
Abstract
Cyclin-dependent kinase 5 (Cdk5)-mediated phosphorylation plays an important role in proper synaptic function and transmission. Loss of Cdk5 activity results in abnormal development of the nervous system accompanied by massive disruptions in cortical migration and lamination, therefore impacting synaptic activity. The Cdk5 activator p35 associates with δ-catenin, the synaptic adherens junction protein that serves as part of the anchorage complex of AMPA receptor at the postsynaptic membrane. However, the implications of Cdk5-mediated phosphorylation of δ-catenin have not been fully elucidated. Here we show that Cdk5-mediated phosphorylation of δ-catenin regulates its subcellular localization accompanied by changes in dendritic morphogenesis and synaptic activity. We identified two Cdk5 phosphorylation sites in mouse δ-catenin, serines 300 and 357, and report that loss of Cdk5 phosphorylation of δ-catenin increased its localization to the membrane. Furthermore, mutations of the serines 300 and 357 to alanines to mimic nonphosphorylated δ-catenin resulted in increased dendritic protrusions accompanied by increased AMPA receptor subunit GluR2 localization at the membrane. Consistent with these observations, loss of Cdk5 phosphorylation of δ-catenin increased the AMPA/NMDA ratio. This study reveals how Cdk5 phosphorylation of the synaptic mediator protein δ-catenin can alter its localization at the synapse to impact neuronal synaptic activity.
Introduction
Cyclin-dependent kinase 5 (Cdk5) phosphorylation of various presynaptic and postsynaptic proteins is important in the influence of synaptic function and transmission. Exocytosis is regulated by direct Cdk5 phosphorylation of synapsin I, Munc-18, and the voltage-dependent calcium channel, as well as the indirect regulation of Pctaire-1 (Matsubara et al., 1996; Shuang et al., 1998; Fletcher et al., 1999; Cheng et al., 2002; Tomizawa et al., 2002). Synaptic vesicle endocytosis is also regulated through Cdk5 phosphorylation and subsequent modulation of phosphoinositide signaling pathways (Lee et al., 2004). Postsynaptically, phosphorylation of the neuregulin receptor ErbB3 by Cdk5 and subsequent neuregulin signaling pathways as well as the phosphorylation of the postsynaptic density protein-95 (PSD-95) places the kinase in a pivotal role on both sides of the synapse (Fu et al., 2001; Morabito et al., 2004). The phosphorylation of PSD-95 suppresses multimerization of the postsynaptic scaffold, causing a reduction in NMDA receptor and potassium channel clustering, two important determinants of neuronal synaptic activity (Morabito et al., 2004). Organizational defects found in the Cdk5 knock-out nervous system lead to incorrect or incomplete neuronal connections being formed and the subsequent disruption to neuronal synaptic activity influencing memory and cognition (Chae et al., 1997; Ko et al., 2001).
To better understand the role of Cdk5 and its activators in synaptic activity and cognition, we used a yeast two-hybrid screen using p35 as “bait.” One protein of interest identified was δ-catenin, a neuron-specific adherens junction protein first identified through its interaction with presenilin-1, a protein found to be most frequently mutated in familial Alzheimer's disease (Zhou et al., 1997; Tanahashi and Tabira, 1999). δ-Catenin belongs to the p120–catenin family of proteins that is characterized by Armadillo (ARM) repeats that bind cadherins to possibly coordinate cadherin–cytoskeletal interactions at the cell membrane (Lu et al., 1999). For this reason, δ-catenin has been called a synaptic adherens junction protein (Ide et al., 1999; Lu et al., 1999; Kosik et al., 2005). δ-Catenin has been found to be involved in adhesion as well as spine morphogenesis and dendritic branching (Martinez et al., 2003; Arikkath et al., 2009). Importantly, deletions in δ-catenin and hemizygosity of the allele closely correlate with severity of mental retardation observed in Cri-du-Chat syndrome (Medina et al., 2000). Mice lacking δ-catenin, although viable, display severe impairments in cognitive function, especially in hippocampal-mediated long- and short-term plasticity and spatial learning (Israely et al., 2004).
Although both δ-catenin and Cdk5 have been shown to regulate synaptic signaling in neurons, the Cdk5-mediated phosphorylation of δ-catenin and its effect on synaptic activity have not been fully described. Our study aims to determine the effects of Cdk5-mediated phosphorylation of δ-catenin and its role in the regulation of synaptic activity and signaling. Through the identification of Cdk5-mediated phosphorylation at two novel sites, serine 300 and serine 357, we were able to determine the importance of Cdk5-mediated phosphorylation in regulating the subcellular localization of δ-catenin in neurons and how this affects neuronal and synaptic function.
Materials and Methods
Animal handling.
All animal experimentation was performed according to approved protocols of the Office of Institutional Animal Care and Use Committee of the National University of Singapore.
Plasmids.
Mouse δ-catenin–EGFP–C2 plasmid was a generous gift of Dr. Qun Lu (East Carolina University, Greenville, NC). Dominant-negative (DN) Cdk5 lentiviral plasmid was used as described previously (Zheng et al., 2005). Serines 300 and 357 were mutated to alanines to mimic a phosphorylation-deficient mutant, δ-cateninAla, using the QuikChange Mutagenesis kit (Stratagene). All mutants were then sequenced to verify site changes.
Antibodies.
Antibodies used were anti-δ-catenin (611537; BD Transduction Laboratories), anti-phospho-Ser300/357 δ-catenin antibody (produced by 21st Century Biochemicals), anti-GluR2 (MAB397; Millipore Corporation), anti-GluR1 clone C3T (05-855; Millipore Corporation), anti-N-cadherin (ab18203; Abcam), anti-GRIP (06-986; Millipore Corporation), anti-PSD-95 (MA1 046; Thermo Fisher Scientific), anti-synaptophysin (SC-9117; Santa Cruz Biotechnology), monoclonal anti-Cdk5 (J3, SC-6247; Santa Cruz Biotechnology), polyclonal anti-Cdk5 (C8, SC-173; Santa Cruz Biotechnology), and anti-p35 (C19, SC-820; Santa Cruz Biotechnology). Secondary antibodies used were Alexa Fluor 488, Alexa Fluor 543, and Alexa Fluor 633 (Invitrogen).
Primary cortical neuronal cultures.
Primary mouse (Mus musculus) cortical neurons were cultured using the same protocol described previously (Kesavapany et al., 2004). Mouse embryonic day 16 (E16) to E18 cortical neurons were cultured in Neurobasal medium with B27 supplement (Invitrogen) containing 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2 mm glutamine. Cells cultured were almost exclusively neurons as described previously by us and others (Nikolic et al., 1996). For transfection of cortical neurons, neurons were plated onto poly-l-lysine-coated glass coverslips and transfected using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. At 7 d in culture (DIC), neurons were transfected with green fluorescent protein (GFP) tagged δ-cateninWt and δ-cateninAla and immunostained at 19–21 DIC.
Immunocytochemical analysis.
