
Abstract
Death-associated protein kinase (DAPK) is a key player in multiple cell death signaling pathways. We report that DAPK is regulated by DANGER, a partial MAB-21 domain-containing protein. DANGER binds directly to DAPK and inhibits DAPK catalytic activity. DANGER-deficient mouse embryonic fibroblasts and neurons exhibit greater DAPK activity and increased sensitivity to cell death stimuli than do wild-type control cells. In addition, DANGER-deficient mice manifest more severe brain damage after acute excitotoxicity and transient cerebral ischemia than do control mice. Accordingly, DANGER may physiologically regulate the viability of neurons and represent a potential therapeutic target for stroke and neurodegenerative diseases.
Introduction
DANGER is a novel protein identified on the basis of its binding to inositol 1,4,5-trisphosphate receptors (IP3R) (van Rossum et al., 2006). It is a membrane-associated protein that contains a partial MAB-21 domain and is expressed in a wide range of tissues. DANGER is evolutionarily conserved but rapidly evolving (Nikolaidis et al., 2007). It physiologically binds to IP3R, enhances calcium-mediated inhibition of IP3R-dependent calcium release, and regulates neuronal process outgrowth. To clarify the physiological function of DANGER, we conducted yeast two-hybrid analysis paying special heed to candidate proteins that are regulated by calcium. In the process, we identified death-associated protein kinase (DAPK) as an interacting protein.
DAPK is a calcium/calmodulin-regulated protein kinase that regulates cell death. It was identified in a screen for genes that influence interferon-induced cell death in HeLa cells (Deiss et al., 1995) and plays an essential regulatory role in multiple cell death signaling pathways (Bialik and Kimchi, 2006). DAPK stimulates caspase-linked apoptosis (Jin and Gallagher, 2003) and p53-mediated cell death (Raveh et al., 2001). It also enhances autophagy-associated cell death via interaction with Beclin-1, a BH3-domain-only protein (Zalckvar et al., 2009). DAPK is critically involved in neuronal cell death, as its activity is increased in response to hypoxic ischemia (Schumacher et al., 2002; Shamloo et al., 2005), and a specific peptide inhibitor of DAPK significantly attenuates brain injury after ischemic stroke (Velentza et al., 2003). DAPK also acts as a tumor suppressor, as its gene silencing via promotor methylation is implicated in tumorigenesis (Kissil et al., 1997). DAPK possesses a death domain that mediates its interactions with a number of other proteins such as the extracellular signal-regulated kinase (ERK) (Chen et al., 2005) and the UNC5H2-dependent receptor (Llambi et al., 2005). DAPK is calcium/calmodulin dependent, autophosphorylates at serine 308 (Feinstein et al., 1995), and phosphorylates diverse substrates, including the myosin light chain (Cohen et al., 1997), beclin-1 (Zalckvar et al., 2009), Zipper-interacting protein kinase (ZIPk) (Shani et al., 2004; Mukhopadhyay et al., 2008), CaMKK (calmodulin-regulated protein kinase kinase) (Schumacher et al., 2004), and syntaxin-1A (Tian et al., 2003). The exact molecular mechanisms accounting for DAPK-associated cell death, particularly in neurons, are not completely understood.
We now report the direct binding of DANGER to DAPK, demonstrate that DANGER inhibits DAPK catalytic activity, and show that DANGER knock-out mice exhibit augmented neuronal and non-neuronal cell death both in vivo and in vitro. Thus, DANGER appears to modulate cell death physiologically through its inhibition of DAPK signaling.
Materials and Methods
Generation and maintenance of DANGER knock-out mice.
