
Abstract
Auditory perception is mediated through a finite number of mechanosensory hair cells located in a specialized sensory epithelium within the inner ear. The formation of the appropriate number of hair cells and the location of those cells is crucial for normal auditory function. However, the factors that regulate the formation of this epithelium remain poorly understood. Truncating mutations in the transcription factor GLI3, a downstream effector of the Hedgehog (HH) pathway, lead to a partial loss of HH signaling and cause Pallister-Hall syndrome (PHS). Here, we report that cochleae from a mouse model of PHS (Gli3Δ699), which produces only the truncated, repressor form of GLI3, have a variably penetrant phenotype that includes an increase in the size of the sensory epithelium and the development of large ectopic sensory patches in Kölliker's organ (KO). Consistent with the mouse model, some PHS individuals exhibit hearing loss across a broad range of frequencies. Moreover, inhibition of HH signaling in vitro results in an increase in the size of the prosensory domain, a precursor population that gives rise to the sensory epithelium, whereas treatment with Sonic hedgehog (SHH) inhibits prosensory formation. Finally, we demonstrate that HH signaling within the cochlea regulates expression of prosensory markers and that the effects of HH in KO are dependent on activation of Notch, an inducer of prosensory fate. These results suggest that HH signaling plays a key role in the specification, size, and location of the prosensory domain, and therefore of hair cells, within the cochlea.
- cochlea
- hair cell
- development
- Pallister-Hall syndrome
- hedgehog
- cell fate
Introduction
The mammalian auditory sensory epithelium, referred to as the organ of Corti (OC), comprises a narrow stripe of cells that extends along the length of the cochlear spiral. The OC consists of mechanosensory hair cells and associated supporting cells arranged in a regular cellular mosaic. All of the cells that comprise the OC are believed to develop from a prosensory domain, a pool of progenitor cells that is normally restricted to the lateral half of the cochlear duct. Although the development of the elaborate cellular pattern within the OC depends on signaling pathways such as Notch (Lanford et al., 1999; Kiernan et al., 2005a) and fibroblast growth factor (Colvin et al., 1996; Mueller et al., 2002; Pirvola et al., 2002), it is unclear how the position and size of the prosensory domain is initially defined.
The highly conserved Hedgehog (HH) signaling pathway plays a role in the development of virtually all vertebrate organ systems. A secreted hedgehog protein, such as Sonic hedgehog (SHH), binds to its receptor, Patched1 (PTCH1), leading to activation of the transcription factors GLI1, GLI2, and GLI3 (for review, see Jacob and Briscoe, 2003; Hooper and Scott, 2005; Huangfu and Anderson, 2006; Wang et al., 2007). GLI1 acts only as a transcriptional activator, but GLI2 and GLI3 can be proteolyzed to truncated, repressor forms from their full-length, activator states (Dai et al., 1999; Sasaki et al., 1999). However, the ability of Gli1 to compensate for loss of Gli2 suggests GLI2 normally acts as an activator (Bai and Joyner, 2001). GLI3, in contrast, appears to be bifunctional. In the absence of SHH, GLI3 exists in a truncated form that represses SHH targets; when SHH is present, GLI3 is found primarily in the full-length activator form (Dai et al., 1999; Wang et al., 2000). Mutations in GLI3 cause distinct human pleiotropic malformation syndromes such as Grieg cephalopolysyndactyly (GCPS) and Pallister-Hall syndrome (PHS) (Vortkamp et al., 1991; Kang et al., 1997). These two syndromes correlate strongly with the type and position of the GLI3 gene mutation (Johnston et al., 2005). PHS is caused by truncating mutations downstream of the DNA-binding domain, likely resulting in constitutive GLI3 repressor activity and a partial loss of HH function.
The role of hedgehog signaling in the development of the OC is unknown. Shh expression in the ventral midline of the midgestation embryo is required for morphogenesis of the cochlear duct (Riccomagno et al., 2002), and Shh interacts with dorsal Wnt signaling to initially pattern the cochlear and vestibular compartments of the inner ear (Riccomagno et al., 2005; Bok et al., 2007). However, the complete absence of the cochlea in Shh mutants has precluded an analysis of the role of HH signaling in development of the OC.
In this study, we took advantage of the partial loss of HH signaling in GLI3/Gli3 truncating mutants along with in vitro HH modulation to demonstrate that HH signaling plays a key role in the size and position of the auditory prosensory domain and in overall auditory function.
Materials and Methods
Mice.
Mice carrying the Gli3Δ699 allele were maintained on either a C57BL/6 or C3H/He background and genotyped as previously described (Böse et al., 2002). No difference in the cochlear phenotype was observed between backgrounds. We obtained mutant and littermate control cochleae from timed pregnancies between embryonic day 13 (E13) and postnatal day 0 (P0). Gli3Δ699 mice were kept according to guidelines stated in either the German animal protection act or the National Institutes of Health Animal Care and Use Committee. Pregnant ICR or C57BL/6 mice used to obtain embryonic cochleae for explants or in situ hybridization expression analysis were from Charles River Laboratories or Harlan; Atoh1LacZ/+ embryonic cochleae were obtained from crosses of Atoh1LacZ/+ and ICR mice, as described previously (Bermingham et al., 1999; Woods et al., 2004). All mice were maintained per the National Institutes of Health Animal Care and Use Committee.
Cochlear explant cultures.
Pregnant ICR or C57BL/6 mice were killed on E13.5, according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and cultured as described previously (Montcouquiol and Kelley, 2003). All explants were maintained in culture for 6 d unless otherwise noted. SHH and Hedgehog-interacting protein (HIP) were obtained from R&D Systems, and 3-keto-N-(aminoethyl-aminocaproyl-dihydrocinnamoyl)cyclopamine (KAAD-cyclopamine) and N-[N-(3,5-difluorophenacetyl)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT) from Calbiochem.
Immunohistochemistry.
Whole-mount cochleae or cochlear explant cultures were fixed in 4% paraformaldehyde before immunostaining. Antibodies used were as follows: anti-Myosin VI (MyoVI; 1:2000; Proteus Biosciences), anti-Sox2 and anti-p75 neurotrophin receptor (p75NTR); 1:1000; Millipore), anti-neurofilament (2H3; 1:200; Developmental Studies Hybridoma Bank), anti-Jagged1 (Jag1; 1:300; Santa Cruz Biotechnology), anti-βgal (1:250; Promega), and anti-p27kip1 (1:100; Lab Vision). All secondary antibodies (1:1000) and labeled phalloidin (1:200) were from Invitrogen. Cell death was assayed using the Live/Dead Viability/Cytotoxicity Kit from Invitrogen. Proliferating cells were identified using anti-phospho-histone H3 antibody (Millipore).