At 19–21 DIC, neurons were fixed with 4% paraformaldehyde (PFA) for 30 min, permeabilized with 0.1% Triton X-100 for 20 min, and blocked for 30 min with 5% fetal bovine serum (FBS) in PBS. Neurons were incubated with the primary antibody prepared in 5% FBS/PBS for 1 h at room temperature, followed by addition of the appropriate secondary antibody for 1 h at room temperature. Nuclei were stained with Hoescht 33342 (Sigma). Coverslips were washed with PBS between incubations and before mounting on glass slides in fluorescence mounting medium (Dako). Fluorescent images were captured with a Carl Zeiss LSM-510 laser-scanning confocal microscope. Photomicrograph figures shown are representative images of the data obtained in which at least 20 transfected neurons were analyzed per trial and all experiments were replicated at least four times.
Immunohistochemistry.
Embryonic brain paraffin sections of Cdk5 knock-out and wild-type (Wt) mouse littermates (E16) were processed and produced by NeuroScience Associates. Sections were deparaffinized with xylene and gradually rehydrated with graded decreases in ethanol. Sections were permeabilized, blocked, and then incubated with primary antibody overnight at 4°C in PBS with 5% FBS. Sections were washed in PBS before incubation with secondary Alexa Fluor 594 antibody for 1 h at room temperature. Nuclei were stained with 4,6-diamidino-2-phenylindole before slides were mounted on cover glasses in fluorescent mounting medium (Dako). Confocal images were taken at 20× and 63× magnifications.
Electron microscopy.
Adult 3-month-old male C57BL/6J wild-type and p35 knock-out mice were anesthetized with the mixture of ketamine (75 mg/kg) and medepomidin (1 mg/kg) and transcardially perfused with freshly made 4% PFA and 0.1% glutaraldehyde. Brains were removed and further fixed in the same fixative for 6 h at 4°C. Microtome sections of 100 μm thickness were collected and washed thoroughly with 0.1 m phosphate buffer (PB). Sections were treated with 1% sodium borohydride solution for 15 min to inactivate residual aldehyde groups and washed with PB several times until the solution was clear of bubbles. Sections were then rinsed with PBS three times and permeabilized with PBS containing 0.1% Triton X-100. Sections were blocked for 30 min with 5% FBS in PBS and incubated in primary antibody, mouse anti-δ-catenin (1:10) diluted with PBS containing 1% BSA overnight at 4°C. After three washes with PBS/0.8% BSA, sections were incubated in gold conjugate (Protein A, 10 nm), diluted (1:50) with/0.8%BSA for 3 h, and then rinsed with PBS three times. The labeled sections were postfixed with 2% glutaraldehyde for 10 min and washed with PBS three times before incubation in silver enhancement solution for 5 min. After three washes with PBS for 10 min each, a last postfixation was performed with 1% osmium tetra oxide for 1 h. The sections were trimmed into pieces of ∼5 mm2 and washed in PBS for 10 min twice at room temperature before dehydration through an ascending ethanol series at room temperature (25, 50, 75, 95, and 100%). The dehydrated sections were infiltrated and embedded in Araldite resin and then polymerized at 60°C for 24 h. These processed tissues were cut into ultrathin sections (1 μm) and stained with lead citrate and uranium acetate for 10 min before being examined under the transmission electron microscope (JEM 1220; Jeol).
In vitro phosphorylation of δ-catenin by Cdk5.
Brain lysates of E16 wild-type and Cdk5 knock-out mice were produced through 40 strokes of a dounce homogenizer in ice-cold immunoprecipitation lysis buffer containing 50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 mm EGTA, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 5 μg/ml pepstatin, and 1 mm PMSF. Protein, 500 μg, was precleared with protein G-agarose beads (Sigma) at 4°C for 1 h. Cdk5 was immunoprecipitated from precleared lysates with anti-Cdk5 (C8) antibody with overnight incubation at 4°C and then incubated for 4 h at 4°C with protein A-agarose beads. Immunoprecipitates were washed three times with lysis buffer and once with kinase buffer (20 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 1 mm EDTA, 10 μm NaF, and 1 μm Na2VO3) and resuspended in 30 μl of water. Ten microliters of 5× kinase assay mixture (100 mm Tris-HCl, pH 7.4, 50 mm MgCl2, 5 mm EDTA, 50 μm NaF, 5 μm Na2VO3, and 5 mm DTT) and 10 μg of histone H1 (Sigma), 0.2 μm neurofilament-H (NF-H) peptide (VKSPAKEKAKSPVK), or δ-catenin peptides 1–7 (synthesized by 21st Century Biochemicals) were added to the immunoprecipitates. Recombinant Cdk5/p35 (Millipore Corporation) was also used in the assay. Kinase assays were performed at 30°C for 30 min by adding 5 μCi of [γ-32P]ATP. The assay was stopped by adding a 50% trichloroacetic acid to precipitate proteins. To detect δ-catenin phosphorylation, 10 μl aliquots of trichloroacetic acid supernatant were transferred onto P81 phosphocellulose paper (spotted in duplicates), air dried, and washed five times for 15 min each in 75 mm phosphoric acid and once in 95% ethanol. After air drying, squares were transferred to vials containing Bio-Safe II scintillation fluid (Research Products International) for counting in a Beckman Coulter scintillation counter.
In vivo phosphorylation of δ-catenin by Cdk5.
Brain lysates of E16 wild-type and Cdk5 knock-out embryos were separated on 4–20% polyacrylamide gels followed by transfer onto nitrocellulose membranes. Phosphorylated species were detected using the commercially synthesized anti-phospho-Ser300/357 δ-catenin antibody (21st Century Biochemicals), after which membranes were stripped and the presence of δ-catenin was confirmed using the anti-δ-catenin antibody. Interference from the IgG (25 and 55 kDa) was not seen in the Western blot signal because δ-catenin runs at ∼130 kDa (endogenous) and 160 kDa (GFP tagged). HEK293 cells were transfected with δ-cateninWt + p35, δ-cateninWt + p35 + Cdk5, δ-cateninWt + p35 + DNCdk5, and δ-cateninAla + p35 + Cdk5. Cell lysates were prepared, separated by SDS-PAGE, and subjected to Western blotting with the anti-phospho-Ser300/357 δ-catenin antibody.
Back-phosphorylation assay and stoichiometric measurements.
For phosphorylation time course experiments, 50 pmol of δ-catenin peptide (1 and 6) was diluted in kinase buffer supplemented with 0.352 μm [γ-32P]ATP, 0.352 mm cold ATP, and 1.5 μg recombinant Cdk5/p25 and incubated at 30°C for between 5 and 240 min. Reactions were terminated by addition of similar volume of 10% trichloroacetic acid to precipitate the protein and scintillation counted to determine total 32P incorporation. Molar amount of 32P incorporated per moles of peptide were calculated and plotted as a function of time. For the brain samples, the total protein of E16 Cdk5 wild-type and knock-out lysates (500 μg) were precleared with 10 μg of normal mouse IgG and thereafter incubated with similar amount of the anti-δ-catenin antibody. Immunoprecipitates were then back-phosphorylated in vitro with recombinant Cdk5/p25 for 60 min, reaction was terminated by adding similar volume of 10% trichloroacetic acid, and 50 μl of mixture was scintillation counted. Specific radioactivity per moles of [γ-32P]ATP was determined by counting diluted aliquots of the stock, and this conversion value was used to calculate the molar amount of 32P incorporated and the molar ratio of 32P to δ-catenin peptide.
Membrane and cytosolic preparations.