The gene encoding mouse DANGER, Danger, is located on chromosome 19 and consists of three exons, the first two of which are exclusively noncoding. DANGER+/− mice were generated at Ozgene. The targeting construct was based on the sequence of the C57BL/6 strain Danger gene (GenBank accession number AC126679). The PGK-neo selection cassette was inserted downstream of exon 3. The PGK-neo cassette was flanked by flippase recognition target (FRT) sites and can be deleted with enhanced flippase recombinase. Exon 3 was flanked by loxP sites and can be deleted with Cre recombinase. This strategy removes the entire coding region of the gene as exon 3 contains both the ATG start codon and the TAA stop codon. All mice were maintained on a C57BL/6 background. Mice were housed in a 12 h light/dark cycle at an ambient temperature of 22°C and fed standard rodent chow. Animal protocols, approved by the Institutional Animal Care and Use Committee of Johns Hopkins University, were used in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Genotyping and reverse transcription-PCR analysis.
Mice were genotyped by PCR analysis of genomic DNA from tail biopsies. Primer sets P1 (GCTCCTGTCCTTTGCTGTCT) and P2 (AGGGAATACTCGCTAATGAG) yielded a 500 bp product from the wild-type (DANGER+) allele, and primer sets P3 (TCCAGGGCAAAAGGTCCT) and P4 (AGAGACTGTGTGGTCACTGTC) yielded a 250 bp product from the knock-out (DANGER−) allele. Reverse transcription-PCR (RT-PCR) analysis was used to assess whether the DANGER transcript was missing in the knock-out mice. RNA was prepared from brain tissues obtained from DANGER knock-out mice and their wild-type littermates by using TRIZOL reagent (Invitrogen). cDNA was prepared by using Oligo(dT) primers (Invitrogen) and the Omniscript reverse transcriptase kit (Qiagen). PCR was performed by using cDNA templates and primers P1 and P2 as above.
Generation of mouse embryonic fibroblasts.
Wild-type (DANGER+/+) and mutant (DANGER−/−) embryonic day 14 (E14) embryos were isolated from a single heterozygous female that had been paired with a heterozygous male. The head and abdominal organs were removed, and the remaining carcasses were minced and incubated in trypsin to obtain single cells. These primary fibroblasts were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gemini Bio-Products), 2 mm l-glutamine (Invitrogen), and 100 U/ml penicillin/100 μg/ml streptomycin (PS; Invitrogen) at 37°C with 5% CO2 atmosphere in a humidified incubator. Experiments with primary mouse embryonic fibroblasts (MEFs) were performed at no later than passage 8.
Cell culture and transfection.
HEK 293T cells were maintained in DMEM with 10% FBS, 2 mm l-glutamine, and PS. Transfection was performed with Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions.
Mouse hippocampal neuron culture.
The dissected hippocampi of E18 DANGER knock-out and wild-type mice were dissociated by papain. The tissue was washed and dissociated by repeated passage through a constricted Pasteur pipette. Cells were plated on polylysine-coated plates in Neurobasal medium with B-27 supplement (Invitrogen) at 5000 cells/cm2 and maintained at 37°C in a humidified atmosphere containing 5% CO2. The medium was changed every 3–4 d until the cells were used for experiments.
Cell lysates and immunoprecipitations.
Cells were washed with cold PBS and lysed in cold lysis buffer [50 mm Tris, pH 7.5, 150 mm NaCl, 0.5 mm EDTA, 0.5% Triton X-100, Complete protease inhibitor tablet (Roche), and PhosphoStop phosphatase inhibitor mixture (Roche)]. For immunoprecipitation experiments, 1 mg of protein extract was incubated for 2 h to overnight at 4°C with indicated antibodies and protein A/G agarose beads (Pierce) or glutathione-Sepharose beads (GE Healthcare) when glutathione S-transferase (GST)-fusion protein was used. Immunoprecipitates were washed three times with lysis buffer and twice with PBS and subjected to SDS-PAGE and Western blotting analysis. Total protein (50 μg) was loaded as an input.
In vitro binding assay.
GST-DAPK, GST, and TAP-DANGER were overexpressed in HEK293T cells and purified with glutathione-Sepharose beads or the Tandem Affinity Purification (TAP) kit (Stratagene). The purified proteins were eluted with glutathione (for GST-fusion protein) or biotin (for TAP-fusion protein) and desalted with a desalting column (Bio-Rad). Immunoprecipitated proteins were quantified by running samples on gels and comparing the Coomassie blue staining of the bands to a quantified standard protein run in parallel. TAP-DANGER (100 ng) was incubated in binding buffer with GST-DAPK (100 ng) or GST for 2 h. Glutathione-Sepharose beads were added to the mix for 30 min, and then beads were washed three times with binding buffer and subjected to SDS-PAGE and Western blotting analysis.
