
Abstract
Episodic ataxia type 2 (EA2) is a hereditary cerebellar ataxia associated with mutations in the P/Q-type voltage-gated calcium (Ca2+) channels. Therapeutic approaches for treatment of EA2 are very limited. Presently, the potassium (K+) channel blocker 4-aminopyridine (4-AP) constitutes the most promising treatment, although its mechanism of action is not understood. Here we show that, in contrast to what is commonly believed, therapeutic concentrations of 4-AP do not increase the inhibitory drive of cerebellar Purkinje cells. Instead, 4-AP restores the severely diminished precision of pacemaking in Purkinje cells of EA2 mutant mice by prolonging the action potential and increasing the action potential afterhyperpolarization. Consistent with this mode of action, the therapeutic efficacy of 4-AP was comparable, and not additive, to chlorzoxazone, an activator of Ca2+-dependent K+ channels that also restores the precision of Purkinje cell pacemaking. The likely target of 4-AP at the concentrations used are the Kv1 family of K+ channels, possibly the Kv1.5 subtype. Because at higher concentrations 4-AP blocks a large array of K+ channels and is a proconvulsant, use of selective Kv1 channel blockers is likely to be a safer substitute for treatment of cerebellar ataxia.
Introduction
Episodic ataxia type 2 (EA2) is a form of hereditary neurological disorder caused by cerebellar malfunction and is characterized by interictal ataxia and frequent attacks of dyskinesia, vertigo, and imbalance that last for hours to days (Jen et al., 2007). EA2 is associated with mutations in the Cav2.1α1 pore-forming subunit of the P/Q-type voltage-gated calcium (Ca2+) channel, which reduce its current density (Fletcher et al., 1996; Ophoff et al., 1996; Dove et al., 1998). These channels are widely expressed in the CNS and are particularly enriched at synaptic nerve terminals (Stea et al., 1994) and in cerebellar Purkinje cells (Mori et al., 1991; Usowicz et al., 1992).
Current treatments for EA2 are limited (Jen et al., 2007; Strupp et al., 2007). Recently, the potassium (K+) channel blocker 4-aminopyridine (4-AP) was used for treatment of this disorder (Strupp and Brandt, 2006; Strupp et al., 2008) in humans and also in animal models of EA2 such as the tottering (tg/tg) mouse (Weisz et al., 2005). The mode of action of 4-AP is not understood. 4-AP has been prescribed based on the assumption that, because of the reduced P/Q-type Ca2+ current density, the Purkinje cell inhibitory drive to neurons of deep cerebellar nuclei (DCN) is reduced (Glasauer et al., 2005; Kalla et al., 2007; Strupp et al., 2008). Because in Purkinje cells 4-AP decreases the latency of Ca2+ spikes evoked with large depolarizing pulses (Etzion and Grossman, 2001), it was thought that 4-AP would restore their inhibitory drive onto DCN neurons (Strupp and Brandt, 2006; Jen et al., 2007; Strupp et al., 2007, 2008). However, several recent findings are inconsistent with these assumptions. Unlike that postulated, the firing rates of mutant Purkinje cells in animal models of EA2 are not lower (Hoebeek et al., 2008; Ovsepian and Friel, 2008). Moreover, despite the presence of morphological synaptic abnormalities, stimulation of Purkinje cells inhibits DCN neurons of tg/tg mice with the same efficacy (mean latency and pause duration) as it does in the wild type (wt) (Hoebeek et al., 2008).
Because as a K+ channel blocker 4-AP is a relatively potent proconvulsant (Bever et al., 1994; Judge et al., 2006), understanding its mode of action is necessary if we are to supersede it by offering safer alternatives. We thus sought to delineate the therapeutic mode of action of 4-AP. We found that at therapeutic concentrations 4-AP neither increased the firing rate nor excitability of Purkinje cells or synaptic drive to DCN neurons. Instead, 4-AP effectively restored the precision of pacemaking in the mutant tg/tg Purkinje cells by increasing the duration of action potentials and the amplitude of afterhyperpolarizations (AHPs). 4-AP's efficacy in restoring the precision of Purkinje cell pacemaking was mimicked by selective blockade of Kv1.5 channels. Consistent with a mode of action on Purkinje cell pacemaking, the therapeutic benefits of 4-AP in the tg/tg mice were not additive to those of chlorzoxazone (CHZ), which also restores the precision of Purkinje cell pacemaking.
Materials and Methods
Cerebellar slices.
All procedures employed were in accordance with the policies established by the Animal Institute Committee of the Albert Einstein College of Medicine.
Postnatal day 12 (P12)–P19 Wistar rats and 2- to 3-month-old tg/tg or age-matched C56BL/6 mice were (wt) anesthetized with halothane and decapitated. The brains were quickly removed and placed in cold extracellular solution containing the following (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 10 glucose, pH 7.4 when gassed with 5% CO2/95% O2. The cerebellums were dissected and mounted on a modified Oxford vibratome and 300-μm-thick sagittal slices were made. The slices were kept in oxygenated extracellular solution at 34°C for 1 h, and then at room temperature until use (1–5 h).
Extracellular recordings.
Slices were placed in a recording chamber on the stage of a Zeiss Axioskop microscope. Purkinje cells were visually identified using a 40× water-immersion objective with infrared optics. The slices were superfused with the extracellular recording solution at a rate of 1.5–2 ml/min and the temperature adjusted to 35 ± 1°C. Extracellular recordings were obtained from single neurons using a homemade differential amplifier and glass pipette electrodes filled with extracellular solution (tip size, 0.3–1 μm). The pipette tip was positioned just above, or lightly touching, the cell body near the axon hillock where the largest potential changes were usually recorded. To isolate the Purkinje cell intrinsic activity, synaptic transmission was blocked using 5 mm kynurenic acid (Spectrum Chemical MFG), a broad-spectrum ionotropic glutamate receptor antagonist (Stone, 1993); 100 μm picrotoxin (Sigma), a GABAA receptor antagonist (Yoon et al., 1993); and 1 μm CGP55845 [(2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl] (phenylmethyl) phosphinic acid; Tocris Bioscience], a GABAB receptor antagonist (Davies et al., 1993). These antagonists were present in all experiments except in those which examined synaptic transmission. 4-AP was obtained from Sigma.
Data were sampled at 10 kHz using an analog-to-digital converter (PCI-MIO-16XE-10; National Instruments), and acquired and analyzed using custom software written in LabView (National Instruments).
In each experiment, interspike interval histograms were constructed using a long stretch of spontaneous activity (>5 min). These histograms were used to calculate the predominant (instantaneous) firing rate, defined as the reciprocal of the interspike interval most frequently observed (the peak of the histogram, i.e., its mode), and the maximum (instantaneous) firing rate, defined as the reciprocal of the shortest interspike interval which accounted for at least 5% of interspike intervals (Womack and Khodakhah, 2003).
Whole-cell recordings.
Whole-cell voltage-clamp recordings were performed using an Optopatch amplifier (Cairn Research) with electrodes pulled from borosilicate glass (1–3 MΩ resistance when filled with intracellular solution). The intracellular solution used was composed of the following (in mm): 70 Cs-gluconate, 10 CsF, 20 CsCl, 10 EGTA, 10 HEPES, and 3 Na2ATP, pH 7.4 (CsOH). Recordings obtained from deep cerebellar neurons were performed using a high-chloride intracellular solution containing the following (in mm): 138 CsCl, 10 CsF, 10 HEPES, 5 NaCl, and 3 MgATP, pH 7.2 (CsOH). For whole-cell current-clamp recordings, the intracellular solution contained the following (in mm): 140 K-methyl sulfate, 10 KCl, 5 NaCl, 4 MgATP, 0.01 EGTA, and 10 HEPES, pH 7.33 (KOH).