E16 Wild-type and Cdk5 knock-out embryonic mice brains were homogenized and placed on ice for 10 min before centrifugation at 1000 × g for 5 min at 4°C. The pellet containing nuclear material was washed three times with lysis buffer containing 0.1% NP-40. The supernatant fraction was centrifuged at 150,000 × g for 30 min to yield a cytosolic and membrane fraction. Membrane fractions were washed three times in lysis buffer and were solubilized in 4× SDS sample buffer containing β-mercaptoethanol. Cytosolic fractions were prepared for Western blotting by addition of 10× SDS sample buffer containing β-mercaptoethanol. Membrane protein loading was balanced through immunodetection of cadherin, and cytosolic protein loading was balanced using tubulin. Densitometric δ-catenin signals were normalized to the two marker signals for quantitation. Experiments were repeated three times using littermate wild-type and Cdk5 knock-out embryos.
Immunoprecipitation and coimmunoprecipitation.
Brain samples from 3-month-old male wild-type and p35 knock-out adult mice were homogenized in radio immunoprecipitation assay buffer containing 50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 1 mm sodium fluoride, and protease inhibitors (Complete Mini protease inhibitor EDTA-free tablet). Homogenates were placed on ice for 30 min before centrifugation at 16,000 × g for 20 min at 4°C. After centrifugation, the supernatant was precleared with protein G-Sepharose beads (Sigma) for 1 h at 4°C. Precleared protein, 500 μg, was used for immunoprecipitation by incubation for 3–4 h at 4°C with 3 μg of anti-δ-catenin; this was followed by addition of protein G-Sepharose beads to the lysates for overnight incubation at 4°C. The beads were washed three times with lysis buffer and solubilized in 2× SDS sample buffer containing β-mercaptoethanol. Coimmunoprecipitated proteins were then detected by immunoprobing with the appropriate antibodies by Western blot.
Immunostaining of surface GluR2.
To immunostain endogenous surface GluR2, 19–21 DIC mouse neurons were stained live by incubation with the anti-GluR2 N-terminal primary antibody prepared in culture medium at 37°C for 40 min, after which, coverslips were fixed with 4% formaldehyde for 30 min at room temperature and blocked for 30 min with 5% FBS. The cells were incubated with the secondary antibody for 1 h at room temperature, after which the nuclei were stained with Hoescht 33342 (Sigma). Coverslips were washed with PBS between incubations and before mounting. Coverslips were mounted using the fluorescence mounting medium onto the glass slides.
Cell surface biotinylation and immunoprecipitation.
Cell surface biotinylation was performed on HEK293 cells plated onto 10 cm2 dishes using the Pierce Cell Surface Protein Isolation kit (Thermo Fisher Scientific). The HEK293 cells were transfected with δ-cateninWt + p35 + GluR1 + GluR2 and δ-cateninAla + p35 + GluR1 + GluR2. The biotin-labeled surface proteins were immunoprecipitated and separated by SDS-PAGE. Surface proteins were detected by immunoprobing with the respective antibodies by standard Western blotting procedures.
Lentivirus production.
Lentiviruses of empty vector and DNCdk5 were prepared essentially as described previously (Zheng et al., 2005). Briefly, HEK293FT cells were cotransfected with DNCdk5-LV and ViraPower Packaging Mix using Lipofectamine 2000 following the methods described in the ViraPower Lentiviral Expression system (Invitrogen). Viral supernatants were collected after 48 and 72 h and cleared of cell debris by centrifugation. Supernatants were then filtered with a 0.45 μm filter and concentrated ∼40-fold by ultracentrifugation through Amicon Ultra-15 centrifugal filter unit with Ultracel-100 membrane (Millipore Corporation). Viral titers, expressed as the percentage of total cells expressing the target gene of interest, were determined by immunocytochemistry using rabbit anti-Cdk5 (C8) antibody after 72 h after transduction of 7 DIC cortical neurons.
Sodium imaging.
The sodium ion flux of transduced neurons was monitored using a Na+ binding dye, sodium-binding benzofuran isophthalate (SBFI)-AM (Invitrogen), as described previously but with slight modifications (Baartscheer et al., 1997). Transduced 10–14 DIC cortical neurons were washed twice with physiological buffer containing the following (in mm): 156 Na+, 4.7 K+, 1.3 Ca2+, 2.0 Mg2+, 150.6 Cl−, 4.3 HCO3−, 1.4 HPO4−, 17 HEPES, and 11 glucose, pH 7.3. The cells were then loaded with 10 μm SBFI in the presence of 1% FBS. The loaded cells were washed twice with physiological buffer after 2 h incubation at 37°C. An additional 15 min incubation at 37°C was performed to ensure complete de-esterification of all residual intracellular SBFI. The SBFI-loaded cortical neurons were treated with 10 μm glutamate or vehicle only. SBFI fluorescence was excited at 340 nm, and the emitted signal was recorded at 420 nm. Recordings were taken at 20 s intervals over 5 min using the Infinite M200 microplate reader (Tecan Group). The fluorescence signal of each individual well of cells was normalized to its respective well total protein concentrations using the BCA Protein Assay kit (Pierce).
Electrophysiology.
To measure the AMPA/NMDA ratio, whole-cell patch-clamp recordings technique was used. Whole-cell patch-clamp recordings were made at room temperature. The transfected neurons were identified by the expression of GFP. The external solution during recording contained the following (in mm): 110 NaCl, 5 KCl, 2 CaCl2, 0.8 MgCl2, 10 HEPES, and 10 d-glucose, pH 7.4. The internal solution contained the following (in mm): 135 CsCl2, 10 HEPES, 2 MgCl2, 4 NaATP, 0.4 NaGTP, and 0.5 EGTA, pH 7.2. Picrotoxin (200 μm) was included in the external solution to block GABAergic IPSCs, whereas 0.5 μm tetrodotoxin was added to prevent action potential-evoked EPSCs. Glutamate (100 μm) was local fast applied by gravity feeding in the presence of glycine (20 μm), and the whole-cell currents (Vh of −80 or +60 mV) were recorded. To confirm that the responses are AMPA or NMDA mediated, we treated the cells with 6-cyano-7-dinitroquinoxaline-2,3-dione, a selective antagonist of AMPA/kainate glutamate receptors, or 2-amino-5-phosphonovaleric acid, for NMDA-type glutamate receptors (data not shown). Pipette resistance for these experiments were typically 3–5 MΩ, and series resistance was 15–20 MΩ. At least 10 neurons were subjected to the recording from each experiment. Data were presented as mean ± SEM from five experiments. Statistical analysis was performed using Prism software (GraphPad Software).
[3H]AMPA radioligand binding assays.