Human audiometry.
We reviewed the medical records of a previously reported cohort of subjects with GLI3 mutant phenotypes (Johnston et al., 2005) for the results of previous audiological evaluations. The evaluations had not been performed prospectively. They included pure-tone and speech audiometry, and middle ear immittance testing in sound-treated booths meeting American National Standards Institute standards. Air- and bone-conduction response thresholds were used to determine the type and degree of hearing loss (Szymko-Bennett et al., 2001). These studies were performed under a protocol reviewed and approved by the National Human Genome Research Institute Institutional Review Board.
Gene expression analysis.
For in situ hybridization, inner ears were dissected from ICR embryos individually staged according to the developmental series presented by Kaufman (1992). Cochleae were cryoprotected in sucrose and OCT and sectioned at 12 μm. Digoxigenin-labeled RNA probes were generated from published plasmids, and in situ hybridization was performed according to standard protocols (Murtaugh et al., 1999). β-Galactosidase activity from the Atoh1-LacZ reporter gene was detected by using standard techniques (Hogan, 1994), substituting 4% paraformaldehyde for glutaraldehyde in fixation solutions.
For quantitative PCR, cochlear epithelia were isolated from E14 Gli3Δ699 mutants and wild-type littermates as described previously (Montcouquiol and Kelley, 2003). Total RNA was isolated using the PicoPure RNA Isolation Kit from Arcturus and treated with DNase 1 (Qiagen). cDNA was synthesized using random hexamers and the Superscript III kit (Invitrogen). Real-time PCR was conducted using the SYBR Green PCR Master Mix (Applied BioSystems), gene-specific primers (sequences available on request), and the ABI Prism 7000 system. All reactions were performed in triplicate. Relative levels of expression were determined using the comparative CT method; 36B4 mRNA was used as the invariant control (Yabe et al., 2003).
Statistical analysis.
Significant differences in cochlear lengths, number of hair cells, and levels of gene expression were determined by one-tailed t test. Ectopic hair cells were defined as MyoVI-positive cells more than three cell diameters from the endogenous sensory epithelium. For KAAD-cyclopamine and HIP experiments, treated cochleae were paired with control cochleae from the same embryo. Data are represented as SEM.
Results
Gli3Δ699/Δ699 cochleae are short and contain additional and ectopic hair cells
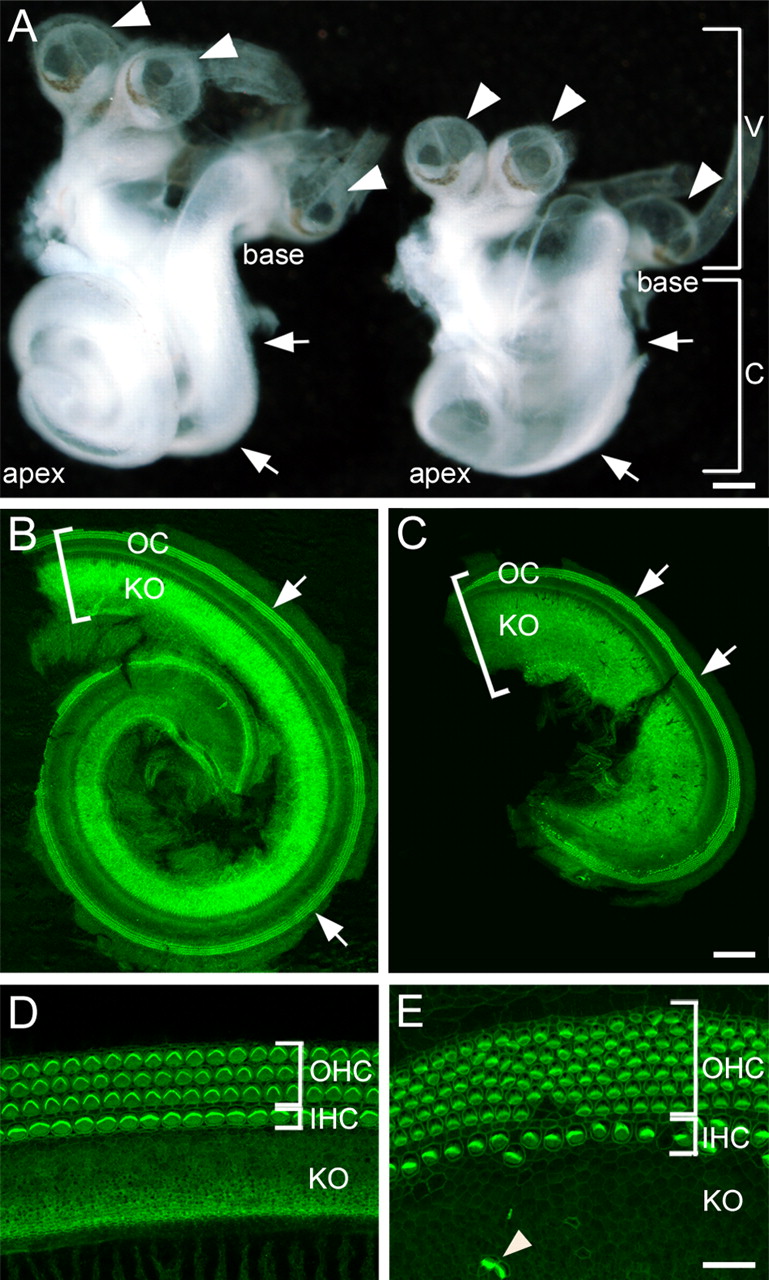
To investigate the role of HH signaling in cochlear development, we examined inner ears from Gli3Δ699 mice, a model for PHS (Böse et al., 2002). Gross examination of Gli3Δ699/Δ699 inner ears at P0 indicated that the vestibular portion appeared normal in size and morphology; phalloidin staining of the vestibular sensory patches also showed a normal distribution and appearance of hair cells (data not shown). In contrast, the cochleae of Gli3Δ699/Δ699 mutants were broader and significantly shorter relative to wild-type or heterozygous littermates (Fig. 1A–C), consistent with the results of Bok et al. (2007). The average length of Gli3Δ699/Δ699 cochleae at P0 was 2.5 ± 0.2 mm, approximately half that of unaffected littermates (4.9 ± 0.2 mm; p < 0.0005).