Data were sampled at 20 kHz using an analog-to-digital converter (PCI-MIO-16XE-10; National Instruments), and acquired and analyzed using a custom software written in LabView (National Instruments).
Electrical stimulation.
Parallel fibers (PFs) were electrically stimulated by a glass pipette (filled with extracellular solution) placed in the molecular layer, above the Purkinje cell under study. In every trial, a single 100 μs current pulse (2–20 μA intensity) was delivered using a constant current isolated stimulator (DS3; Digitimer LTD). To measure the paired-pulse ratio, two identical electric pulses were delivered separated by a 50-ms-long interval. To stimulate mossy fibers, the stimulation pipette was placed in the white matter tract underneath the granule cell layer that targeted the Purkinje cell under study.
To stimulate Purkinje cells inputs to DCN neurons, the stimulation pipette was positioned between the cerebellar cortex and the nuclei to prevent direct effects on nuclear cells.
Behavioral analysis.
We used the accelerating rotarod test as a paradigm to examine motor performance (Walter et al., 2006; Crawley, 2008). The apparatus consisted of a 3-cm-diameter rotating rod (Rotamex-5; Columbus Instruments) elevated 55 cm above a covered platform. Each trial started from stationary position accelerating at a rate of 0.1 cm/s every second. The animals' speed and latency to fall were automatically recorded by an interfaced computer. Every day, tg/tg mice and wt littermates (4–5 months old) were tested in 10 consecutive trials.
To quantify the frequency, severity, and duration of episodes of dyskinesia observed in tg/tg mice, the overall motor behavior was scored every 10 min before and after each rotarod session for up to 2 h. The scoring followed a scale published previously (Weisz et al., 2005) as follows: 0, normal motor behavior; 1, slightly slowed or abnormal movements; 2, mild impairments, limited ambulation unless disturbed; 3, moderate impairment, limited ambulation even when disturbed, frequent abnormal postures; 4, severe impairment, almost no ambulation, sustained abnormal postures; 5, prolonged immobility in abnormal postures.
The stress and exertion associated with the rotarod trials resulted in long-lasting episodes of paroxysmal dyskinesia. The severe attacks (scores of 4–5) typically last for 30–60 min and are very stereotyped. These attacks start with the involuntary extension of hind limbs followed by lowering of the hips and extending the knees, ankles, and paws. Throughout these movements, the back is abnormally arched. This posturing then spreads to the rest of the body, with particularly severe contractions of the neck and face muscles (Campbell et al., 1999). Most of these characteristics are notable every time a tg/tg mouse has a severe attack, making the severe episodes (scores of 4–5) unambiguously distinguishable from milder motor impairments (scores of 2–3).
Scoring of the severity of the symptoms was performed by one of the authors with previous knowledge of the treatment implemented. To ascertain that this scoring was not biased, a few colleagues who were blind to the treatment were also asked to provide dyskinesia scores by reviewing a number of videotaped episodes. There were no significant differences between the scores assigned by five such observers blind to the treatment of the mice and the scores obtained by the authors.
CHZ and 4-AP were orally administered to tg/tg and wt mice by adding them to their drinking water. The CHZ solution was prepared fresh every day by adding CHZ to a 0.1% solution of hydroxypropyl-β-cyclodextrin (Tocris Bioscience) and then adding a few drops of 1N NaOH until the CHZ was fully dissolved. The solution was supplemented with 10% sucrose to improve its taste and thus ensure its consumption. The 4-AP solution was also prepared daily from a 10× frozen stock. To improve the accuracy of the water consumption measurements, the large water bottles in the cages were replaced with graduated 15 ml plastic tubes. The weight of the animals and the extent of their water intake were monitored daily throughout the experiment.
All data are reported as mean ± SEM. To determine statistical significance, one-way ANOVA was followed by Bonferroni's post test for multiple comparisons or a paired t test (for Fig. 2H only). Differences were considered to be statistically significant only at p < 0.05.
Results
Therapeutic concentrations of 4-AP do not alter the rate of spontaneous activity of cerebellar Purkinje cells
Although the output of Purkinje cells onto DCN neurons is dynamically regulated by their synaptic inputs, Purkinje cells are intrinsically active and fire regularly in the absence of any synaptic input (Hausser and Clark, 1997; Raman and Bean, 1999; Womack and Khodakhah, 2002). Therapeutic concentrations of 4-AP may thus increase the inhibitory synaptic drive of Purkinje cells onto DCN neurons by elevating the rate of their intrinsic pacemaking. In humans, 4-AP has a relatively short half life of 3–4 h (Hayes et al., 2003), and its beneficial effects in alleviating motor symptoms in EA2 and cerebellar and ocular motor disorders have been obtained within an hour of administration of a single 10 mg dose, or with 5 mg doses given thrice daily (Strupp et al., 2004; Kalla et al., 2007). Given its pharmacokinetics in humans, the maximum plasma concentration of 4-AP after a single 10 mg dose corresponds approximately to a concentration just shy of 1 μm. Although throughout this study we explored the effects of 1, 5, and 10 μm 4-AP to more clearly delineate trends in potential physiologic effects, it should be noted that, therapeutically, only the 1 μm concentration is of relevance particularly since higher doses are proconvulsant (Bever et al., 1994).
To explore whether therapeutic concentrations of 4-AP affect the rate of spontaneous activity of Purkinje cells, we noninvasively monitored their activity with extracellular recordings in acutely prepared cerebellar slices. After recording the activity of individual Purkinje cells for several minutes to obtain their baseline firing rate, we applied 4-AP. Bath perfusion of neither 1 nor 5 μm 4-AP affected the rate of spontaneous activity of Purkinje cells (control firing rate, 42.6 ± 2.4 spikes per second; 1 μm 4-AP, 43.8 ± 4.5 spikes per second, p > 0.1; 5 μm 4-AP, 42.6 ± 5.8 spikes per second, p > 0.1; n = 13 cells) (Fig. 1A–C). 4-AP at 10 μm marginally reduced the firing rate (33.9 ± 3.2 spikes per second; p < 0.05), presumably by increasing GABAergic drive onto Purkinje cells, because 4-AP did not affect the firing rate of Purkinje cells when synaptic transmission was pharmacologically blocked (Fig. 1D,F). Comparable results were obtained when GABAergic synaptic transmission was selectively blocked (data not shown). Thus, the data demonstrate that therapeutic concentrations of 4-AP do not affect intrinsic pacemaking of Purkinje cells. By inference, given that at these concentrations 4-AP did not reduce the firing rate of Purkinje cells in experiments where inhibitory synaptic transmission was intact, it is likely that 4-AP also does not appreciably change the firing rate of the molecular layer interneurons.