Frozen 3-month-old male p35 knock-out and wild-type mouse brains were thawed on ice and dissected free of meninges and white matter and then placed in 10 vol of ice-cold lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EGTA, 1 mm Na2EDTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, and 1 μg/ml leupeptin), with 2 mm 4-(2-aminoethyl) benzenesulfonyl fluoride and Complete Mini protease inhibitor EDTA-free tablet added before being passed through 10 strokes in a Teflon–glass homogenizer. The crude homogenate was then washed twice with 10 vol of 50 mm Tris-HCl buffer, pH 7.4, followed by centrifugation (15,000 × g, 10 min, 4°C). The final pellet was stored at −80°C until assay. Binding of mouse brain homogenates to [3H]AMPA was based on published methods with slight modifications (Olivera et al., 1999). Briefly, frozen pellets were resuspended in 10 vol of 50 mm Tris-HCl buffer, pH 7.4, and then incubated at 37°C for 30 min to facilitate breakdown of endogenous glutamate. Thereafter, aliquots of homogenates were added in triplicate to 20 nm [3H]AMPA (specific activity, 40 Ci/mmol; PerkinElmer Life and Analytical Sciences) in a total volume of 400 μl of buffer with 100 mm KSCN and incubated for 60 min at 4°C. To determine nonspecific binding, parallel series of binding assays were set up with the addition of 2 mm l-glutamate. At the end of incubation, the assays were terminated by vacuum filtration onto 0.01% polyethylenimine-treated Whatman GF/B filters using a cell harvester (Molecular Diagnostics), followed by washing with ice-cold sodium phosphate buffer. Filters were then dried and punched into vials, and aliquots of scintillant (Optiphase HiSafe 2; PerkinElmer Life and Analytical Sciences) were added for measurement of bound radioactivity (expressed in disintegrations per minute) using a Wallac beta counter. Binding was measured in mean ± SEM disintegrations per minute per microgram of protein. Total counts (T) were determined in the presence of 20 nm [3H]AMPA, and nonspecific counts (NS) were determined by 20 nm [3H]AMPA plus 2 mm l-glutamate. For specific counts, S = T − NS.
Results
Cdk5 phosphorylates δ-catenin on serines 300 and 357
Examining the mouse δ-catenin sequence, we found in total 20 proline-directed serine/threonine sites of which seven of them represented putative Cdk5 phosphorylation sites according to the consensus sequence favored by the kinase (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We synthesized these seven peptides and used them “blind” in in vitro kinase assays using active kinase isolated from adult mouse brain (Fig. 1A), as well as recombinant purified kinase (Fig. 1B). These assays identified Ser300 (peptide 1-PKQS300PSRLAK) and Ser357 (peptide 6-ATLS357PTKRLVH) as the main sites of Cdk5 phosphorylation. These data were reconfirmed by using wild-type and Cdk5 knock-out brains as the sources of the kinase in which the phosphorylation of these two sites were abolished in the Cdk5 knock-out assays (Fig. 1C). We also performed back-phosphorylation assays to determine stoichiometry of phosphorylation at these sites of δ-catenin: serines 300 and 357 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Our results validate that both sites are indeed sites of Cdk5 phosphorylation. Using the information from the in vitro work, we produced a phospho-specific δ-catenin antibody directed to these two sites and examined whether δ-catenin phosphorylation was reduced in wild-type and Cdk5 knock-out brain lysates. The Western blot analysis of these lysates with this antibody showed that the signal was completely abolished in knock-out lysates compared with the robust signal obtained in the wild-type samples (Fig. 2A). To confirm that the antibody did indeed detect Cdk5 phosphorylated δ-catenin on the two sites, we cotransfected HEK293 cells with wild-type δ-catenin with p35 + Cdk5 and p35 + DNCdk5 and found that there was no signal in the latter transfections (Fig. 2B). To further characterize the phospho-δ-catenin antibody, we transfected cortical neurons with δ-cateninAla (δ-catenin double alanine mutant at Ser300 and Ser357) (Fig. 2C) and with DNCdk5 (Fig. 2D). We immunostained for phosphorylated δ-catenin and found a reduction in signal in neurons overexpressing DNCdk5 and δ-cateninAla.
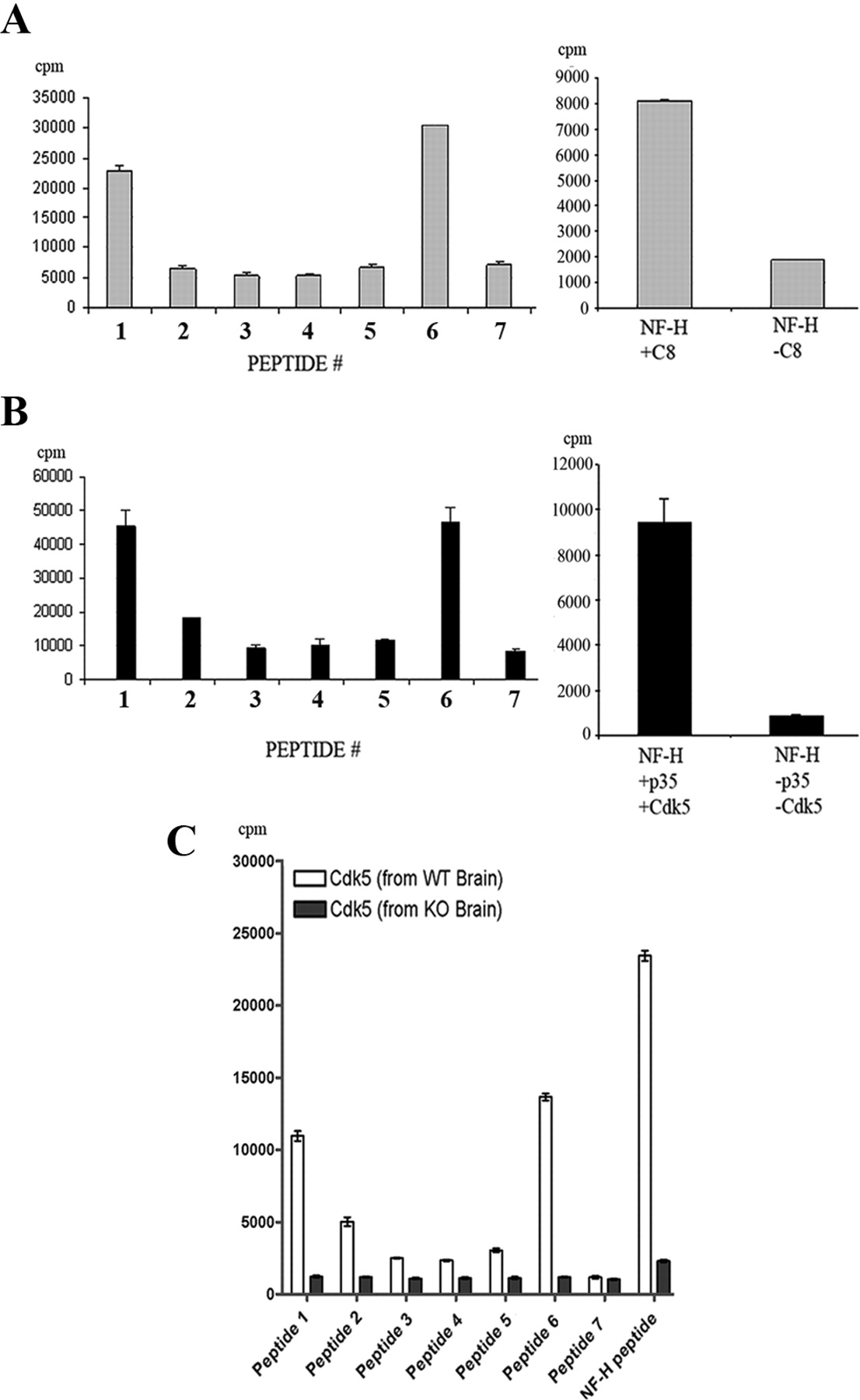
Cdk5 phosphorylates δ-catenin on serines 300 and 357. A, Peptides representing seven putative Cdk5 consensus phosphorylation sites used in in vitro kinase assays in which the source of the kinase was immunoprecipitated from adult mouse brain. The bar graph on the left showed phosphorylation of peptides 1 and 6, and the bar graph on the right showed the positive control NF-H peptide phosphorylation. B, Peptides representing seven putative Cdk5 consensus phosphorylation sites used in in vitro kinase assays in which the source of the kinase was recombinant p35/Cdk5. Specific activity was understandably higher in these assays. The bar graph on the left showed phosphorylation of peptides 1 and 6, and the bar graph on the right showed the positive control NF-H peptide phosphorylation. C, Peptides representing seven putative Cdk5 consensus phosphorylation sites used in in vitro kinase assays in which the source of the kinase was Cdk5 immunoprecipitated from Cdk5 knock-out (KO) and wild-type lysates (WT). The last two bars represent the positive control NF-H peptide phosphorylation.