Antibodies.
The following antibodies were used for Western blotting: polyclonal anti-DANGER antibody (1:5000) (van Rossum et al., 2006); polyclonal anti-GST HRP-conjugated antibody (1:10,000; Sigma); monoclonal anti-DAPK antibody (1:1000) (Sigma); monoclonal anti-phospho-DAPK antibody (1:1000; Sigma); and monoclonal anti-phospho S19-MLC antibody (1:1000; Cell Signaling Technology).
Kinase assay.
In vitro DAPK kinase assay was performed essentially as described previously (Cohen et al., 1997). Cells were lysed, and proteins were immunoprecipitated as described above. The immunoprecipitates were washed three times with lysis buffer and twice with kinase reaction buffer. The precipitated proteins were then incubated with kinase reaction buffer (50 mm HEPES, pH 7.5, 10 mm MgCl2, 10 mm MnCl2, 150 mm NaCl, and 0.1 mg/ml bovine serum albumin). Kinase assay was performed for 10 min in 50 μl of reaction buffer at room temperature. Calmodulin/CaCl2 was added at indicated concentrations.
Thiazolyl blue tetrazolium bromide assay.
To quantitatively measure cell death after stimulation with various agonists, we used the thiazolyl blue tetrazolium bromide (MTT) assay. A 5 mg/ml stock of MTT (Sigma) was diluted 1:20 in culture medium and added to cells after treatments with the indicated drug or dimethylsulfoxide (DMSO) for 20 h. Cells were incubated at 37°C for 2 h, during which healthy cells with intact mitochondria will convert the yellow-colored MTT reagent to purple crystals. Then the medium was removed, and the cells were lysed in DMSO. The samples were transferred to 96-well plates and read in a 96-well plate reader at optical density (OD) 560 and 630 nm. The OD 560–630 nm reading was normalized to vehicle-treated control and expressed as a percentage of cell viability.
Caspase and lactate dehydrogenase assay.
The caspase-3 fluorescence assay and lactate dehydrogenase (LDH) assay were performed according to the manufacturer's instructions (Cayman Chemical).
Hoechst staining and cell viability assessment by nuclear morphology.
Cells were fixed with 4% paraformaldehyde (PFA), washed in PBS, and stained with Hoechst 33258 (1:1000 in PBS; Invitrogen). The morphology of the cells' nuclei was observed with a Zeiss fluorescence microscope at excitation wavelength 350 nm. Nuclei were considered to have the normal phenotype if they glowed brightly and homogenously. Apoptotic nuclei were identified by the condensed chromatin gathering at the periphery of the nuclear membrane or a total fragmented morphology of nuclear bodies. More than 150 cells in each sample were counted, and the percentage of apoptotic nuclei was determined.
NMDA-induced neurotoxicity.
Adult male wild-type and knock-out mice were used for NMDA-mediated cytotoxicity measurement. Animal well being was monitored by measuring weight and rectal temperature before the surgical procedure. Mice were anesthetized with 3.0% isoflurane, maintained with 1.0% isoflurane, and mounted on a stereotaxic frame (Stoelting). The skull was exposed, and 15 nmol of NMDA was slowly injected into the striatum (0.5 mm anterior, 2 mm lateral from bregma, and 2.5 mm ventral relative to dura) with a 1.0 μl Hamilton syringe. After each injection, mice were placed in a thermoregulated chamber maintained at 31 ± 0.5°C and returned to their cages after full recovery from anesthesia. The rectal temperatures were monitored and maintained at 37.0 ± 0.5°C during the experimental procedure. At 48 h after NMDA injection, mice were perfused transcardially with 0.1 m PBS and fixed with 4% PFA. Brains were harvested, soaked in 4% PFA overnight, and equilibrated with 30% sucrose. Sequential brain sections of 25 μm obtained on a cryostat were stained with cresyl violet to estimate the lesion volume as described previously (Ahmad et al., 2006; Ahmad et al., 2007). One-way ANOVA followed by Tukey's post hoc analysis was used to calculate the difference between the groups.