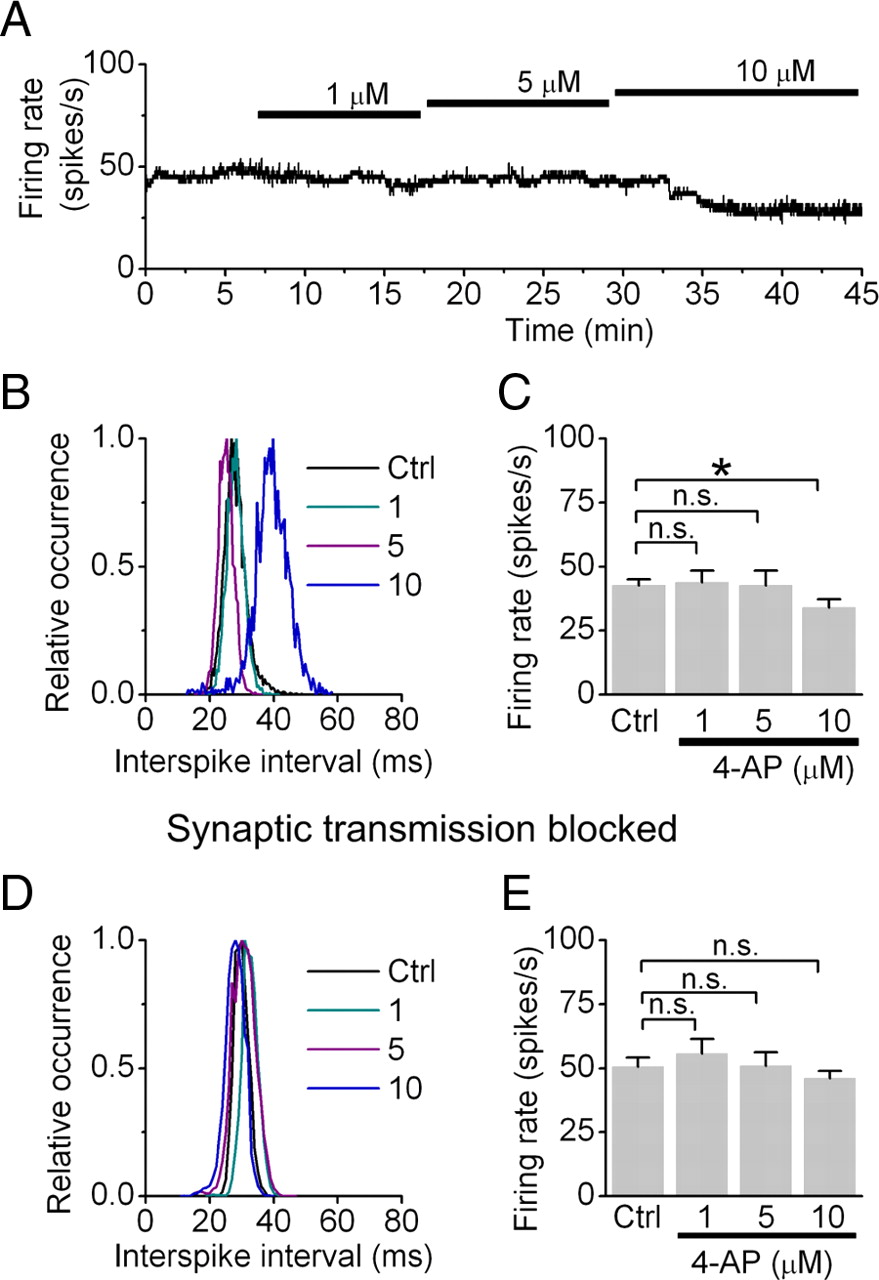
Therapeutic concentrations of 4-AP do not affect the activity in Purkinje cells. A, The firing rate of a cerebellar Purkinje cells was monitored by extracellular recording and sequentially higher concentrations of 4-AP were bath applied. B, Histograms of the interspike interval distributions obtained from the cell shown in A in each condition. C, Average Purkinje cell firing rates in control condition and in the presence of 4-AP. *p < 0.05. D, Distribution of interspike intervals from a Purkinje cell recorded in the presence of blockers of excitatory and inhibitory synaptic transmission under control conditions and when the cell was exposed to 4-AP. E, Average Purkinje cell firing rates in control condition and in the presence of 4-AP with synaptic transmission blocked. n.s., Not significant.
Therapeutic concentrations of 4-AP do not increase release probability at parallel fiber synapses or affect the response of Purkinje cells to synaptic input
We next examined whether therapeutic concentrations of 4-AP potentiate the response of Purkinje cells to excitatory synaptic inputs. Purkinje cells receive excitatory inputs from >150,000 PF synapses. In principle, 4-AP can potentiate PF-evoked responses in Purkinje cells by increasing either the presynaptic or postsynaptic excitability (or both). At some synapses, even low micromolar concentrations of 4-AP can increase neurotransmitter release by broadening the action potential, thus leading to greater Ca2+ influx (Sacco and Tempia, 2002). 4-AP can also increase the response of a Purkinje cell to the same synaptic input if it blocks dendritic or perhaps even somatic K+ channels. We explored these possibilities by first quantifying the response of spontaneously firing Purkinje cells to extracellular electrical stimulation of PFs. The intensity of the electrical stimulation was adjusted such that the maximum firing rate of the target Purkinje cells increased to about 100 spikes per second (mean, 113.4 ± 9.1 spikes per second; n = 8 cells). 4-AP, in a concentration-dependent manner, potentiated these responses (Fig. 2A,B) such that PF-evoked responses marginally increased to 129.1 ± 16.4 spikes per second in the presence of 1 μm 4-AP (p > 0.1), to 174.6 ± 28.8 spikes per second with 5 μm 4-AP (p < 0.05), and to 228.6 ± 37.7 spikes per second with 10 μm 4-AP (p < 0.001).

Therapeutic concentrations of 4-AP do not alter neurotransmitter release probability at parallel fiber synapses. A, Example traces of the response of an extracellularly recorded Purkinje cell to the electrical stimulation of PFs, in control conditions (Ctrl) and in increasing concentrations of 4-AP. B, Average Purkinje cell maximum firing rate in response to PF stimulation under various conditions (n = 8 cells). *p < 0.05, ***p < 0.001 (one-way ANOVA with Bonferroni's post test for multiple comparisons). C, Parallel fiber-evoked EPSCs recorded from a voltage-clamped Purkinje cell, under control conditions and sequential application of increasing concentrations of 4-AP. D, Average EPSC amplitudes in each condition reported above (n = 12 cells). *p < 0.05, ***p < 0.001. E, A comparison of the efficacy of 4-AP in increasing the amplitude of PF-evoked EPSCs versus PF-evoked increases in the firing rate of Purkinje cells. F, Paired-pulse ratio of the amplitudes of the two EPSCs evoked by paired activation of PFs (50 ms apart) in voltage-clamped Purkinje cells. 4-AP did not alter this ratio at any of the concentrations examined. G, Examples of mossy fiber (MF)-evoked EPSCs in voltage-clamped Purkinje cells under control conditions and in the presence of 5 μm 4-AP. H, Average of the MF-evoked EPSCs before and after application of 5 μm 4-AP. n.s., Not significant. p > 0.85 (paired t test).
To delineate the mechanism by which 4-AP potentiates PF-evoked responses in Purkinje cells, we examined its effect on PF-evoked EPSCs in voltage-clamped Purkinje cells. Use of a Cs-based internal solution to block K+ channels ensured a better space clamp and eliminated the possibility of a postsynaptic action of 4-AP. Similar to that seen with PF-evoked increases in the firing rate of Purkinje cells, 4-AP potentiated PF-evoked EPSCs in a concentration-dependent manner (Fig. 2C,D) (n = 12 cells). The average peak amplitude of EPSCs was 318 ± 24 pA under control conditions. With 1 μm 4-AP it increased to 399 ± 46 pA, although this value was not statistically different from control (p > 0.1). The average EPSC amplitude increased further to 473 ± 59 pA with 5 μm (p < 0.05), and to 614 ± 59 pA with 10 μm 4-AP (p < 0.001). Moreover, the extent to which 4-AP augmented PF-evoked EPSCs was the same as its potentiating effects on PF-evoked increases in the firing rate (Fig. 2E). Because in Purkinje cells the increase in the firing rate is a linear function of the charge injected by the PF-evoked EPSCs (Walter and Khodakhah, 2006), our data demonstrate that therapeutic concentrations of 4-AP do not affect integration of synaptic inputs by Purkinje cells by altering their dendritic or somatic excitability. Instead, the effects of low concentrations of 4-AP are solely restricted to increasing the amplitude of EPSCs.
