by removal of these four subjects from analyses.
One of our subjects in the former smoker group had a schizophrenic episode. First, he could not have contributed to increased adenylate cyclase and PKA activity levels in the smokers group. Second, although D1 dopamine receptor “coupling” to adenylate cyclase was increased in schizophrenic subjects (Memo et al., 1983), basal activity levels of adenylate cyclase itself were not altered (Carenzi et al., 1975; Memo et al., 1983). Most importantly, adenylate cyclase and PKA activity levels were not altered by removal of this subject from analyses.
Although alterations within the cAMP signal transduction pathway have not been previously examined in the brains of smokers, they have been previously detected in users of other addictive drugs. In heroin addicts, levels of several isoforms of adenylate cyclase protein and enzyme activity are lower in cortex but unaltered in nucleus accumbens (Shichinohe et al., 1998, 2001). In alcoholics, levels of various isoforms of adenylate cyclase are also lower in cortex, whereas levels are variably reported to be elevated or unaltered in nucleus accumbens and amygdala (Hashimoto et al., 1998; Sohma et al., 1999; Yamamoto et al., 2001a). In methamphetamine users, dopamine-stimulated adenylate cyclase activity is less in nucleus accumbens, caudate, and putamen (Tong et al., 2003). Levels of G-proteins capable of inhibiting adenylate cyclase activity are less in nucleus accumbens and cortex of heroin addicts, alcoholics, and methamphetamine users (Saito et al., 1994; McLeman et al., 2000). Thus, across several classes of addictive drug users, cAMP signal transduction levels are less in nucleus accumbens and cortex, whereas cAMP signal transduction levels are higher in nucleus accumbens and ventral midbrain dopaminergic region of smokers in our study.
Higher levels of cAMP signal transduction activity in smokers, however, correspond better with the effects of nicotine and other addictive drugs on cAMP signal transduction in animal studies (Nestler, 2001). Chronic nicotine treatments tend to increase cAMP signal transduction in rat brain. Continuous and intermittent nicotine administration and environmental tobacco exposure increase adenylate cyclase activity in the brains of young rats, including the striatum (Slotkin et al., 2001; Abreu-Villaca et al., 2003). However, chronic nicotine treatment did not alter adenylate cyclase activity levels in the caudate or nucleus accumbens of adult rats (Tzavara et al., 2002). These results suggest that young rats can be used in future studies as animal models of human smokers to examine a causal role for upregulation of cAMP signal transduction on nicotine-related behaviors.
The lack of studies on the role of cAMP signal transduction in nicotine-related behaviors forces us further to consider studies with other addictive drugs. Chronic treatment with other addictive drugs also upregulates cAMP signal transduction (for review, see Nestler, 2001). Levels of adenylate cyclase and PKA activity are increased in nucleus accumbens, but not ventral tegmental area, after chronic administration of morphine (Parenti et al., 1982; Terwilliger et al., 1991; Schoffelmeer et al., 1996), heroin (Self et al., 1995), cocaine (Terwilliger et al., 1991; Izenwasser et al., 1996; Lu et al., 2003; Hope et al., 2005), and ethanol (Ortiz et al., 1995). Although levels of adenylate cyclase and PKA were not altered in the VTA after chronic administration of these drugs, altered levels of another component of cAMP signal transduction has been observed. Chronic cocaine treatment reduces the level and function of the G-protein Gi that inhibits adenylate cyclase activity in the ventral tegmental area (Nestler et al., 1990). Because the G-protein Gs, which activates adenylate cyclase activity, is not altered in this brain region, the expected net effect of decreasing Gi is to increase adenylate cyclase activity levels. Overall, chronic treatment with morphine, heroin, cocaine, or ethanol increases cAMP signal transduction similar to that seen with chronic nicotine in young rats and in human smokers. In addition, the locomotor activating and rewarding effects of these drugs and nicotine are mediated in large part by the same mesolimbic dopamine system. Thus, animal models that use morphine, heroin, cocaine, or ethanol might be used to infer the behavioral effects of upregulation of cAMP signal transduction on nicotine-related behaviors in human smokers.
Overall, the cAMP signal transduction pathway is greater in nucleus accumbens of smokers and in the ventral midbrain dopaminergic region nucleus of smokers and former smokers long after they have ceased smoking. Similar increases in cAMP signal transduction have been observed in animal models after repeated treatment with other addictive drugs such as morphine, heroin, cocaine, and ethanol and are thought to contribute to alterations in the rewarding and locomotor activating effects of these drugs (Self et al., 1998; Nestler, 2001; Beninger and Gerdjikov, 2004). This implies that an enhanced cAMP signal transduction pathway in human smokers and former smokers may contribute to long-lasting alterations in nicotine-induced reward and addiction in humans.
Footnotes
- Received August 23, 2006.
- Revision received January 12, 2007.
- Accepted January 18, 2007.
-
This work was supported by the Intramural Research Program of the National Institutes of Health/National Institute on Drug Abuse.
- Correspondence should be addressed to Dr. Bruce T. Hope, The National Institute on Drug Abuse, Behavioral Neuroscience Branch, 5500 Nathan Shock Drive, Building C, Baltimore, MD 21224. bhope{at}intra.nida.nih.gov
- Copyright © 2007 Society for Neuroscience 0270-6474/07/271964-09$15.00/0