Transient focal ischemia.
Mice were anesthetized with 1.5% isoflurane in 25% oxygen-enriched air. Transient focal ischemia was induced by middle cerebral artery occlusion (MCAO) via the intraluminal suture technique (Saleem et al., 2009). A reduction in blood flow over the ipsilateral parietal cortex of 87–90% was used as confirmation of successful occlusion. Blood flow was monitored with a laser-Doppler flowmeter (Moor Instruments), and body temperature was maintained at 37.0 ± 0.5°C in all mice during the experiment. The filament was removed from the lumen 90 min after occlusion to allow the blood to return to the ischemic region of the brain. All mice were allowed to recover from anesthesia before being returned to their cages. All animals were killed at 48 h after MCAO. Brains were harvested, sliced into five 2-mm-thick coronal sections, mounted onto slides, and stained with 1% triphenyltetrazolium chloride (TTC; Sigma). Infarct areas of all slices were traced with the image analysis software (SigmaScan pro 5; Systat). The infarct area of the ipsilateral hemisphere of each brain was integrated into cubic millimeters and translated into a percentage of the contralateral hemisphere of the same brain with correction for swelling.
Results
To elucidate a physiologic role for DANGER, we conducted yeast two-hybrid analysis using full-length DANGER fused to the GAL4 DNA binding domain and a rat whole-brain cDNA library fused to the GAL4 DNA activation domain. Of 19 identified interactors, one of the most prominent was DAPK (data not shown). We confirmed the DANGER–DAPK interaction by transient transfection of GST-DAPK and TAP-DANGER into HEK293T cells followed by a GST pull-down experiment, which revealed selective binding of DAPK and DANGER (Fig. 1A). Using an antibody against DAPK, we coprecipitated endogenous DANGER with DAPK from HEK293T cells, which basally express both proteins (Fig. 1B). The in vitro binding assay with purified recombinant proteins demonstrates direct interaction between DANGER and DAPK (Fig. 1C). In an effort to map DANGER–DAPK binding, we examined binding interactions of various fragments of these two proteins (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Multiple fragments of both DANGER and DAPK display binding to the other protein, indicating that the conformation of the full-length proteins is required for physiological binding. Three members of DAPK family proteins have been reported (Kawai et al., 1998; Inbal et al., 2000). Although our studies focused on DAPK1, we also observed similar binding of overexpressed DANGER to DAPK2 (DAPK-related protein-1) and DAPK3 (ZIPk) in HEK293T cells (data not shown).

DANGER physiologically interacts with DAPK. A, HEK293T cells were transfected with either GST or GST-DAPK and a TAP-tagged DANGER construct. DANGER specifically interacts with DAPK. B, Immunoprecipitation experiments were performed with HEK293T cell lysate and a DAPK antibody showing coprecipitation of DANGER. C, In vitro binding experiments were performed with GST/GST-DAPK and TAP-DANGER purified from transfected HEK293T cells. DANGER binds to GST-DAPK but not to GST alone.
To investigate the physiologic consequence of DANGER–DAPK binding, we monitored the influence of DANGER on DAPK catalytic activity. DANGER inhibits DAPK activity toward myosin light chain in a concentration-dependent manner (Fig. 2A). Maximal inhibition is achieved with equimolar concentrations of DANGER and DAPK. Whereas the catalytic activity of DAPK is calcium/calmodulin dependent, inhibition of DAPK activity by DANGER occurs similarly at 0.1 and 1.0 μm calmodulin concentrations (Fig. 2B). Thus, DANGER does not appear to act via influencing calmodulin's binding to DAPK. Because DAPK is a protein kinase, we also examined whether DANGER is a direct substrate of DAPK. We performed in vitro DAPK assays using DANGER as a potential substrate but did not observe any phosphorylation of DANGER (data not shown).