In the experiments reported above, a number of PFs were activated by a brief current applied to an extracellular stimulation electrode placed in the molecular layer. With this stimulation paradigm, the increase in the evoked-EPSC amplitudes in the presence of 4-AP could be attributable to either augmented neurotransmitter release from the same number of PFs, an increase in the number of PFs activated by each stimulation, or both. The former can occur if 4-AP blocks a sufficient number of K+ channels in PF terminals to broaden the presynaptic action potential. This will yield a larger Ca2+ influx, thus increasing the release probability at each synapse. Alternatively, if blockade of axonal K+ channels with 4-AP were to increase their input resistance, more PFs might be brought to threshold with each stimulation. To delineate the potential contribution of each of these mechanisms to the increased EPSC amplitudes in the presence of 4-AP, we examined the paired-pulse ratio of two successive EPSCs before and after application of 4-AP. At the PF-to-Purkinje cell synapse changes in the paired-pulse ratio are faithfully correlated with changes in release probability (Foster et al., 2002). Under control conditions, when PFs were electrically activated with a pair of stimulation pulses 50 ms apart, the ratio of the peak amplitude of the second EPSC to the first was on average 1.43 ± 0.03 (Fig. 2F). This paired-pulse ratio was not significantly different in the presence of 1 μm 4-AP (1.43 ± 0.14; p > 0.3), 5 μm 4-AP (1.36 ± 0.04; p > 0.3), or even 10 μm 4-AP (1.48 ± 0.03; p > 0.3). As a control we increased the concentration of 4-AP even further. As reported previously (Buckle and Haas, 1982), with 100 μm 4-AP the paired-pulse ratio reduced to 1.14 ± 0.12, and with 1 mm 4-AP the paired-pulse facilitation changed to significant paired-pulse depression (data not shown). These results indicate that at therapeutically relevant concentrations, 4-AP does not change the probability of neurotransmitter release at PF synapses. Instead, under our experimental conditions, 4-AP simply increased the number of PFs that are brought to threshold with each stimulation (although at the therapeutically relevant concentration of 1 μm, the effects were small and not statistically significant). These effects of 4-AP are unlikely to be of physiological significance and possibly reflect the necessarily artifactual manner in which PFs were activated in these experiments. This is because in vivo granule cells, the neurons whose axons form PFs, are brought to threshold for firing of an action potential even by single quanta released from their mossy fiber synaptic inputs (Chadderton et al., 2004), and thus with therapeutic concentrations of 4-AP one would not anticipate a significant increase in the number of PFs activated under physiological conditions. In agreement with this supposition, as shown in Figure 2, G and H, 5 μm 4-AP failed to potentiate PF-evoked EPSCs in voltage-clamped Purkinje cells when the stimulation electrode was positioned in the white matter to activate the mossy fiber inputs to the granule cells (control EPSC amplitude, 159 ± 26 pA vs 5 μm 4-AP, 162 ± 30 pA; p > 0.85, paired t test; n = 3 cells). Similarly, the paired-pulse ratios of EPSCs evoked by mossy fiber stimulation were not affected after addition of 5 μm 4-AP (control, 1.18 ± 0.05 vs 4-AP, 1.21 ± 0.4; p > 0.1).
Comparable experiments demonstrated that therapeutic concentrations of 4-AP do not alter release probability at the Purkinje cell to DCN synapses (see supplemental material 1, available at www.jneurosci.org).
Therapeutic concentrations of 4-AP increase the precision of Purkinje cell pacemaking in tottering mice
The experiments described above show that therapeutic concentrations of 4-AP do not increase the excitability or rate of activity of Purkinje cells. Moreover, 4-AP does not increase the release probability at the PF-to-Purkinje cell or the Purkinje cell-to-DCN synapses. Combined, in contrast with what is widely believed (Strupp and Brandt, 2006; Jen et al., 2007; Strupp et al., 2007, 2008), our data suggest that it is unlikely that the therapeutic mode of action of 4-AP in EA2 is by increasing the inhibitory drive of Purkinje cells onto DCN neurons. An alternative hypothesis regarding the therapeutic mode of action of 4-AP proposed by Otis and Jen (2006) is that 4-AP restores the precision of Purkinje cell pacemaking by broadening the duration of action potentials. This mode of action is based on the findings that loss in the precision of Purkinje cell pacemaking has been suggested to contribute to the motor symptoms of EA2 (Walter et al., 2006). Indeed, in vivo cerebellar perfusion of the KCa channel activator 1-ethyl-2-benzimidazolinone (EBIO) restores the precision of Purkinje cell pacemaking and significantly improves motor function in mouse models of EA2 (Walter et al., 2006). This potential therapeutic mode of action of 4-AP, although not experimentally examined, has been dismissed.
To test this hypothesis, we examined the activity of Purkinje cells in acutely prepared cerebellar slices from the tg/tg mice. These mice suffer from a spontaneous mutation in the pore-forming subunit of the P/Q-type Ca2+ channel, which reduces their current density (Fletcher et al., 1996), and are one of the most studied and best established models of EA2 (Jinnah et al., 2005; Pietrobon, 2005). Similar to EA2 patients, the tg/tg mice show baseline ataxia and episodes of severe dyskinesia, which are triggered by stress, caffeine, and ethanol (Fureman et al., 2002; Shirley et al., 2008). Although the mutation in tg/tg mice results in a very large decrease in the P/Q Ca2+ current density and more severe episodes of dyskinesia than typically seen in EA2 patients, these mice have served as a very useful and reliable model of EA2 both for examining the etiology of the disorder and also for surveying therapeutic options. In fact, acute administration of 4-AP is as effective in alleviating the motor symptoms of the tg/tg mice as it is in humans (Weisz et al., 2005).