δ-Catenin is phosphorylated by Cdk5 in vivo on serines 300 and 357. A, Representative images to show detection of Cdk5-mediated phosphorylation of δ-catenin in Cdk5 knock-out (KO) and wild-type (WT) embryonic lysates using the commercially produced phospho-specific antibody (anti-phospho-Ser300/357 δ-catenin). Two different amounts of δ-catenin lysates were used and immunoblotted for total δ-catenin (top). The same nitrocellulose membrane was immunoprobed for phospho-δ-catenin (middle). Identical amounts of lysates were immunoprobed using preimmune serum (bottom). B, Western blot of HEK293 cell lysates transfected with δ-cateninWt + p35 + Cdk5 or p35 + DNCdk5 (top) and δ-cateninAla + p35 + Cdk5 as well as δ-cateninWt + p35 (bottom) and immunoprobed for phospho-δ-catenin. It was clear that the Ala mutant was not immunodetected by phospho-δ-catenin antibody, whereas the presence of p35 that could complex with endogenous Cdk5 in these cells was sufficient to phosphorylate δ-catenin. C, Cortical neurons transfected with GFP-tagged δ-cateninWt (a–c) and phospho-deficient mutant (δ-cateninAla) (d–f) and immunodetected using the anti-phospho-Ser300/357 δ-catenin (red). Arrow indicates transfected neurons, and the respective nontransfected neurons are indicated by arrowhead. The δ-cateninWt-transfected neurons showed a greater signal with the phospho-δ-catenin antibody. In contrast, the transfection of δ-cateninAla exhibited no increase in staining. Scale bars, 10 μm. D, Cortical neurons transfected with a DNCdk5 construct; the representative figure shows that the DNCdk5-overexpressing neuron (red) did not exhibit phospho-δ-catenin staining (green). Arrow indicates DNCdk5-transfected neuron, and arrowhead indicates the nontransfected neuron. Scale bars, 10 μm. All figures are representations of at least three independent experiments.
Cdk5 phosphorylation mediates subcellular localization of δ-catenin in neurons
To investigate the effect of Cdk5-mediated phosphorylation on the subcellular localization of δ-catenin, we examined cytosolic and membrane preparations of Cdk5 knock-out and wild-type control brain lysates. We found that cytosolic δ-catenin levels decreased in Cdk5 knock-out preparations compared with wild-type samples (Fig. 3A). This was accompanied by increased membranous δ-catenin levels in Cdk5 knock-out samples compared with wild-type samples. To confirm this by immunohistochemistry, we performed δ-catenin immunostaining on paraffin-embedded Cdk5 knock-out and wild-type embryonic brain sections. δ-catenin localization mirrored the biochemical results found earlier, with δ-catenin primarily localized to the cell periphery in Cdk5 knock-out sections accompanied by reduced cytosolic staining (Fig. 3B,C). To ensure that this was not an immunohistochemical artifact, we immunostained wild-type and Cdk5 knock-out primary cortical neurons with anti-δ-catenin antibody, and, after 5 DIC, the neurons exhibited lower cytosolic levels of δ-catenin accompanied by accumulations in membrane staining (Fig. 3D). Additionally, the transfection of DNCdk5 into neurons displayed similar results (Fig. 3E). To investigate this phenomenon ultrastructurally, we used immunoelectron microscopy (EM) to determine localization of δ-catenin. In p35 knock-out samples, δ-catenin primarily localized to the membrane with a reduction of cytosolic δ-catenin (number of gold conjugate particles observed per 10 μm: membrane, 186 ± 8.25; cytosol, 68 ± 5.5; p < 0.001) compared with wild-type sections, which showed higher δ-catenin localization to the cytosol than the membrane (membrane, 78 ± 9.8; cytosol, 171 ± 11; p < 0.001) (Fig. 4A,B). Sections without primary anti-δ-catenin antibody are shown to illustrate background staining. We had to use p35 knock-out tissue for these experiments so that proper fixation for EM was possible. Immuno-EM of brain tissue from Cdk5 knock-out mice was not of sufficient quality, probably because of the lack of proper fixation of these samples (data not shown). These results suggest that Cdk5-mediated phosphorylation of δ-catenin regulates its subcellular localization with increased accumulations of the nonphosphorylated δ-catenin to the membrane of neurons.

Cdk5 phosphorylation of δ-catenin mediates its subcellular localization. A, Cytosolic and membrane preparations produced from E16 Cdk5 wild-type (+/+) and knock-out (−/−) mouse brain immunodetected for δ-catenin levels. It was found that δ-catenin levels derived from Cdk5 knock-out samples were reduced in cytosolic but increased in membrane preparations compared with wild-type preparations. δ-Catenin levels were densitometrically scanned and represented as mean density/tubulin (cytosolic) or N-cadherin (membrane) levels (***p < 0.001). Figure shows representative from at least three independent experiments. B, Paraffin-embedded sections of Cdk5 knock-out and wild-type (+/+) embryonic brain sections immunostained for δ-catenin. Cdk5 knock-out (−/−) sections exhibited reduced cytosolic staining (asterisks) and increased membranous staining (arrows). Confocal images were taken at 20× magnification. Scale bar, 20 μm. DAPI, 4′,6′-diamidino-2-phenylindole. C, Higher magnification of images in B. Cdk5 knock-out (−/−) sections exhibited reduced cytosolic staining and increased membranous staining compared with the wild-type (+/+) sections. Confocal images were taken at 63× magnification. Scale bars, 20 μm. D, Primary cortical neurons from Cdk5 wild-type (WT) and Cdk5 knock-out (Cdk5−/−) were fixed and immunostained with anti-δ-catenin antibody at 5–7 DIC. It is clear that wild-type neurons have increased cytosolic staining of δ-catenin, whereas Cdk5 knock-out neurons have increased membranous staining with reduced cytosolic δ-catenin (arrowheads). Scale bars, 20 μm. E, Wild-type cortical neurons transfected with dominant-negative Cdk5 at 10 DIC and immunostained for δ-catenin (red). Arrows indicate nontransfected neurons, and arrowhead indicates DNCdk5-transfected neuron (DN). Nontransfected neurons displayed increased cytosolic δ-catenin, whereas DNCdk5-transfected neurons displayed increased membranous staining accompanied by reduced cytosolic staining. Figure shows representative neurons from at least three independent experiments. Scale bar, 20 μm.