Cochlear development is disrupted in Gli3Δ699/Δ699 mutants. A, Isolated membranous labyrinths from Gli3Δ699/+ (left) and Gli3Δ699/Δ699 (right) littermates at P0. The vestibular morphology, including the development of the three semicircular canal cristae (arrowheads) appears normal in both, but the cochlea (arrows) is significantly shorter and appears wider in the mutant. Scale bar, 250 μm. B, Dissected Gli3Δ699/+ cochlear duct from A stained with phalloidin (green) to mark filamentous actin. The basal end of the cochlear duct is located in the top left-hand corner of the image. Hair cell stereociliary bundles in the OC (arrows) are intensely stained with phalloidin. No hair cells are present in KO. The bracket indicates the width of the cochlear duct. C, Gli3Δ699/Δ699 cochlea from A stained and oriented as in B. By comparison with the control cochlea, the duct is noticeably shorter and wider (bracket indicates width). Stereociliary bundles are present within the OC (arrows). D, High-magnification view of the OC and adjacent KO from a Gli3Δ699/+ cochlea. The organ of Corti is comprised of one row of IHCs and three rows of OHCs. No hair cells are present in KO. E, Comparable view as in D, from a Gli3Δ699/Δ699 mutant. The OC is disorganized and as many as seven rows of OHCs are present. Ectopic hair cells are also visible in KO (arrowhead). Scale bars: (in C) B, C, 100 μm; (in E) D, E, 20 μm. V, Vestibular portion; C, cochlea.
At the cellular level, phalloidin staining of F-actin in the cochlear duct revealed additional defects in Gli3Δ699/Δ699 mutants. Cochleae from unaffected littermates contained the normal complement of one row of inner hair cells (IHCs) and three rows of outer hair cells (OHCs) (Fig. 1D). In contrast, we observed at least one extra row of OHC in all mutant cochleae, with as many as seven rows of OHCs in severely affected mutants (4 of 16 Gli3Δ699/Δ699 cochleae examined at E18.5 or P0). Moreover, the highly ordered arrangement of cells was disorganized in all Gli3Δ699/Δ699 mutants examined (Fig. 1E). Finally, mutant cochleae also contained patches of ectopic hair cells in Kölliker's organ, a transient embryonic structure located medial to the sensory epithelium, which normally contains only nonsensory epithelial cells (Figs. 1E, 2A,B, arrowheads). The size and position of the ectopic patches of hair cells varied among mutants, but were present in 14 of 16 cochleae. In addition, ectopic hair cells were surrounded by cells positive for Jag1, a marker of supporting/prosensory cells (Fig. 2C–E). These results indicate that ectopic patches contain the same cell types that are normally observed within the sensory epithelium.

Ectopic, vestibular-like hair cells are present in Gli3Δ699/Δ699 cochleae. A, Phalloidin-stained E18.5 Gli3Δ699/Δ699 cochlea illustrating the normal rows of endogenous HCs (arrow) and patches of ectopic HCs within KO (arrowheads). B, High-magnification view of the ectopic indicated by the green arrowhead in A. Stereociliary bundles of ectopic HCs are long and wavy (arrowheads) and appear similar to stereociliary bundles found in vestibular HCs. C, MyoVI staining of E16.5 Gli3Δ699/Δ699 endogenous (labeled IHC) and ectopic HCs (arrow). D, Same region as in C. Jag1 labels supporting cells adjacent to MyoVI-positive HCs in both the sensory epithelium (IHC) and ectopic hair cell patch (arrow). E, Merge of C and D. F, Phalloidin staining of an ectopic hair cell patch. G, Anti-neurofilament (2H3) staining of the same patch as in F, illustrating large, spherical neuronal terminals within the patch. H, Merge of F and G. Each neuronal terminal encloses an ectopic hair cell. Arrows in F–H indicate two HCs clearly surrounded by 2H3 staining. I, Z-stack confocal reconstruction illustrates the calyceal-shaped neurofilament staining around an ectopic HC (arrow). J, No calices are present surrounding endogenous HCs within the organ of Corti of the same cochlea as in I. Scale bars: A, 50 μm; B, E (for C–E), H (for F–H), 10 μm; (in I) I, J, 5 μm.
Ectopic hair cells in Gli3Δ699/Δ699 mutants have vestibular characteristics
Additional examination of the patches of ectopic HCs located in Kölliker's organ in Gli3Δ699/Δ699 mutants indicated that the structure of the stereociliary bundles on these HCs appeared strikingly different from that of normal cochlear HCs. In contrast to the normal V- or W-shaped bundles of cochlear HCs, those found on the ectopic HCs were long and wispy, reminiscent of the bundles found on HCs in the sensory patches of the vestibular system (Fig. 2B, supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To determine whether these HCs had other vestibular characteristics in addition to their bundle morphology, we examined the morphology of afferent synapses. In contrast to cochlear HC synapses, vestibular type I hair cells are surrounded by a calyceal synapse (Wersall, 1956). From the surface, antineurofilament staining of neurons surrounding ectopic hair cells (Fig. 2F–H) indicated that a neuronal terminal that was as wide as the hair cell body surrounded many of the HCs. Z-stack confocal reconstructions revealed calyceal-like terminals surrounding ectopic HCs (Fig. 2I). In contrast, no calyx-like endings were observed on endogenous cochlear hair cells in Gli3Δ699/Δ699 cochleae (Fig. 2J).
To further explore the vestibular nature of these ectopic HCs, cochleae from E15.5 Gli3Δ699/+ and Gli3Δ699/Δ699 animals were examined. Because vestibular hair cells develop before cochlear hair cells (Zheng and Gao, 1997; Xiang et al., 1998), we wanted to determine whether the development of ectopic HCs was more comparable with vestibular or cochlear HCs, By E15.5, multiple ectopic sensory patches were already evident in Gli3Δ699/Δ699 cochleae and development of ectopic HCs was more advanced by comparison with inner HCs located in the organ of Corti. In contrast, ectopic HC development appeared comparable with the development of HC located in the sacculus (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
In summary, Gli3Δ699/Δ699 cochleae have multiple defects including a shortened duct, defects in cellular patterning in the OC including a marked widening, and the presence of variable patches of ectopic HCs and supporting cells within Kölliker's organ. In addition, the phenotype and developmental profile for these ectopic hair cells is consistent with vestibular rather than cochlear hair cells.