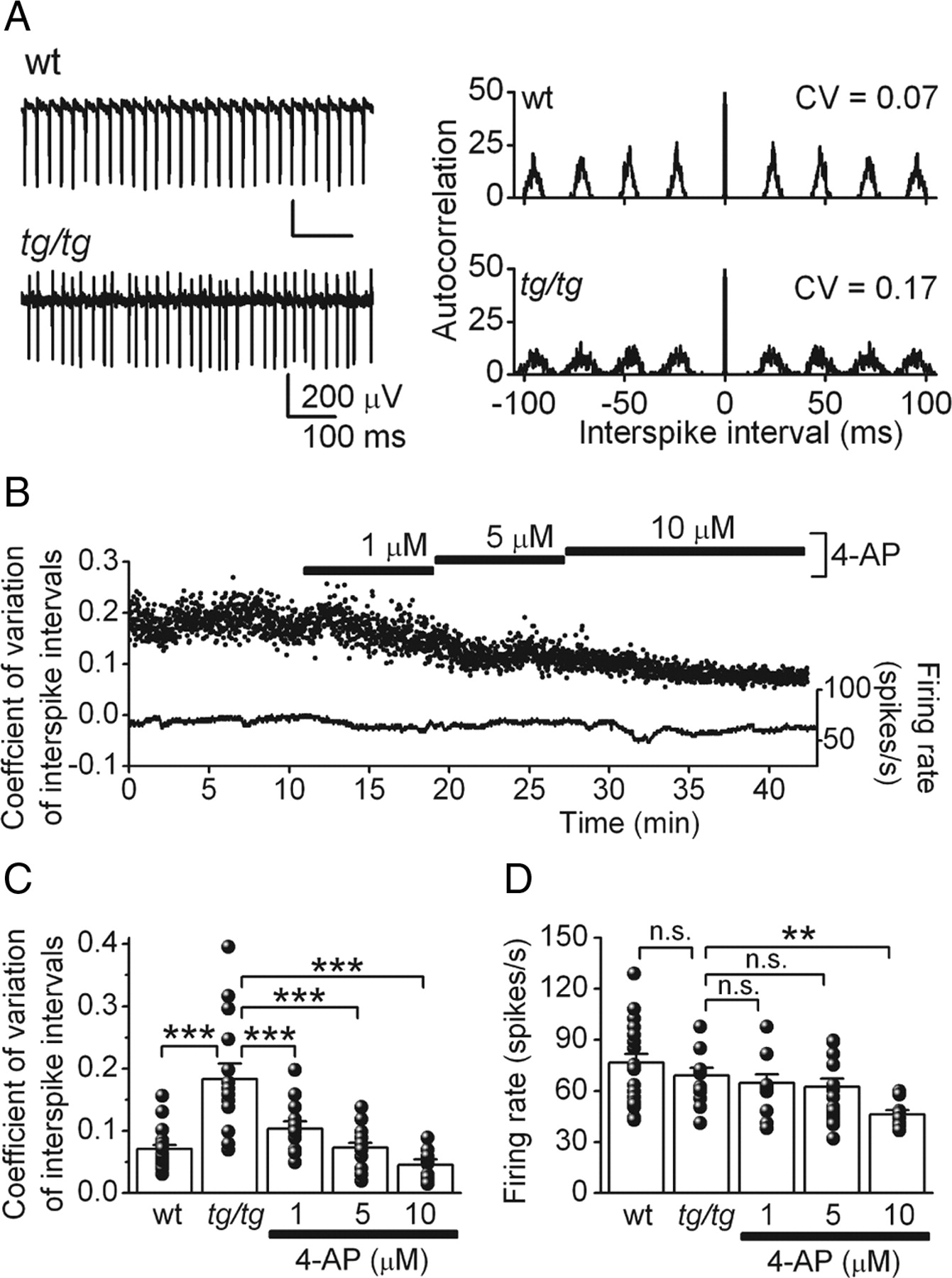
Consistent with observations in other mutant animal models of EA2 such as the ducky and leaner mice (Walter et al., 2006; Ovsepian and Friel, 2008), we found that the intrinsic pacemaking of tg/tg Purkinje cells was also highly erratic (Fig. 3A–C). Thus, the coefficient of variation (CV) of the interspike intervals in tg/tg Purkinje cells was 0.18 ± 0.03 (Fig. 3C) (n = 15 cells), a value significantly higher than 0.07 ± 0.01 seen in age-matched wt Purkinje cells (p < 0.001; n = 22 cells). Application of 4-AP, in a concentration-dependent manner, made the firing of tg/tg Purkinje cells more regular (Fig. 3B,C). Even at a concentration of 1 μm, 4-AP reduced the coefficient of variation of the interspike intervals to 0.10 ± 0.01 (p < 0.001; n = 15 cells). 4-AP at 5 μm reduced the coefficient of variation of the interspike intervals to 0.07 ± 0.01 (p < 0.001; n = 20 cells), and at 10 μm to 0.05 ± 0.01 (p < 0.001; n = 12 cells). The latter values were not statistically different from that seen in age-matched wt Purkinje cells (p > 0.1). Whereas 10 μm 4-AP reduced the firing rate of tg/tg Purkinje cells from an average of 69.2 ± 4.5 spikes per second to 46.1 ± 2.7 spikes per second (p < 0.01), the effects of 1 and 5 μm 4-AP on the firing rate were minor and not statistically significant (1 μm 4-AP, 64.6 ± 5.1 spikes per second; 5 μm 4-AP, 62.4 ± 4.8 spikes per second; p > 0.1) (Fig. 3D). Thus, 4-AP at the therapeutically relevant concentration of 1 μm significantly improves the precision of pacemaking of tg/tg Purkinje cells. In a separate set of studies, we used low concentrations of cadmium to block Ca2+ channels to decrease the precision of pacemaking in rat Purkinje cells (Walter et al., 2006). We found that the precision of pacemaking in these cells was restored with the same concentrations of 4-AP that were effective in tg/tg mice (see supplemental material 2, available at www.jneurosci.org).

Therapeutic concentrations of 4-AP restores the precision of Purkinje cell pacemaking in the ataxic tg/tg mouse. A, Left, Raw traces from extracellular recordings obtained from tg/tg and wt Purkinje cells. Right, The corresponding autocorrelogram of the spontaneous activity of each cell. Note the increased variability in the interspike interval in the tg/tg cell. B, Coefficient of variation of interspike intervals and the firing rate of an extracellularly recorded tg/tg Purkinje cell under control conditions, and with sequential application of therapeutic concentrations of 4-AP. C, Average and individual values of coefficient of variation of interspike intervals in tg/tg Purkinje cells in control conditions and in the presence of 4-AP. The coefficient of variation of wt Purkinje cells is plotted for comparison. D, Average and individual values of the corresponding firing rates for the same cells shown in C. **p < 0.01; ***p < 0.001 (one-way ANOVA followed by Bonferroni's post test for multiple comparisons). n.s., Not significant.
Therapeutic concentrations of 4-AP broaden the action potential and increase the amplitude of afterhyperpolarization
Activators of Ca2+-dependent K+ channels restore the precision of Purkinje cell pacemaking in EA2 animal models by increasing the amplitude of action potential AHP (Walter et al., 2006). We tested the possibility that by blocking a subset of K+ channels 4-AP restores the precision of Purkinje cell pacemaking by broadening their action potentials, thus increasing the amplitude of their AHPs.
We first examined the extracellularly recorded spikes. These voltage deflections correspond to the derivative of the actual membrane potential of the target Purkinje cell. Thus, the start of the negative voltage deflection corresponds to the start of the rising phase of the action potential, and the positive peak of the signal relates to the time at which the rate of membrane potential repolarization is the greatest, that is, the time at which the net outward ionic current is at its maximum. The positive voltage thereafter corresponds with the continuation of the down stroke of the action potential and its AHP. Compared with the direct examination of the action potential width with whole-cell recordings, using the extracellular signals has the advantage that it avoids dialysis of the cell and the associated changes in Ca2+ buffering and homeostasis. Quantification of the data demonstrated that 4-AP broadens the action potential in a concentration-dependent manner (Fig. 4A,B). The time to the peak positive voltage deflection (measured as the time taken from the beginning of the negative deflection to the peak of the positive deflection) increased from an average of 0.75 ± 0.04 ms under control conditions to 0.94 ± 0.05 ms in 1 μm 4-AP (p < 0.05), and to 1.06 ± 0.03 ms and 1.12 ± 0.06 ms with 5 and 10 μm 4-AP, respectively (p < 0.001 for both cases). Consistent with the hypothesis that broadening of the action potential was the cause of improved pacemaking, there was a clear linear relationship between the average duration and the coefficient of variation (Fig. 4C). In agreement with the notion that 4-AP prolongs the AHP, in the presence of 4-AP, the positive deflections of the extracellular signals were longer (Fig. 4A, inset).