DANGER inhibits DAPK catalytic activity. A, DANGER inhibits DAPK in a concentration-dependent manner. Data are mean ± SE. B, Inhibition of DAPK by DANGER is calmodulin independent. The degree of inhibition was similar with 0.1 and 1.0 μm calmodulin in the DAPK assay.
To examine potential roles of DANGER in regulating DAPK-associated cell death, we generated mice with targeted gene deletion of DANGER. We used a targeting vector to delete the complete open reading frame of DANGER (Fig. 3A). PCR genotyping and RT-PCR confirmed the complete genomic deletion of DANGER and the absence of DANGER mRNA in brain tissues (Fig. 3B). The mutant mice appear to be grossly normal. They do not differ from wild-type in fecundity, locomotor activity, body size, or weight. Gross anatomic dissection reveals no apparent abnormalities in any organ of adult knock-out mice. Histological examination with hematoxylin and eosin stain of brain, liver, lung, kidney, heart, and spleen tissues indicates no apparent abnormalities in the mutant mice (data not shown). We also examined organs of mice at various ages and failed to detect any obvious developmental alterations. Additionally, cell growth rate is not altered in the mutant mice (data not shown).

DANGER knock-out cells exhibit increased DAPK activity and augmented cell death. A, Schematic representation of the targeting vector used to generate DANGER knock-out mice. B, Confirmation of DANGER deletion. DNA from mouse tails was extracted and genotyped by PCR with primers specific for wild-type and knock-out alleles, respectively. Total RNA was isolated from the brains of wild-type and knock-out animals. RT-PCR analysis was performed with primers specific for DANGER mRNA. C, Endogenous DAPK activity was increased in DANGER knock-out MEFs. Endogenous DAPK was immunoprecipitated from wild-type and DANGER knock-out MEFs with an anti-DAPK antibody, and the DAPK kinase assay was performed by using purified myosin light chain (MLC) as substrate. Phosphorylation of MLC was detected with a phosphor-S19-MLC antibody. Phospho-S308-Akt, Phospho-GSK3β, and Phospho-ERK1/2 levels were not changed in DANGER knock-out MEFs. D, Caspase-3 activity and LDH activity were higher in tunicamycin-treated DANGER knock-out MEFs than in wild-type MEFs. E, Cell death was augmented in tunicamycin-treated DANGER knock-out MEFs compared with wild-type MEFs, as determined by MTT assay. F, Cell death was augmented in C6-ceramide-treated DANGER knock-out hippocampal neurons compared with wild-type cells, as determined by MTT assay. G, Depletion of DANGER enhances NMDA-mediated excitotoxicity of hippocampal neurons. Primary hippocampal neurons derived from DANGER knock-out mice demonstrated a higher level of neurotoxicity 20 h after treatment with NMDA compared with wild-type controls. Data are mean ± SE. HET, heterozygous; WT, wild-type; KO, knock-out.
Physiological inhibition of DAPK activity would predict augmented catalytic activity of DAPK in DANGER knock-out mice. MEFs are primary cells widely used as an in vitro model to study cell death. MEFs derived from DANGER knock-out mice exhibit a 35% increase in DAPK activity (Fig. 3C). To ascertain that the increase in DAPK activity is specific, we also examined the basal activity of three other kinases (ERK1/2, GSK3β, and Akt) using phospho-specific antibodies against activated forms of these kinases. We observed no changes in the levels of phospho-ERK1/2, phospho-GSK3β, or phospho-Akt in DANGER knock-out MEFs.
To determine whether the augmented DAPK activity in DANGER knock-out MEFs is associated with alterations in cell death, we monitored cell death events in tunicamycin-treated MEFs. Using assays based on caspase-3 activity, LDH activity (Fig. 3D), and MTT assays (Fig. 3E), we observed significantly augmented cell death in DANGER knock-out MEFs.