Low-frequency hearing loss is associated with PHS
Because of the neonatal lethality of Gli3Δ699/Δ699 mice, hearing function could not be assessed. However, a retrospective medical record review of a previously reported cohort of subjects with GLI3 mutant phenotypes (Johnston et al., 2005) identified 12 of 51 PHS patients and 3 of 58 GCPS patients who had had comprehensive audiologic evaluations. Six of these 12 PHS subjects had sensorineural hearing loss confirmed by bone-conduction testing (Fig. 3) (Szymko-Bennett et al., 2001); in contrast, none of the three GCPS subjects exhibited hearing loss. The PHS subjects' losses ranged from mild to severe/profound, and primarily, but not exclusively, affected low frequencies (Fig. 3). Word recognition scores and auditory brainstem responses were consistent with a peripheral (cochlear) origin of the hearing loss (data not shown). Because the cochlea is tonotopically organized such that high-frequency stimuli are detected toward the base and low-frequency stimuli are detected toward the apex (for review, see Davis, 2003), low-frequency hearing loss is consistent with the observed loss of the apical portion of the cochlea in Gli3Δ699/Δ699 mutants. In contrast, the variable occurrence of high-frequency hearing loss could be a result of cellular mispatterning within the OC or of the presence of ectopic HCs.

Auditory deficits in patients with PHS. The dashed lines indicate 90th centile sex-averaged air-conduction thresholds for normal individuals grouped by age decile (International Organization for Standardization, 1984). Solid lines indicate pure-tone air-conduction thresholds for the worse-hearing ear of six PHS subjects. Line color indicates the subject's age decile at the time of testing. A low-frequency hearing loss of at least 35 dB was observed in all subjects. In contrast, hearing sensitivity at high frequencies was only affected in a subset of the subjects. Bone-conduction threshold testing confirmed the hearing losses as sensorineural.
Inhibition of hedgehog signaling in vitro results in expansion of the organ of Corti and ectopic sensory patches
To determine whether the changes observed in the cochleae of Gli3Δ699/Δ699 mutants are the result of a direct role for HH signaling in cochlear patterning, we treated cochlear explant cultures with the HH-inhibitor KAAD-cyclopamine. Wild-type cochlear explant cultures were established on E13 as described previously (Montcouquiol and Kelley, 2003) and exposed to KAAD-cyclopamine or vehicle control. KAAD-cyclopamine-treated explants showed a significant increase in the number of ectopic HCs formed within Kölliker's organ (100 ± 16 in KAAD-cyclopamine-treated vs 30 ± 13 in control, p < 0.0005) (Fig. 4A,B). These ectopic HCs, unlike those in Gli3Δ699/Δ699 cochleae, were mostly found as scattered individual cells or in very small patches (Fig. 4B,C). However, the cells appeared very similar to those observed in Gli3Δ699/Δ699 mutants, including the presence of surrounding Jag1-positive cells (Fig. 4C), innervation by spiral ganglion neurites (Fig. 4D), and the development of rudimentary stereocilia bundles (Fig. 4E). Ectopic hair cells expressed the pro-hair-cell transcription factor Atoh1, demonstrating that HH signaling acts at or upstream of hair cell commitment (Fig. 4F–H).

Inhibiting HH signaling in vitro results in extra hair cells. A, B, Paired C57BL/6 cochlear explants with hair cells labeled with MyoVI. A, Control explant, established at E13 and maintained for 6 d in vitro (DIV). B, Explant treated with 5 μm KAAD-cyclopamine for 6 DIV. The total number of hair cells in the sensory epithelium is increased, and ectopic MyoVI-positive cells are present within Kölliker's organ (arrows). C, High magnification of a group of ectopic HCs from a cyclopamine-treated explant. HCs are labeled with MyoVI (red). Cells adjacent to the MyoVI-positive cells express higher levels of the supporting cell marker Jag1 (green; arrowheads). D, Ectopic HCs attract neurites. 2H3-positive neurites (blue) extend toward MyoVI-positive cells (red) located in Kölliker's organ. E, Z-stack confocal reconstruction of two ectopic MyoVI-positive cells. Phalloidin staining (green) shows that ectopic hair cells have stereociliary bundles at their lumenal surfaces (arrowheads). F, Ectopic MyoVI-positive HCs in an explant established from an Atoh1-LacZ reporter mouse (arrowheads). G, The same explant as in F, stained for β-galactosidase. All ectopic and endogenous HCs are positive for β-galactosidase expressed by the Atoh1-LacZ reporter. H, Merge of F and G showing coincidence of anti-MyoVI and anti-β-galactosidase staining.
In addition to the formation of ectopic HCs, inhibition of HH signaling in vitro by KAAD-cyclopamine caused a significant increase in the number of HCs within the endogenous sensory epithelium (1502 ± 118 in KAAD-cyclopamine-treated vs 1093 ± 61 in control, p < 0.0005) (Fig. 4A,B). We observed similar increases in HC number in explants treated with a soluble form of the antagonist HIP (1844 ± 49 in HIP-treated vs 1425 ± 60 in control, p < 0.005). Corresponding increases in supporting cells were also observed. However, treated explants were not significantly shorter than paired controls (data not shown), suggesting that the increased number of HCs observed in Gli3Δ699/Δ699 mutants is not a result of shortening of the cochlear duct, and instead that HH signaling directly regulates sensory cell formation within the OC.
Exogenous SHH represses HC development in wild-type cochlear explants
The above results were consistent with the hypothesis that HH signaling is required for repression of ectopic sensory cell formation within Kölliker's organ and, therefore, that HH signaling acts as a sensory cell repressor within the cochlea. To test this hypothesis, we increased activation of the HH pathway in vitro using exogenous SHH protein. Cochlear explants were established on E13 and treated with different concentrations of SHH. Consistent with a role in repression of sensory cell development, exogenous SHH resulted in a dramatic reduction in the number of HCs compared with controls (Fig. 5, compare A,B and C,D). The reduction in HC number in response to SHH treatment occurred in a dose-dependent manner, with significant decreases at SHH concentrations of 25 nm or greater (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Within SHH-treated explants, a greater number of HCs were observed at the base relative to the apex (Fig. 5C, arrowhead). Because HCs within the cochlea develop in a gradient that progresses from base to apex (Sher, 1971), the greater number of HCs in the basal region suggests that addition of exogenous SHH likely occurred after the period of HH sensitivity in this region of the cochlea. This suggests that endogenous SHH signaling affects sensory development at a relatively early stage; in fact, addition of SHH to cultures at or after E15 had no effect on hair cell number (data not shown).