4-AP broadens action potentials in Purkinje cells. A, Averaged extracellularly recorded action potentials in a tg/tg Purkinje cell under control conditions and in the presence of different concentrations of 4-AP. Inset, The average of the normalized waveforms in each condition. B, Average and individual values of time to peak positive deflection, measured as indicated at the top of the inset in A. C, Comparison of the efficacy of 4-AP in improving the precision of Purkinje cell pacemaking (represented by the coefficient of variation of interspike intervals) and its efficacy in prolonging the duration of the spike. Red trace represents the linear fit (r = −0.99; p = 0.007). D, Averaged action potentials recorded in whole-cell configuration in a wt juvenile rat Purkinje cell, in control conditions, and after the application of therapeutic concentrations of 4-AP. E, Average and individual values of maximum AHP potentials in six Purkinje cells. F, Average and individual values of the AHP amplitudes of the same cells in E. G, Average and individual values of the action potential width of the same cells in E. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA with Bonferroni's post test for multiple comparisons).
As a complementary approach, we also examined the effect of 4-AP on the action potential waveform using whole-cell current-clamp recordings, taking into consideration the caveat that whole-cell recordings do dialyze the cells and affect Ca2+ buffering (and may also alter other intracellular biochemical signaling pathways). As can be noted in the sample traces shown, 4-AP dose-dependently increased the width of the action potential and also the absolute AHP potential (Fig. 4D). The average maximum AHP potential in control conditions was −59.8 ± 1.6 mV (n = 6 cells), which, after application of 1 μm 4-AP, increased to −61.4 ± 0.6 mV (Fig. 4E). 4-AP at 5 and 10 μm increased the maximum AHP potential further to −63.4 ± 1.1 mV (p < 0.05) and −65.8 ± 1.3 mV (p < 0.001), respectively. In addition to the absolute AHP potential, the AHP amplitude (measured from threshold) increased significantly with 4-AP (Fig. 4F) (control, 10.9 ± 0.5 mV; 1 μm 4-AP, 11.3 ± 0.2 mV; 5 μm 4-AP, 13.7 ± 0.6 mV, p < 0.05; 10 μm 4-AP, 14.8 ± 0.9 mV, p < 0.01). Finally, the width of the action potential (measured as the time taken from threshold to 10% peak action potential amplitude during the down stroke of action potential) also increased when 4-AP was added (Fig. 4G) (control, 0.50 ± 0.01 ms; 1 μm 4-AP, 0.51 ± 0.017 ms; 5 μm 4-AP, 0.55 ± 0.03 ms, p < 0.01; 10 μm 4-AP, 0.56 ± 0.015 ms, p < 0.001).
Collectively the data presented demonstrate that at concentrations effective in the treatment of cerebellar-related motor disorders, 4-AP increases the regularity of a Purkinje cell's pacemaking by prolonging the duration of its action potentials. The broader action potentials are accompanied with larger AHPs, presumably because of the greater Ca2+ influx associated with the broader action potentials and the additional activation of Ca2+-dependent K+ conductances. This larger Ca2+ influx is likely to compensate, in part, for the reduced P/Q-type Ca2+ current density associated with EA2 mutations (Fletcher et al., 1996; Dove et al., 1998; Barclay et al., 2001).
Therapeutic concentrations of 4-AP most likely block Kv1.5 potassium channels
Based on the affinity of 4-AP for block of various K+ channel subtypes (Coetzee et al., 1999) and the expression pattern of channels in the cerebellum, and specifically in Purkinje cells (Madeja et al., 1997; Chung et al., 2001, 2005; Martina et al., 2003; Hurlock et al., 2008), the most likely target of therapeutic concentrations of 4-AP are the Kv1 family of K+ channels, specifically the Kv1.5 channels that have the highest affinity for 4-AP. To a much lesser extent, Kv3.1 or Kv3.3 channels could also be the targets of 4-AP. Therefore, we tested whether the Kv1.5 channel blocker 2-isopropyl-5-methylcyclohexyl (DPO) (Lagrutta et al., 2006) could substitute for 4-AP in restoring the precision of pacemaking. We monitored the activity of normal Purkinje cells in slices by extracellular recordings, and as before applied cadmium to partially block Ca2+ channels and make the firing of Purkinje cells irregular. This was reflected as an increase the coefficient of variation of the interspike intervals (Fig. 5A).

Blockade of Kv1.5 channels mimics the effects of 4-AP. A, Coefficient of variation of interspike intervals an extracellularly recorded Purkinje cell under control conditions, and with application of increasing concentrations of cadmium to partially block Ca2+ channels and make the firing rate of the cell irregular. Bath perfusion of the selective Kv1.5 K+ channel blocker DPO restored the precision of pacemaking. Subsequent addition of 10 μm 4-AP did not improve the precision of pacemaking beyond that done by DPO. B, C, Average and individual values of coefficient of variation of interspike intervals (B) and firing rate (C) in Purkinje cells in control conditions, in the presence of DPO, and with subsequent addition of 4-AP. ***p < 0.001 (one-way ANOVA followed by Bonferroni's correction). n.s., Not significant.
Bath application of 5 μm DPO decreased the coefficient of variation Purkinje cell firing to levels comparable to control conditions (Fig. 5A,B) without altering the firing rate (Fig. 5C). The coefficient of variation of interspike intervals in control conditions was 0.052 ± 0.005, which, after application of cadmium, increased to 0.114 ± 0.007 (Fig. 5B) (n = 6–12 cells; p < 0.001). Application of DPO reduced it to 0.071 ± 0.003 (p < 0.001 vs cadmium). Furthermore, application of 10 μm 4-AP after DPO did not change further reduce the coefficient of variation of interspike intervals (CV, 0.07 ± 0.003 after application of 4-AP, p < 0.001 vs cadmium, p > 0.1 vs DPO). This occlusion of effects suggests (but of course does not prove) that the effects of 4-AP were likely mediated by the blockade of Kv1 channels.
The therapeutic actions of 4-AP in vivo are consistent with improving the precision of pacemaking
The data presented above suggest that by restoring the precision of Purkinje cell pacemaking, the therapeutic mode of action of 4-AP in EA2 may be the same as that of EBIO, an activator of Ca2+-dependent K+ (KCa) channels. EBIO improved the precision of Purkinje cell pacemaking in slices of EA2 mutant mice and lessened their ataxia when chronically perfused into their cerebellum in vivo (Walter et al., 2006). Although in vivo perfusion of EBIO could, in principle, affect other cerebellar neurons such as DCN cells, at the concentrations used, their most likely target were cerebellar Purkinje cells (Alvina and Khodakhah, 2008). If the primary mode of action of 4-AP is indeed restoring the precision of Purkinje cell pacemaking, then one would anticipate that the beneficial effects of 4-AP in reducing motor symptoms should not be additive to those of KCa channel activators; i.e., coadministration of 4-AP with a KCa channel activator should only be as effective as the sole administration of either drug with the highest efficacy. We tested this hypothesis by examining whether treatment of tg/tg mice concurrently with both a KCa channel activator and 4-AP was more effective in reducing their motor symptoms than with either one alone.