δ-Catenin localizes to the membrane in p35 knock-out sections. A, Wild-type (p35+/+) and p35 knock-out (p35−/−) sections immunostained for δ-catenin and processed for immuno-EM. Top two panels show δ-catenin staining, and the bottom two panels were not immunostained for δ-catenin (controls). p35 knock-out sections showed increased δ-catenin localized to the membrane. Scale bars, 0.2 μm. B, Quantitation of membrane versus cytosol staining in EM sections by number of gold particles per 10 μm. Wild-type sections (p35+/+) exhibited higher cytosolic but lower membranous δ-catenin immunoreactivity compared with knock-out sections (p35−/−). ***p < 0.001.
Loss of Cdk5 phosphorylation of δ-catenin mediates increased dendritic protrusions
δ-Catenin has been reported previously to modulate dendritic and spine morphogenesis with loss of δ-catenin leading to increased spine formation (Arikkath et al., 2009). To investigate whether the consequence of Cdk5 phosphorylation in neurons affects the morphological change in dendritic structures, we transfected δ-cateninWt and δ-cateninAla constructs into cortical neurons at 7 DIC. The δ-cateninAla-transfected neurons displayed a distinct morphological difference (Fig. 5Ad–Af) compared with the wild-type δ-catenin-transfected neurons (Fig. 5Aa–Ac). There were an increased number of dendritic protrusions along the dendrites with the accumulations of nonphosphorylated δ-catenin in the δ-cateninAla at the membrane compared with δ-cateninWt-transfected neurons (Fig. 5B). Quantitation of the number of dendritic protrusions in the transfected neurons showed an increase in the δ-cateninAla-expressing neurons (number of dendritic protrusions per 10 μm, 8.3 ± 0.25) compared with δ-cateninWt (5.3 ± 0.23; p < 0.001) and empty vector-transfected neurons (5.1 ± 0.25; p < 0.001) (Fig. 5C). To determine whether the dendritic protrusions we observed were synaptic, we immunostained δ-cateninWt- and δ-cateninAla-transfected neurons with synaptophysin and PSD-95 (supplemental Fig. 4A,B, available at www.jneurosci.org as supplemental material). We found that there was colocalization of the presynaptic and postsynaptic markers with the dendritic protrusions, indicating that there was an increase in the synapses along the dendrites of the δ-cateninAla-transfected neurons.

Nonphosphorylated δ-catenin results in increased dendritic protrusions. A, Cortical neurons transfected with GFP-tagged δ-cateninWt and δ-cateninAla at 7 DIC and GFP fluorescence visualized at days 19–21. Wild-type δ-catenin is shown in a–c with inset box in a magnified and shown in b; magnification of inset box in b is shown in c. There was an obvious morphological difference in the neurons transfected with wild-type δ-catenin, which have a much lesser extent of dendritic protrusions compared with δ-cateninAla (d–f). Inset box in d is magnified and shown in e, and magnification of inset box in e is shown in f. Scale bars, 10 μm. Figures are representative of at least 30 transfected neurons from at least three separate experiments. B, Magnified image of δ-cateninWt- and δ-cateninAla-transfected neurons show increased dendritic protrusions along the length of the dendrites in the neurons transfected with δ-cateninAla compared with δ-cateninWt. Scale bars, 10 μm. C, The number of dendritic protrusions per 10 μm dendritic length quantified from 15 randomly selected 19–21 DIC transfected neurons. δ-CateninAla-transfected neurons (δcatAla) displayed an almost twofold increase in the number of protrusions compared with δ-cateninWt-transfected (δcatWt) and empty vector-transfected (EV) neurons (***p < 0.001).
Cdk5 phosphorylation of δ-catenin regulates its interaction with the PDZ-binding proteins and GluR2 trafficking to the membrane
δ-Catenin is known to associate with several proteins [such as glutamate receptor interacting protein (GRIP), PSD-95, and AMPA binding protein (ABP)] at its PSD-95/disc large/zona occludens-1 (PDZ) binding domain as well as with cadherins at the ARM region. To examine whether Cdk5-mediated phosphorylation of δ-catenin regulates its interaction with its associative proteins, GRIP, PSD-95, and cadherin, we immunoprecipitated wild-type and p35 knock-out brain lysates with anti-δ-catenin antibody and compared the protein levels (Fig. 6A). The Western blot results indicate that, in the absence of Cdk5 phosphorylation of δ-catenin, there was an increase in the affinity of δ-catenin with the PDZ-binding proteins GRIP and PSD-95. However, there was no significant change in the interaction between cadherin and δ-catenin in the absence of Cdk5-mediated phosphorylation. Because δ-catenin has been described previously to anchor the GluR2 subunit of the AMPA receptor through an ABP/GRIP interaction at its PDZ-binding domain, we investigated the effect of Cdk5-mediated phosphorylation of δ-catenin on GluR2 (Silverman et al., 2007). GluR2 surface expression in δ-cateninWt- and δ-cateninAla-transfected neurons was determined by immunocytochemical surface staining. Our data showed that the GluR2 subunit demonstrates preferential accumulation at regions along the dendrite with increased dendritic protrusions and δ-catenin accumulations (Fig. 6B). Therefore, in the δ-cateninAla-transfected neurons (Fig. 6Ce–Ch), which have increased dendritic protrusions and nonphosphorylated δ-catenin localization at the membrane, we observed a higher accumulation of GluR2 subunits at the surface along the neurites compared with the δ-cateninWt-transfected neurons (Fig. 6Ca–Cd). To further confirm these results, we performed surface protein isolation in HEK cells transfected with δ-cateninWt + p35 + GluR1 + GluR2 and δ-cateninAla + p35 + GluR1 + GluR2. Probing of the Western blots with anti-GluR2 and GluR1 antibodies showed that there was increased surface expression of GluR1 and GluR2 when δ-catenin was nonphosphorylated (Fig. 6D). This indicates that, in the absence of Cdk5 phosphorylation of δ-catenin, there was increased AMPA trafficking to the membrane.

Phosphorylation of δ-catenin mediated by Cdk5 regulates the interaction with its associative proteins. A, Wild-type (p35+/+) and p35 knock-out (p35−/−) adult mice brain lysates were immunoprecipitated using anti-δ-catenin antibody. Samples were separated by SDS-PAGE and immunoprobed for N-cadherin, GRIP, and PSD-95. In the absence of Cdk5 phosphorylation of δ-catenin in p35 knock-out samples, there was increased GRIP and PSD-95 association with δ-catenin. However, the association of N-cadherin with δ-catenin was not affected by the phosphorylation status of δ-catenin mediated by Cdk5. Equal loading of lysates was shown by reprobing of blot with anti-δ-catenin antibody (bottom). + or − indicates pulldown with or without δ-catenin antibody. B, Magnified images of 19–21 DIC cortical neurons transfected with δ-cateninWt and δ-cateninAla surface immunostained with GluR2. There was increased GluR2 punctate (red) in regions with δ-catenin accumulations and dendritic protrusions (green; arrowhead). Regions that did not contain these protrusions showed obvious reductions in GluR2 subunits (arrow). Scale bars, 10 μm. C, Nineteen to 21 DIC cortical neurons transfected with GFP-tagged δ-cateninWt (a–d) and δ-cateninAla (e–h) immunostained to show surface GluR2 (red). Inset box in a is magnified and shown in b–d, and inset box in e is magnified and shown in f–h. The δ-cateninAla-transfected neurons showed higher GluR2 immunostaining along the surface of its neurites compared with δ-cateninWt-transfected neurons. The phospho-deficient δ-catenin (δ-cateninAla), which was localized at the dendritic protrusions, showed higher accumulations of GluR2 at these sites. Scale bars, 10 μm. D, HEK293 cells transfected with δ-cateninWt + p35 + GluR1 + GluR2 and δ-cateninAla + p35 + GluR1 + GluR2 were surface biotinylated and immunoprecipitated to isolate the surface proteins. Western blot of lysates showed that there was an increase in GluR1 and GluR2 surface expression in HEK293 cells transfected with the δ-catenin alanine mutant (δcatAla) compared with wild-type δ-catenin (δcatWt). Equal transfection efficiencies were shown by total protein lysates probed with GluR1 and GluR2 antibodies (bottom). E, Quantitation of Western blot in D represented as bar graphs. There was increased surface localization of GluR1 and GluR2 in the HEK293 cells transfected with the δ-cateninAla + p35 + GluR1 + GluR2 (δcatAla) compared with δ-cateninWt + p35 + GluR1 + GluR2 (δcatWt) (*p < 0.05,**p < 0.01).