DAPK is a known positive regulator of neuronal cell death (Schumacher et al., 2002). To investigate a role of DANGER on neuronal viability, we monitored cell survival of hippocampal neurons after treatment with C6-ceramide (Fig. 3F). DANGER knock-out neurons are substantially more sensitive to C6-ceramide-induced cell death, with knock-outs displaying only 28% the survival of wild-type. Overactivation of NMDA receptors leads to excitotoxicity, which has been implicated in vascular stroke and neurodegenerative diseases (Choi, 1994) and is readily monitored in brain cultures (Koh and Choi, 1988; Dawson et al., 1991). We assessed NMDA-induced neuronal cell death by monitoring nuclear morphology after Hoechst staining. In 8- to 10-d-old wild-type hippocampal neurons, survival of knock-out cells is only half that of wild type (Fig. 3G).
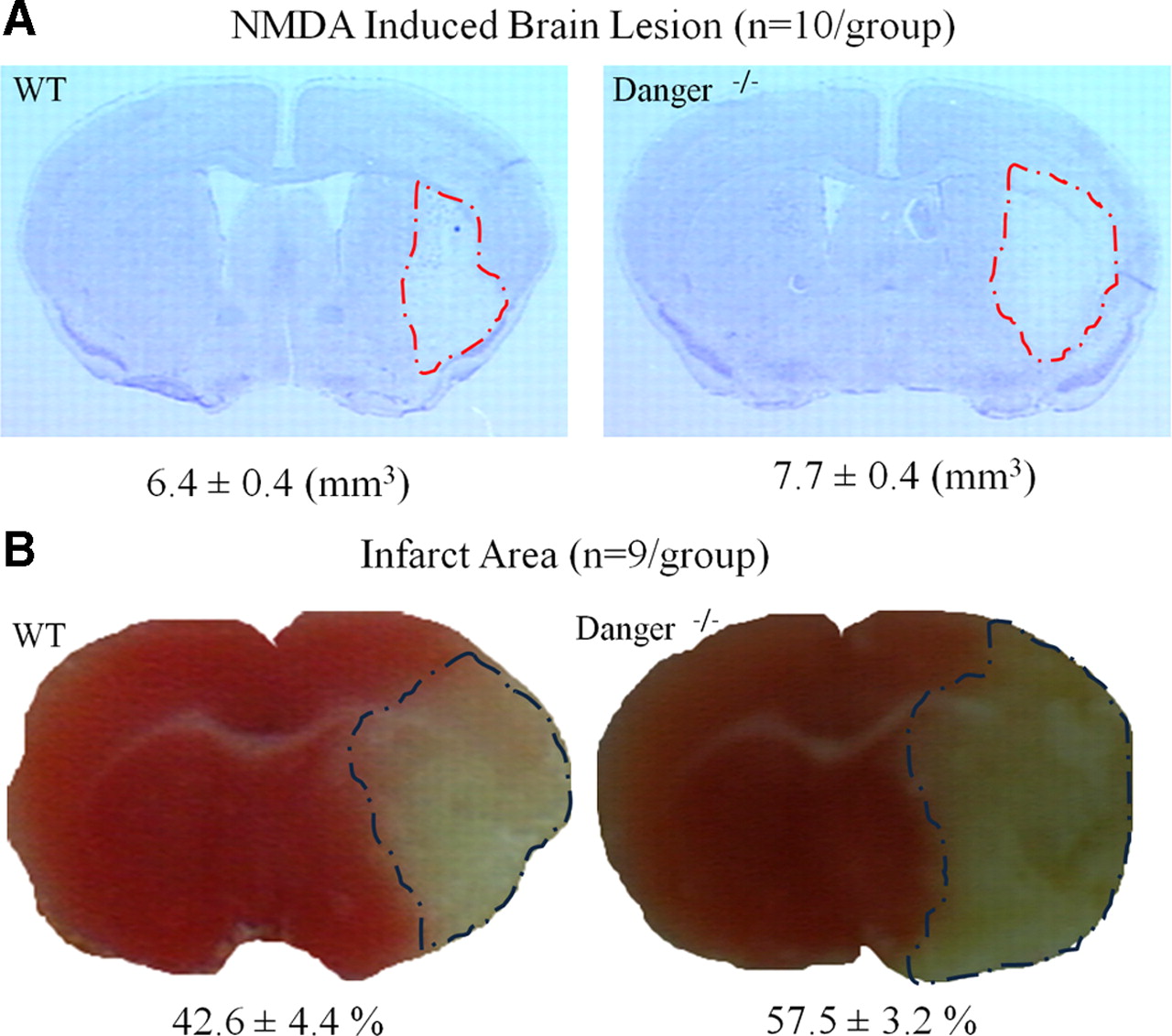
We wondered whether DANGER regulates neuronal viability in intact brain. Inhibition of DAPK attenuates brain injury after ischemic stroke (Velentza et al., 2003). We monitored brain damage in mice after NMDA injections (Ahmad et al., 2006; Ahmad et al., 2007) or cerebral ischemia elicited by MCAO (Kaundal et al., 2006; Saleem et al., 2009). NMDA-induced brain lesions are significantly greater in DANGER knock-out than in wild-type animals (Fig. 4A). Likewise, after MCAO, infarct volume is significantly greater in DANGER knock-out brains than in wild-type brains (Fig. 4B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
DANGER knock-out mice exhibit enhanced susceptibility to brain injury. Brain injury volume was greater in DANGER knock-out mice than in wild-type mice after NMDA injection (A) and MCAO (B). Brain sections were stained with cresyl violet after NMDA injection and with TTC after MCAO to estimate the lesion volume. Infarct areas of all slices were traced and integrated. Data are mean ± SE.
Discussion
In the present study, we have established that DANGER binds physiologically to DAPK. DANGER inhibits DAPK activity in a manner that appears to be physiologically relevant, as DAPK activity is substantially increased in DANGER knock-out mice. We also detected significantly augmented cell death rates in multiple DANGER-deficient cells, including MEFs and primary hippocampal neurons. In addition, DANGER knock-out mice displayed greater neurotoxicity elicited by