We first calculated the concentration of 4-AP that needed to be added to the drinking water of tg/tg mice to achieve the therapeutic concentration of 4-AP found in the plasma of patients (∼50 ng/ml). Given that in rodents the half-life of 4-AP is ∼2 h (Capacio et al., 1996), and considering that the tg/tg mice weigh an average of 20 g, we estimated that a daily 4-AP consumption of ≈0.1 mg will result in an equivalent, but temporally uniform, 4-AP plasma concentration in mice. Since, on average, the tg/tg mice consume ∼2.5 ml of water daily, we supplied them with a 425 μm solution of 4-AP, which they readily drank.
Patients affected by EA2 not only have episodic attacks of dyskinesia, but also show mild baseline ataxia that progress in severity with time (Jen et al., 2004). Thus, we examined the efficacy of oral administration of 4-AP in alleviating baseline ataxia in the tg/tg mice using an accelerating rotarod paradigm (Walter et al., 2006; Crawley, 2008). The performance of tg/tg (n = 13) but not the wt mice (n = 14) increased on the rotarod when 4-AP was added to their drinking water (Fig. 6A). During the pretreatment period, the performance of the tg/tg on the rotarod reached on average 10.9 ± 0.8 rpm (Fig. 6B), whereas during treatment with 4-AP it increased to 13.8 ± 0.9 rpm (p < 0.05). 4-AP had no effect on the performance of the wt mice (pretreatment, 34.5 ± 0.9 rpm; 4-AP, 34.9 ± 1.4 rpm; p > 0.1).

The therapeutic efficacy of orally administered 4-AP in improving basal motor performance in tg/tg mice is not additive to that of CHZ. A, The performance of tg/tg (n = 13) and wt mice (n = 14) was measured using the accelerating rotarod paradigm. After 8 d of receiving normal drinking water, the drinking water was supplemented with 4-AP, 4-AP and CHZ combined, and CHZ alone as denoted with colored vertical bars. Each treatment lasted for 2 weeks. B, Maximum speed achieved on the rotarod during each treatment. The average value was obtained by averaging the second week of each treatment period. **p < 0.01; ***p < 0.001 (one-way ANOVA with Bonferroni's correction). n.s., Not significant.
We then set out to examine whether the beneficial effects of 4-AP were additive to those of the KCa channel activator CHZ (Syme et al., 2000). CHZ is an analog of EBIO (Cao et al., 2001) and crosses the blood–brain barrier (Chou et al., 2004), thus allowing it to be supplied to mice by adding it to their drinking water. In experiments performed in acutely prepared cerebellar slices, we recently determined the concentrations of CHZ that restores the precision of pacemaking in tg/tg Purkinje cells (Alviña and Khodakhah, 2010). Moreover, using this information, and taking into account the pharmacokinetics of CHZ (Wan et al., 2006), we have found that supplementing the drinking water of the tg/tg mice with 15 mm CHZ markedly improves their performance on the rotarod (Alviña and Khodakhah, unpublished observation). We thus examined whether in the same group of mice treated with 4-AP the beneficial effects of CHZ were additive. Although the performance of the tg/tg mice on the rotarod improved further when, in addition to 4-AP, CHZ was also added to their drinking water, this increase was small and just shy of being statistically different (from 13.8 ± 0.9 rpm in 4-AP alone to 16.2 ± 0.9 rpm with the addition of CHZ; p = 0.06). The performance of tg/tg mice continued at its high level when 4-AP was removed and the mice received only CHZ (16.2 ± 0.8 rpm; p > 0.1 vs 4AP and CHZ combined), suggesting that there was little benefit in combining the two drugs. Throughout the various treatments, the performance of wt mice did not change (pretreatment, 34.5 ± 0.9 rpm; 4-AP alone, 34.9 ± 1.4 rpm; 4-AP and CHZ, 34.9 ± 1.1 rpm; CHZ alone 35.0 ± 1.1 rpm; posttreatment, 35.5 ± 1.1 rpm; p > 0.5 for all treatments vs pretreatment). Thus, at least with respect to improving baseline motor function, the data are consistent with the hypothesis that both 4-AP and CHZ may have a common mode of action.
It is plausible that although the mode of action of 4-AP in alleviating baseline ataxia may be the same as that of KCa activators, 4-AP may have a different mechanism of action in preventing the episodes of dyskinesia associated with EA2. We thus similarly compared the efficacy of 4-AP alone with that of its coadministration with CHZ in reducing the frequency, severity, and duration of stress-induced episodes of dyskinesia triggered by the rotarod session in tg/tg mice (Fig. 7A). Both 4-AP and CHZ significantly reduced the average overall severity of symptoms after stress (Fig. 7B), although CHZ produced a slightly larger reduction, and there was little benefit in the coadministration of the two drugs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The therapeutic efficacy of orally administered 4-AP in reducing the frequency and severity of stress-induced episodes of dyskinesia in tg/tg mice is not additive to that of CHZ. A, The stress-triggered attacks of dyskinesia were quantified at 10 min intervals for the same mice examined using the accelerating rotarod paradigm reported in Figure 4. The beginning and end of each treatment is indicated with colored bars on top. B, The average severity of dyskinesia during each period for the same tg/tg mice shown in A. C, The average frequency of occurrence of an episode of dyskinesia after the rotarod session, regardless of the severity of the attack. D, The average severity of all attacks of dyskinesia before, during, and after the three different treatments. E, The duration of all attacks of dyskinesia shown in D. F, The average frequency of occurrence of a severe episode of dyskinesia (dyskinesia score ≥3.5) triggered by the rotarod session. G, The average severity of the attacks shown in F. H, Average duration of severe episodes of dyskinesia. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by Bonferroni's post test). ns, Not significant. I, Summary comparison of the relative efficacies of 4-AP and CHZ in reducing stress-evoked attacks of dyskinesia in tg/tg mice. Frequency of sessions in which tg/tg mice were free of symptoms (no attack) or had an episode of dyskinesia with a score <3.5 (mild) or one with a score ≥3.5 (severe) with various treatments.
To scrutinize the impact of these treatments on frequency, duration, and severity of episodes, we first analyzed all attacks of dyskinesia regardless of their individual severity. 4-AP treatment reduced the frequency of attacks to 81.7 ± 5.2% of the baseline pretreatment values (Fig. 7C) (p < 0.01). Addition of CHZ to 4-AP further reduced the frequency of attacks to 59.6 ± 7.8% (p < 0.001 vs pretreatment). When CHZ was administered without 4-AP, the frequency of all attacks were 50.0 ± 5.4% of baseline (p < 0.001 vs pretreatment), a value statistically different when compared with the 4-AP treatment alone (p < 0.05), but not statistically different from CHZ and 4-AP combined (p > 0.1). After returning mice to normal drinking water, the frequency of attacks in the tg/tg mice increased to its pretreatment values (98.5 ± 1.5%; p > 0.1 vs pretreatment). In addition, treatment with both 4-AP and CHZ comparably reduced the severity (Fig. 7D) and the duration of all attacks of dyskinesia (Fig. 7E).
The occurrence of the most severe attacks (those with a maximum score of ≥3.5) was also reduced by all three treatments (Fig. 7F). Even though both 4-AP and CHZ were effective in reducing the frequency of these severe attacks, we found that CHZ was slightly more effective than 4-AP alone (22.1 ± 3.1% of baseline vs 33.7 ± 3.2%; p < 0.05), and there was no advantage in coapplication of 4-AP with CHZ. In all cases, when severe attacks occurred they were as severe as those before treatment (Fig. 7G), although their duration was significantly and comparably briefer with all three treatments (Fig. 7H).