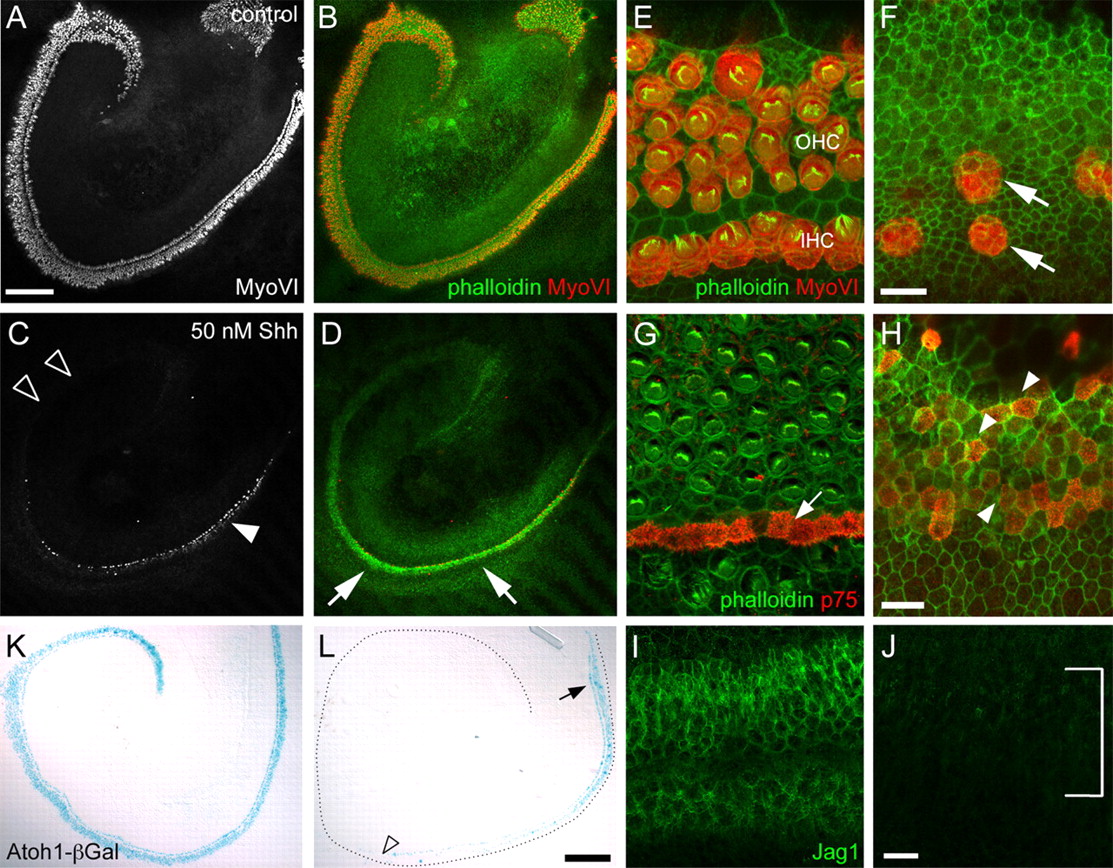
SHH inhibits hair cell formation. A, Low-magnification image of a control cochlear explant established on E13 and maintained for 6 d in vitro. Hair cells have been labeled with anti-MyoVI. The base of the cochlea is located to the right. B, The same image as in A, with anti-MyoVI (red) labeling hair cell bodies and phalloidin (green) labeling stereociliary bundles. C, Low-magnification image of an explant established, maintained, and oriented as in A, with the addition of 50 nm SHH. The number of MyoVI-positive cells is markedly reduced compared with control. Very few MyoVI-positive cells are found at the apex of the SHH-treated explant (open arrowheads), although more are visible at the base (closed arrowheads). D, The same explant as in C except with MyoVI and phalloidin illustrated as in B. Note the upregulation of F-actin in the region of the epithelium that would develop as the organ of Corti (arrows) but the significantly reduced number of MyoVI-positive cells. E, High-magnification view of the organ of Corti from the basal region of a control explant. A single row of IHCs and three rows of OHCs are present (red). Note that each HC has a relatively mature stereociliary bundle (green). F, Comparable view of the organ of Corti in an explant treated with 50 nm SHH. The hair cells (red, arrows) are disorganized, have small lumenal surfaces, and lack mature stereociliary bundles. G, Pillar cells, labeled with p75NTR (red; arrow) are located in an ordered row between IHCs and the first row of OHCs in the apical region of the organ of Corti in a control explant. H, In contrast with G, p75NTR expression is weak and disorganized in the apical region of an explant treated with 50 nm SHH (arrowheads). I, Anti-Jag1 labels supporting cells in the midbasal region of a control explant. J, By comparison, Jag1 labeling is almost completely absent in an explant treated with 25 nm SHH. K, Atoh1 reporter activity (blue) in a control explant established and treated as in A. The base of the cochlea is located in the top right corner of the image. L, Atoh1 reporter activity in an explant as in K, except treated with 50 nm SHH. Atoh1 expression is evident in the base of the cochlea (arrow) but is almost completely lost by the midbasal region (open arrowhead). The outer edge of the explant is indicated by the dotted black line for orientation. Scale bars: (in A) A–D, 200 μm; (in F), E, F, (in H), G, H, 10 μm; (in J) I, J, 20 μm; (in L) K, L, 200 μm.
At a higher magnification, HCs in the midbasal region of cochleae treated with SHH appeared disorganized, with limited development of stereociliary bundles (Fig. 5F, arrows). To determine whether supporting cell development was similarly disrupted in the presence of SHH, the expression of supporting cell markers, including p75NTR and Jag1, was determined. Similar to MyoVI, p75NTR staining was relatively normal in the base where hair cells were present (data not shown), but became increasingly sparse and disorganized toward the apex (Fig. 5G,H). By comparison with p75NTR, Jag1 expression was almost completely absent in SHH-treated explants (Fig. 5I,J). However, p75NTR is initially expressed in progenitors that will develop as both sensory and nonsensory cells (Mueller et al., 2002); therefore, the p75NTR-positive cells in SHH-treated explants may actually indicate undifferentiated progenitor cells as opposed to differentiated supporting cells. Consistent with the effects of KAAD-cyclopamine, treatment with SHH induced a decrease in expression of Atoh1 (Fig. 5K,L). No differences were observed in cell death or proliferation between SHH-treated and control explants (data not shown), again suggesting a role for HH signaling in cell fate.