NMDA and augmented stroke damage compared with wild-type control mice. These findings indicate that loss of DANGER's inhibition of DAPK leads to enhanced cell death. Our protein interaction mapping studies did not identify discrete regions of the two proteins responsible for the binding. This prevented us from developing mutants and dominant-negative constructs to establish definitively that the DANGER–DAPK interaction is responsible for the regulation of cell death. Because DANGER-deficient mice appear to be comparable to wild type in overall health and behavior, it appears unlikely that the enhanced neurotoxicity of the knock-outs can be attributed to general animal debility.
DANGER was first identified as a protein that interacts with IP3 receptors and influences calcium release (van Rossum et al., 2006). Because DAPK is a calcium/calmodulin-dependent enzyme, we conducted binding studies seeking a ternary complex among the three proteins but did not detect such interactions. As DAPK binds to DANGER directly in vitro, IP3 receptors are not required for their interactions and thus may act in pathways distinct from the regulation of DAPK by DANGER. We also performed calcium imaging experiments on bradykinin-treated MEFs but did not observe significant changes of calcium release/entry in the DANGER knock-out cells.
DAPK is a tumor suppressor and a critical player in diverse neurodegenerative diseases, and DANGER is a physiologic inhibitor of the enzyme. Accordingly, compounds that block the interactions between DANGER and DAPK might be effective as antitumor agents. DANGER deletion augments neurotoxicity. Thus, drugs that enhance the inhibitory activity of DANGER on the DAPK signaling pathway might be useful in blocking cell death in vascular stroke and neurodegenerative diseases.
Footnotes
- Received August 11, 2009.
- Revision received October 24, 2009.
- Accepted November 5, 2009.
-
This work was supported by United States Public Health Service Grant DA00266 and Research Scientist Award DA00074 (S.H.S.) and by an American Heart Association Pre-doctoral Fellowship (B.N.K). We thank Drs. A. C. Resnick, R. Bhandari, and K. M. Sixt for helpful discussions.
- Correspondence should be addressed to Solomon H. Snyder, 725 North Wolfe Street, Baltimore, MD 21205. ssnyder{at}jhmi.edu
- Copyright © 2010 the authors 0270-6474/10/300093-06$15.00/0