Phospho-deficient δ-catenin increases functional AMPA and enhances the AMPA-mediated response
Based on the increased GluR2 surface staining at the membrane of neurons transfected with phospho-deficient δ-catenin, we quantitatively measured the differences in the number of functional AMPA receptors using radioligand binding assays on wild-type and p35 knock-out brains. The binding of [3H]AMPA was significantly increased in p35 knock-out membrane preparations compared with wild-type control ([3H]AMPA binding: p35 knock-out, 201 ± 6.5; wild type, 41 ± 6.5; p < 0.01), indicating that there was an increase in the number of functional AMPA receptors in the absence of Cdk5 phosphorylation of δ-catenin (Table 1). To determine whether the increased tethering of GluR2 by the nonphosphorylated δ-catenin was accompanied with an increased AMPA-mediated response, we recorded the amplitudes of AMPA receptor-mediated and NMDA receptor-mediated responses of δ-cateninWt- and δ-cateninAla-transfected neurons, and the normalized ratio of AMPA/NMDA response was determined (Fig. 7). Inhibition of Cdk5 phosphorylation of δ-catenin increased the amplitude of the AMPA-mediated component in δ-cateninAla-transfected neurons (AMPA/NMDA ratio: δ-cateninAla, 2.9 ± 0.68) compared with δ-cateninWt-transfected (1.81 ± 0.35; p < 0.01) and vector-transfected neurons (pcDNA3.1, 1.28 ± 0.76; p < 0.05). The increase in amplitude of AMPA/NMDA ratio in the δ-cateninAla-transfected neurons suggests that Cdk5 phosphorylation of δ-catenin is important for synaptic function by regulation of the surface expression of AMPA receptor and subsequent AMPA receptor-mediated synaptic signaling.
Phosphorylation of δ-catenin by Cdk5 regulates functional AMPA amounts in neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cdk5-mediated phosphorylation of δ-catenin enhances the AMPA-mediated response of neurons. The amplitudes of AMPA receptor-mediated and NMDA receptor-mediated responses were recorded from 19 DIC δ-cateninWt- and δ-cateninAla-transfected neurons, and the normalized ratio of AMPA/NMDA response was determined. Sample traces of AMPA- or NMDA-evoked response in neurons after transfection with different expression constructs were shown (top). It was found that δ-cateninAla-overexpressing neurons (δcatAla) displayed an approximate doubling of the AMPA/NMDA ratio compared with δ-cateninWt-transfected neurons (δcatWt) (bottom). pcDNA3.1-transfected neurons exhibited similar AMPA-mediated response as δ-cateninWt-expressing neurons (*p < 0.05, **p < 0.01).
Together, our results propose a novel mechanism in modulation of synaptic activity by Cdk5-mediated phosphorylation of δ-catenin, which alters the subcellular localization of δ-catenin, subsequently affecting the AMPA subunit GluR2 surface expression.
Discussion
This study reports our findings of how Cdk5-mediated phosphorylation of δ- catenin regulates synaptic activity in neurons. Loss of Cdk5 activity alters the subcellular localization of δ-catenin, resulting in increased accumulations to the membrane. Additionally, there was an obvious increase in dendritic protrusions found along the neurites of the phospho-deficient δ-catenin. These changes in δ-catenin localization and cellular morphology result in increased GluR2 tethering at the membrane, causing higher surface expression of the AMPA receptor. This leads to the regulation of synaptic function by increasing the AMPA-mediated response, thus affecting synaptic activity.
We found that δ-catenin associated with p35 in a yeast two-hybrid screen and coimmunoprecipitated in mammalian systems (supplemental Figs. 1, 3, available at www.jneurosci.org as supplemental material). Muñoz et al. (2007) reported that p35/Cdk5 coimmunoprecipitated with δ-catenin and that this phosphorylation enhanced its interaction with the peptidyl-prolyl cis-/trans- isomerase 1 Pin1 (Muñoz et al., 2007), but the physiological consequence of this interaction was not clearly defined. Having identified the Cdk5 phosphorylation sites on δ-catenin at serines 300 and 357 through peptide-based in vitro kinase and back-phosphorylation assays, we produced a phospho-specific antibody at serines 300 and 357 and showed that δ-catenin was indeed phosphorylated at those two sites in brain in vivo.
Cdk5-mediated δ-catenin phosphorylation regulates δ-catenin subcellular localization into two distinct pools: a cytoplasmic or intraneurite state as well as a pool that is localized to the membrane. In the phospho-deficient state of δ-catenin as seen in the Cdk5 and p35 knock-out brains, there was an obvious distribution of δ-catenin to the membrane with reduction in cytosolic δ-catenin levels. This change in δ-catenin distribution, in the absence of Cdk5 phosphorylation, was clearly distinct even at the single-cell level in the dominant-negative Cdk5 neurons and at an ultrastructural level as revealed by electron microscopy of p35 knock-out brain.
To determine the specific effects of nonphosphorylated δ-catenin, we generated a double alanine mutant at serines 300 and 357 to mimic permanently nonphosphorylated δ-catenin. We further confirmed that there were distinct δ-catenin accumulations at the membrane accompanied by an increase in dendritic protrusions in the δ-cateninAla-transfected neurons. Colocalization between δ-catenin with PSD-95 and synaptophysin at the dendritic protrusions indicate that the majority of these structures are synaptic in nature (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We found phospho-deficient δ-catenin localized to the dendritic protrusions, confirming that δ-catenin is indeed important in synapse morphogenesis. δ-Catenin has been shown previously to be involved in synapse morphogenesis whereby, when its expression was knocked down in hippocampal neurons, neurons exhibited increased numbers of dendritic spines (Arikkath et al., 2009). Previous papers have suggested that phosphorylation of δ-catenin mediated by kinases such as Akt and Abl have resulted in changes of dendritic and cellular morphogenesis (Lu et al., 2002; Kim et al., 2008). Lu et al. (2002) reported that inhibition of Abl tyrosine kinase-mediated phosphorylation of δ-catenin resulted in accelerated neurite extensions. Akt phosphorylation of δ-catenin at threonine 454 was shown to sequester p190RhoGEF from activating Rho signaling, resulting in enhanced dendrogenesis and spine formation (Kim et al., 2008). Our data also suggests that, in addition to its presence, the phosphorylation status of δ-catenin mediated by Cdk5 is important in regulating cellular morphogenesis and dendrogenesis.