Shh signaling factors are expressed in the cochlea during cell fate specification
The phenotype of the Gli3Δ699/Δ699 mutants along with the in vitro results were consistent with a role for HH signaling in the specification of cell fates within the cochlea, an event that normally occurs between ∼E13 and E15. To determine which members of the Hedgehog signaling pathway might be mediating these events, we examined the expression of several HH pathway components during mid to late gestation. Shh had been shown previously to be important for the early morphogenesis of the inner ear (Riccomagno et al., 2002), so we investigated its RNA expression, as well as the transcripts of its receptor, Ptch1, and the downstream transcription factors Gli3, Gli1, and Gli2, between E14 and P0. At E14, Shh RNA was strongly expressed in the spiral ganglion underlying the developing sensory epithelium (Fig. 6A), but no expression was present in the epithelium itself. At the same time, Ptch1 expression was more widespread than Shh, with transcripts detected throughout the ventral cochlear epithelium, the spiral ganglion, and weakly in the surrounding mesenchyme (Fig. 6B). Gli3 expression was similar to Ptch1 at E14 (Fig. 6C). At E16, Shh was still expressed in the spiral ganglion, although the overall level appeared reduced (Fig. 6D). The patterns of expression for Ptch1 (Fig. 6E) and Gli3 (Fig. 6F) also appeared mostly unchanged between E14 and E16, but, like Shh, expression was weaker at E16, becoming barely detectable in the case of Gli3. Gli1 and Gli2 were also expressed in the developing cochlear duct in patterns that were similar to Gli3 (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Both additional HH ligands, Indian hedgehog (Ihh) and Desert hedgehog (Dhh), were found in E14 whole inner ear RNA by reverse transcription (RT)-PCR analysis, but neither Ihh or Dhh could be detected in E14 or E16 cochlear sections by in situ hybridization (data not shown). Based on its much higher level of expression, it seems likely that Shh is the predominant HH ligand active in patterning the cochlear epithelium at these stages.

Expression of Shh pathway components during normal cochlear development. Developmental ages (rows) and probe used for in situ hybridization (columns) are indicated. A, At E14, Shh is strongly expressed in the developing spiral ganglion (SG) but is not expressed in the cochlear duct (dotted line). B, In contrast with Shh, Ptch1 is expressed within the duct at E14, with more intense expression in the modiolar side (MO). Ptch1 expression is also present in the surrounding mesenchyme. C, Gli3 is also expressed in the cochlear duct, although more weakly than Ptch1. D, At E16, Shh expression is maintained in the SG, but the level of expression appears reduced. E, F, Ptch1 and Gli3 continue to be expressed in the cochlear duct and in the SG at low levels. Gli3 expression is barely detectable at E16. Scale bar: (in A) A–F, 50 μm.
Because expression of these genes, with the exception of Shh, occurred at relatively low levels within the cochlear duct, we used quantitative PCR to determine whether the Gli3Δ699 allele resulted in any changes in expression of HH pathway members. Consistent with the previous report of changes in Ptch1 expression in Gli3Δ699 mutant limbs (Hill et al., 2007), we found that expression of Ptch1, a positive target of hedgehog signaling, was significantly reduced in Gli3Δ699/Δ699 cochlear epithelia (p < 0.05) (supplemental figure S4, available at www.jneurosci.org as supplemental material). However, there was no change in the expression of Gli1 or Gli2. Because the overall phenotype of Gli3Δ699 clearly demonstrates that this allele does not completely repress hedgehog signaling, the lack of change in Gli1 expression may reflect continued Shh signaling, or more complex interactions between the GLI3Δ699 protein and other GLI transcription factors, as has been suggested previously (Hill et al., 2007).
Hedgehog signaling represses prosensory formation
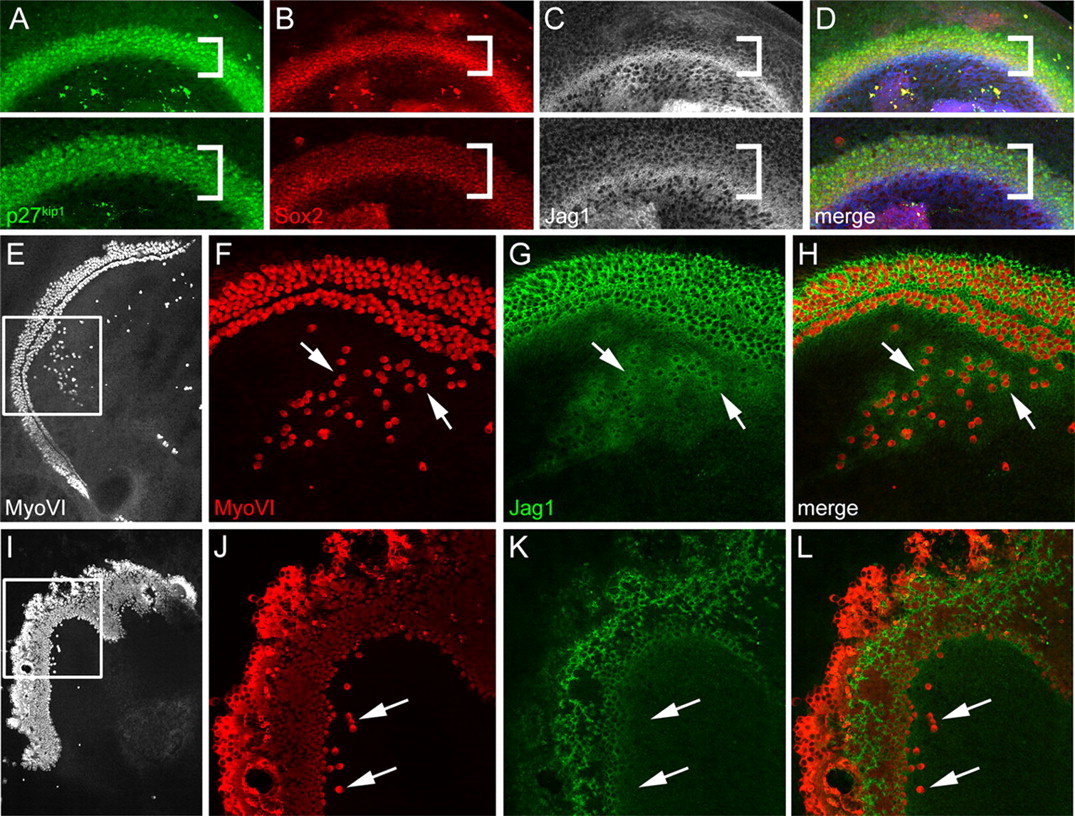
The presence of ectopic sensory patches containing both hair cells and supporting cells in Gli3Δ699/Δ699 mutants and in cyclopamine-treated explants suggested that SHH from the spiral ganglion might act to inhibit the formation of the prosensory cells that give rise to both cell types. Alternatively, because hair cells are known to induce surrounding cells to develop as supporting cells, it could also be the case that Shh signaling acts to repress hair cell formation at the level of Atoh1 expression. We therefore examined expression of prosensory markers in Gli3Δ699 mutant and wild-type cochleae at E13, before HC differentiation. Expression of three prosensory markers, p27kip1 (Chen and Segil, 1999), Sox2 (Kiernan et al., 2005b), and Jagl1 (Kiernan et al., 2006), was expanded in the Gli3Δ699/Δ699 cochlea relative to the wild-type cochlea (Fig. 7A–D, compare top and bottom panels). A broader prosensory domain could result from the shortened cochlea in Gli3Δ699 mutants, but, at E13, the sensory epithelia of mutant cochleae (marked by p27kip1) were significantly wider (55.8 ± 2.0 μm vs 41.1 ± 3.0 μm; p < 0.005) but not significantly shorter than wild-type littermates (947 ± 58.5 μm vs 1016 ± 59 μm; p > 0.2). This suggests that the expansion of the prosensory domain and, thus, of the OC, is a result of a direct role of HH signaling on prosensory formation and not a secondary effect of the shortened cochlea.