The pie charts in Figure 7I directly compare the efficacy of 4-AP, CHZ, and 4-AP with CHZ in reducing the frequency of stress-evoked attacks of dyskinesia in tg/tg mice. Although both 4-AP and CHZ potently reduce the frequency of attacks, CHZ is much more effective in increasing the percentage of attack-free sessions in which stress did not cause any motor impairment whatsoever (50% in CHZ alone vs 18.3% during 4-AP). Moreover, there was no benefit in coapplication of the two compounds.
In summary, our findings are consistent with the hypothesis that both CHZ and 4-AP have a common mode of action. Moreover, it is plausible and perhaps quite likely that their therapeutic efficacies are the consequence of their ability to improve the precision of pacemaking in Purkinje cells.
Discussion
Etiology of EA2
EA2 is the most prevalent form of episodic ataxia (Jen et al., 2007). The symptoms of EA2 patients are mostly cerebellar in origin, and in animal models of EA2 removal of the cerebellum, or genetic elimination of Purkinje cells eliminates the episodes of dyskinesia (Campbell et al., 1999; Grusser-Cornehls and Baurle, 2001). The Ca2+ channels that are affected in EA2 are highly expressed throughout the CNS (Evans and Zamponi, 2006) and are particularly enriched in axon terminals and in cerebellar Purkinje cells (Mori et al., 1991; Usowicz et al., 1992; Stea et al., 1994). Ca2+ influx primarily through these channels mediates synaptic transmission at CNS nerve endings (Evans and Zamponi, 2006) and initial hypotheses regarding the etiology of EA2 focused on the dysfunction of cerebellar synapses. However, although alterations in synaptic transmission at the parallel and climbing fiber synapses are notable in some animal models of EA2, they are relatively minor and not as profound as that anticipated, mainly as a consequence of functional compensation by other voltage-gated Ca2+ channels (Matsushita et al., 2002; Ovsepian and Friel, 2008).
Within the cerebellum P/Q-type Ca2+ channels are particularly enriched in Purkinje cells, where the dendritic ones generate Ca2+ action potentials (Llinas and Sugimori, 1980), and the somatic ones provide the sole source of Ca2+ for activation of KCa channels (Womack et al., 2004). Indeed, in Purkinje cells the net Ca2+-dependent current associated with each action potential is outward (Raman and Bean, 1999), and the P/Q-channel-mediated activation of KCa channels is required to maintain the precision of their pacemaking (Womack and Khodakhah, 2004; Walter et al., 2006). In animal models of EA2, the precision of Purkinje pacemaking is significantly deteriorated, and there is little evidence for the presence of compensatory mechanisms (Hoebeek et al., 2005; Walter et al., 2006). It has therefore been suggested that irregular firing of cerebellar Purkinje cells contributes to motor symptoms associated with EA2 (Walter et al., 2006).
Therapeutic approaches to EA2
There are presently few therapeutic options available for EA2 patients, with acetazolamide (ACTZ) and 4-AP constituting the most commonly used drugs (Jen et al., 2007; Strupp et al., 2007). Many patients respond well to ACTZ, which both improves some of the baseline symptoms and reduces the frequency of episodes of dyskinesia (Zasorin et al., 1983; Friedman and Hollmann, 1987; Harno et al., 2004). However, with time, many patients become nonresponsive to ACTZ treatment (Jen et al., 2007; Strupp et al., 2007). The mechanism of action of ACTZ in the treatment of EA2 is not understood. Because ACTZ is a carbonic anhydrase inhibitor (Maren, 1967), it is suggested that ACTZ prevents elevations in the intracellular pH (Strupp et al., 2007), although it is not clear why this should be helpful.
More recently 4-AP has been used to reduce the symptoms of EA2 (Strupp et al., 2004; Lohle et al., 2008), and it is found to be effective in both improving baseline motor coordination and in reducing the frequency and severity of the episodic attacks (Glasauer et al., 2005; Strupp et al., 2007; Lohle et al., 2008). However, 4-AP has to be used with caution since as a K+ channel blocker it can be epileptogenic (Bever et al., 1994; Judge and Bever, 2006).
The therapeutic mode of action of 4-AP
4-AP was first prescribed based on the notion that, because of the reduced Ca2+ current density associated with EA2 mutations, Purkinje cells must be less active (Strupp et al., 2004, 2005; Glasauer et al., 2005). The remarkable efficacy of 4-AP in alleviating cerebellar motor symptoms in several patients has indirectly substantiated this presumed mode of action (Strupp et al., 2004; Glasauer et al., 2005; Kalla et al., 2007; Lohle et al., 2008). The data presented here demonstrate that therapeutic concentrations of 4-AP do not alter the rate of activity of Purkinje cells. Nor does 4-AP, at the relevant concentrations, alter synaptic transmission at the parallel fiber-to-Purkinje cell or the Purkinje cell-to-DCN synapses. The only functional consequence of application of 4-AP that we could discern was that of restoring the precision of pacemaking in the EA2 mutant Purkinje cells by blocking K+ channels and prolonging the AP and increasing its AHP.
The therapeutic mode of action of 4-AP described here is consistent with the finding that in mouse models of EA2, direct perfusion of the KCa channel activator EBIO into the cerebellum to improve the precision of Purkinje cell pacemaking lessens their motor symptoms (Walter et al., 2006). The fact that the beneficial effects of CHZ and 4-AP were not additive also corroborates the hypothesis that the mode of action of 4-AP might be the same as EBIO (although this does not by any means constitute proof). The greater efficacy of CHZ compared with 4-AP in alleviating motor symptoms of the tg/tg mice (even though in slices their efficacies in restoring the precision of Purkinje cell pacemaking is comparable) may be attributable to differences in their pharmacokinetics, in their ability to cross the blood–brain barrier, or differences in their potential side effects in vivo.
Kv1 channel blockers as K+ therapeutic agents in EA2
Based on its affinity for K+ channels, the most likely target of therapeutic concentrations of 4-AP is the Kv1 family of K+ channels (Coetzee et al., 1999), and possibly the Kv1.5 K+ channels that are thought to be expressed in Purkinje cells (Madeja et al., 1997; Chung et al., 2001, 2005). The efficacy of the Kv1.5 channel blocker DPO (Lagrutta et al., 2006) in restoring the precision of pacemaking in Purkinje cells substantiates the hypothesis that therapeutic concentrations of 4-AP target a member of the Kv1 family of K+ channels. A higher concentrations 4-AP blocks a wide range of K+ channels and is a potent proconvulsant. Thus, selective blockers of Kv1 channels (and pending further confirmation with more specific channel blockers particularly the Kv1.5 subfamily) may constitute safer and more effective candidates for treatment of EA2 and perhaps other cerebellar ataxia.
Footnotes
- Received July 23, 2009.
- Revision received March 17, 2010.
- Accepted April 8, 2010.
-
This work was supported by the National Institutes of Health. Studies on the effects of 4-AP on Purkinje cells were initiated by Dr. Simin Khavandgar during her tenure in K.K.'s lab. We are particularly grateful to her, and also thank members of K.K.'s lab for help in scoring the motor behavior of the mice and for discussions.
- Correspondence should be addressed to Kamran Khodakhah, Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Kennedy Center, Room 506, 1410 Pelham Parkway South, Bronx, New York 10461. k.khodakhah{at}einstein.yu.edu
- Copyright © 2010 the authors 0270-6474/10/307258-11$15.00/0