The PDZ-binding domain and ARM region of δ-catenin are important for its role as a synaptic adherens junction protein, positioning δ-catenin as the link between the scaffolds and signaling molecules. The PDZ domain of δ-catenin at the C terminal is responsible for its interaction with proteins such as GRIP, ABP, and PSD-95 (Silverman et al., 2007). Both the Cdk5 phosphorylation sites serines 300 and 357 are situated at the opposite end to the PDZ-binding domain (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Interestingly, however, our coimmunoprecipitation data showed that, in the absence of Cdk5-mediated phosphorylation of δ-catenin, there was increased affinity between δ-catenin with the PDZ-binding proteins PSD-95 and GRIP. Because of the poor performance of the commercially available ABP antibody, we could not ascertain the role of the ABP protein during δ-catenin phosphorylation. It is likely that the localization of δ-catenin to the membrane accompanied by increased dendritic protrusions in the phospho-deficient δ-catenin-transfected neurons contributed toward the organization of the postsynaptic density and modulation of the interaction with its PDZ-binding proteins. In contrast, however, our data suggest that phosphorylation of δ-catenin by Cdk5 at serines 300 and 357 did not affect its interaction with cadherin at the ARM domain. The association between δ-catenin and cadherin, although important for the cadherin–δ-catenin cell adhesion complex, does not appear to be involved in the role of δ-catenin controlling synaptic and dendritic morphogenesis, as also reported previously by Arikkath et al. (2009).
The PDZ-binding proteins of δ-catenin, GRIP and ABP, form the scaffold for AMPA receptors GluR2 and GluR3 (Srivastava et al., 1998). δ-Catenin has been reported to act as part of the anchorage complex for GluR2 at the membrane via its PDZ-binding domain interaction with ABP/GRIP (Silverman et al., 2007). Silverman et al. (2007) has shown that the δ-catenin mutant lacking the PDZ-binding motif resulted in reduced surface GluR2 levels. Because phosphorylation of δ-catenin by Cdk5 was shown to affect its interaction with the PDZ-binding proteins, this could contribute to the increased surface localization of GluR2 observed in the δ-cateninAla-transfected neurons especially at regions with accumulated nonphosphorylated δ-catenin at the dendritic protrusions. Cycling of GluR2 has also been shown to depend on the presence of δ-catenin, in which gene silencing and dominant-negative δ-catenin-expressing neurons exhibited reduced surface levels of GluR2 (Silverman et al., 2007; Ochiishi et al., 2008). Our results show that the phosphorylation state of δ-catenin mediated by Cdk5 is also important in the cycling of GluR2 at the membrane. In addition to GluR2, we also investigated the effects of Cdk5-mediated phosphorylation of δ-catenin on GluR1, which also has been extensively studied on its role in synaptic plasticity. The GluR1 surface levels were increased in the presence of phospho-deficient δ-catenin. Although δ-catenin is not associated with GluR1 at its PDZ-binding domain by interaction with GRIP/ABP, it has been reported previously that heteromerization of GluR1/2 may be responsible for increased surface GluR1 with upregulation of GluR2 surface expression (Misra et al., 2010). It would also be interesting to determine whether the other AMPA receptors GluR3 and GluR4 are affected by this phenomenon as a result of heteromerization with GluR2.
The increase in dendritic protrusions and the “locking” of GluR2 and δ-catenin at the membrane resulted in an increase in the number of functional AMPA receptors as well as enhanced AMPA/NMDA ratio. The AMPA-mediated component is of obvious importance for message transduction in neurons; however, this process has to be tightly regulated for proper function. The Na+ flux in neurons was also affected with loss of Cdk5-mediated phosphorylation of δ-catenin, which resulted in an increased sodium influx into neurons transduced with DNCdk5 lentivirus (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Loss of Cdk5 activity and subsequent locking of δ-catenin into the membrane for sustained AMPA-mediated firing and enhanced Na+ influx may actually be detrimental to the neuron. The increased AMPA-mediated response may not be beneficial because Cdk5 knock-out neurons ultimately die, and this could be part of a mechanism that precedes improper synaptic function (Ohshima et al., 1996). A previous report described that modulation of Cdk5 activity in a conditional knock-out in adult mice did not result in significant changes in the AMPA-mediated activity in a mature nervous system (Hawasli et al., 2007). However, our findings of AMPA-mediated changes related to posttranslational modifications of δ-catenin are representative of a developmental paradigm. In both paradigms in which δ-catenin cannot be phosphorylated or its expression is reduced, spine formation and excitatory synaptic currents are increased, but it is uncertain whether these are linked (Arikkath et al., 2009).
The roles of the other 18 proline-directed serine–threonine sites that span across δ-catenin should also be investigated. It is likely that, in addition to Cdk5 phosphorylation, other proline-directed kinases could also target these sites with specific effects, which will expand the role of δ-catenin dependent on phosphorylation. Phosphorylation of the sister molecule β-catenin by other kinases, such as GSK3β and protein kinase A, influences its function and degradation, although p35/Cdk5 phosphorylation does not regulate δ-catenin degradation, but perhaps phosphorylation by either Cdk5 and/or other kinases on the other sites may perform this function (Hart et al., 1998; Ikeda et al., 1998; Hino et al., 2005).
The phosphorylation of δ-catenin by Cdk5 introduces a novel molecular mechanism to regulate localization of the GluR2 subunit of the AMPA receptor on the postsynaptic neuronal membrane and to mediate subsequent neuronal synaptic activity. Our data suggest that exit of δ-catenin from the membrane is facilitated by Cdk5, whereas its transit to the membrane could be facilitated by an unknown phosphatase. Data from this report increase the knowledge on how δ-catenin and Cdk5 coordinate synaptic activity in neurons and how this process, when perturbed through the loss of Cdk5 activity, leads to synaptic dysfunction that could be applicable in the study of mental retardation and to fully understand the development of the nervous system.
Footnotes
- Received December 7, 2009.
- Revision received April 4, 2010.
- Accepted April 19, 2010.
-
This work was supported by National Medical Research Council and Provost Matching Component Grant WBS 183-000-171-214/171-133, Biomedical Research Council Grant 183-000-214-305, and the Intramural Research Program of the National Institute of Neurological Disorders and Stroke/National Institutes of Health. We thank Dr. Qun Lu for the generous gift of EGFP–δ-catenin plasmid. We also thank Pan Ning and Prof. Ong Wei Yi of the Immunohistochemistry Core Facility, Neurobiology Programme, Life Science Institute, National University of Singapore, for expert technical assistance. For assistance with electron microscopy, we thank Prof. Mary Ng, Mickey Leong, and Tan Suat Hoon of the Electron Microscopy Unit, National University of Singapore. We acknowledge the Life Science Institute Start-Up Grant to A/Prof. C. Chen and use of facilities of the Dementia Research Laboratory, National University Health System.
- Correspondence should be addressed to Sashi Kesavapany, Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, 8 Medical Drive, MD7, #02-03, Singapore 117597. sashikesavapany{at}gmail.com
- Copyright © 2010 the authors 0270-6474/10/308457-11$15.00/0