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HH signaling regulates prosensory formation through the Notch signaling pathway. A, p27kip1 staining (green) in E13 Gli3Δ699/+ (top) and Gli3Δ699/Δ699 cochleae (bottom) (midbasal region). The p27kip1-positive domain is wider in the mutant cochlea (brackets). B, Sox2 staining (red) of the same cochleae as in A indicates a similar expansion in the mutant versus heterozygous littermate. C, The bright band of Jag1 staining is similar in both cochleae, but there is a higher level of Jag1 staining (white) in the prosensory region of the Gli3Δ699/Δ699 cochlea (bracketed region). D, Merge of A–C. Overlapping staining of all three prosensory markers indicates the increased size of the prosensory domain in the Gli3Δ699/Δ699 cochlea. Jag1 staining is shown in blue. E, I, Low-magnification views of paired C57BL/6 explants stained with anti-MyoVI. The explant in E was treated with 5 μm KAAD-cyclopamine; the explant in I was treated with 5 μm KAAD-cyclopamine and 5 μm DAPT. F, J, Higher-magnification views of boxed regions in E and I show many ectopic MyoVI-positive cells (red) in F, although only a few ectopic cells are apparent in J (arrows). Nearly all cells within the sensory epithelium in J are MyoVI-positive because of the disruption of lateral inhibition resulting from DAPT treatment. G, Jag1 staining in supporting cells near both ectopic (arrows) and endogenous HCs. K, Very little Jag1 staining is detectable in the DAPT-treated explant. H, L, Merge of F, G, and J, K, respectively. Jag1-positive cells surround MyoVI-positive HCs in H but not L.
To confirm that the ectopic HCs formed within KO also resulted from expanded or ectopic prosensory domains, we examined prosensory markers in cochlear explants. Explants were treated with KAAD-cyclopamine as described but were fixed after only 24 or 48 h, a time point before the formation of hair cells within ectopic patches. Individual cells that were positive for Sox2 were observed within Kölliker's organ, and the Jag1-positive domain was expanded (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Similar Sox2 or Jag1-positive cells were not observed in Kölliker's organ in control explants. These results were consistent with a role in prosensory formation. Culturing isolated cochlear epithelia, without the underlying mesenchyme or Shh-expressing spiral ganglion, did not result in a significant increase in ectopic HCs (data not shown). However, previous work has shown that there is a positive, HC-inducing signal, yet unidentified, present in cochlear mesenchymal tissue (Montcouquiol and Kelley, 2003). Isolation of the cochlear epithelium presumably removes both this positive signal and the repressive Shh signal. Whether Ihh or Dhh, both detectable in the inner ear by RT-PCR, play a role in inhibiting prosensory development in the absence of the spiral ganglion is still to be determined.
Activation of Notch signaling is required for the formation of inner ear prosensory domains (Brooker et al., 2006; Kiernan et al., 2006), but once prosensory domains have been specified, Notch acts as an inhibitor of the hair cell fate (Kelley, 2003). Therefore, we hypothesized that if HH signaling acted at the level of prosensory formation, then inhibition of Notch signaling would act to inhibit KAAD-cyclopamine-induced ectopic sensory patches. In contrast, if HH signaling acts at the level of hair cell induction, then inhibition of Notch signaling should not affect the ability of KAAD-cyclopamine to induce ectopic hair cells. To block Notch activity in vitro, cochlear explants were treated with the γ-secretase inhibitor DAPT, which prevents cleavage of Notch proteins and, thus, the translocation of the Notch intracellular domain to the nucleus (De Strooper et al., 1999; Hadland et al., 2001). As reported previously (Takebayashi et al., 2007), treatment of cochlear explants with DAPT alone results in a significant increase in the number of hair cells within the sensory epithelium and a corresponding decrease in the number of supporting cells (data not shown). This phenotype is caused by the later developmental requirement for Notch signaling in the process of lateral inhibition that specifies HC or supporting cell fates (Lanford et al., 1999). Therefore, analysis was restricted to ectopic patches located well within KO. In contrast with cyclopamine-treated explants, explants treated with both KAAD-cyclopamine and DAPT had few to no ectopic HCs in Kölliker's organ (4 ± 2 for DAPT/KAAD-cyclopamine-treated vs 86 ± 27 for KAAD-cyclopamine-treated, p < 0.005) (Fig. 7E–L), suggesting that activation of Notch signaling is required for HH-mediated formation of ectopic hair cell patches. These results are consistent with the hypothesis that HH signaling acts at the level of prosensory specification through antagonism of the Notch pathway and suggest that HH signaling may normally act to regulate the size and position of the prosensory domain within the cochlear epithelium.
Discussion
The data presented here demonstrate previously unreported roles for HH signaling in the development and function of the mammalian cochlea. All of the structures within the membranous labyrinth of the inner ear are derived from the epithelial cell-lined otocyst. The factors that specify some regions of the otocyst to develop as prosensory cells, whereas other regions develop as nonsensory, remain mostly unknown. This is a particularly intriguing question for the mammalian cochlear duct. Normally, only a limited percentage of cells within the duct become prosensory cells; however, several previous studies have demonstrated that cells in other regions of the duct possess the ability to develop as sensory cell types, including hair cells (Zheng and Gao, 2000; Kawamoto et al., 2003; present study) and supporting cells (Zheng and Gao, 2000; Kawamoto et al., 2003; Woods et al., 2004). Although activation of the Notch pathway and expression of Sox2 have been implicated as important regulators of prosensory formation, the factors that regulate either of these events are unknown. The results presented here suggest that, at least for the cochlear duct, HH signaling plays an important role in repressing prosensory formation. The presence of an asymmetric source of SHH originating in the medially located spiral ganglion is consistent with a higher level of HH signaling in the medially located Kölliker's organ. Reduction of HH signaling either through the exclusive presence of the repressor form of GLI3 or the addition of KAAD-cyclopamine or HIP leads to a loss of HH-mediated repression, an increase in the size of the endogenous prosensory domain, and the spontaneous formation of ectopic prosensory domains within Kölliker's organ. The formation of these ectopic prosensory domains is dependent on Notch signaling, because significantly fewer ectopic hair cell patches are observed when Notch signaling is blocked by DAPT treatment. This result is consistent with the recent demonstration that prosensory formation is dependent on activation of Notch, presumably by Jagged1 (Kiernan et al., 2006). This result also suggests that the SHH and Notch signaling pathways normally interact antagonistically to define the position and size of the prosensory domain in the developing cochlea. Antagonistic interactions between SHH and Notch signaling have been reported in some systems (Nicolas et al., 2003); however, it is also possible that HH signaling influences prosensory formation upstream from Jagged1/Notch activation. It is possible that the ectopic sensory patches arise as a result of migration of prosensory cells from the endogenous prosensory domain. However, although this possibility cannot be completely discounted, the similarities in the patterns of ectopic Sox2 expression in KAAD-cyclopamine-treated explants after 48 h and ectopic hair cell formation in KAAD-cyclopamine-treated explants after 6 d suggests that ectopic prosensory cells arise spontaneously within Kölliker's organ.
The formation of ectopic hair cells in the presence of reduced HH activity may have intriguing implications regarding the evolution of the mammalian cochlea. In placental mammals, the sensory epithelium comprises a relatively small percentage of the cochlear duct by comparison with other vertebrate classes or in nonplacental mammals. Although the basis for this change has not been determined, it has been suggested that the total size of the sensory epithelium may have become reduced in response to selective pressures related to the perception of high frequencies (Rubel, 1978). Because, as mentioned previously, the cells of Kölliker's organ can form HCs or supporting cells, it seems possible that some regions of Kölliker's organ may have developed as sensory cells in an ancestral cochlear duct. Therefore, HH signaling may have evolved to repress prosensory formation within this region.
Based on stereociliary bundle morphology and the presence of calyceal neuronal terminals, the ectopic HCs in Gli3Δ699/Δ699 mutant cochleae are vestibular in nature. Because the stereociliary bundles on the endogenous cochlear HCs appear normal in these mutants, the vestibular phenotype of the ectopic HCs is unlikely to be caused by a requirement for full-length GLI3 or HH signaling for cochlear HC or stereociliary bundle development. Rather, because mammalian cochlear hair cells are more derived, the ectopic hair cells may be differentiating along an ancestral developmental pathway. As a result of their location, the ectopic cells in Gli3Δ699/Δ699 mutants presumably do not receive all of the instructive signals necessary to form the unique mammalian cochlear HC types, and thus default to an earlier, ancestral form. It would be interesting to compare the role of hedgehog signaling in development of the auditory sensory epithelia in other vertebrates to see if this is indeed the case.
The presence of adjacent supporting cells and neuronal terminals on these ectopic HCs suggests that these cells are probably functional. Among the Gli3Δ699/Δ699 cochleae examined, variable degrees of cellular mispatterning within the organ of Corti and generation of ectopic hair cells within Kölliker's organ were observed. Because overproduction of hair cells can lead to hearing loss (Chen and Segil, 1999), the presence and innervation of both additional rows of hair cells within the OC and ectopic hair cells in Kölliker's organ likely contributes to disruption of sensorineural function. This result is consistent with the variable high-frequency hearing loss observed in many PHS subjects.
Many PHS patients show evidence of auditory defects that are consistent with a shortening of the cochlear duct. Low-frequency hearing loss, indicative of defects in the apical portion of the cochlea, was observed in all PHS subjects with auditory deficits. Although neonatal lethality prevented any assessment of auditory function in Gli3Δ699/Δ699 mice, direct measurement of cochlear length demonstrated a significant shortening of the cochlea relative to WT. The Gli3Δ699/Δ699 phenotype of a shortened cochlea, but unaffected vestibular structures, is consistent with previous work showing that Shh signaling is required for development of the ventral (cochlear) but not the dorsal (vestibular) portion of the inner ear (Riccomagno et al., 2002, 2005; Bok et al., 2005, 2007). GLI activator function is apparently required for transducing positive Shh signaling in this context for the proper outgrowth of the cochlear duct. However, we have not observed shortening of the cochlear duct in magnetic resonance images of the inner ears of a PHS patient (A. Griffith and S. Pryor, unpublished observation).
In summary, we have shown here that HH signaling is necessary for proper development of the cochlea and sensory epithelium in mice, and for auditory function in humans. The results demonstrate a new role for HH signaling as a repressor of prosensory fate within the cochlear duct. The results also suggest that specification of prosensory domains within the inner ear does not represent a purely inductive process, and that at least some regions of the cochlear duct that normally develop as nonsensory, may possess an innate ability to adopt a prosensory fate. These results have intriguing implications in terms of understanding developmental patterning of the otocyst and the potential for spontaneous hair cell formation in the adult inner ear.
Footnotes
- Received January 23, 2008.
- Revision received April 28, 2008.
- Accepted May 21, 2008.
-
This work was supported by the Intramural Program at the National Institute on Deafness and Other Communication Disorders (NIDCD). We thank M. Montcouquiol for assistance with cochlear dissections; D. Wu and H. Arnheiter for comments on a previous version of this manuscript; the audiology unit of the Otolaryngology Branch, NIDCD, for audiologic evaluations of PHS subjects; and A. McMahon and J. Klingensmith for providing probe cDNAs.
- Correspondence should be addressed to Elizabeth Carroll Driver, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Building 35, 2A-205, 35 Convent Drive, MSC 3729, Bethesda, MD 20892. drivere{at}nidcd.nih.gov
- Copyright © 2008 Society for Neuroscience 0270-6474/08/287350-09$15.00/0