
Abstract
Mechanisms of ischemic neuronal and vascular injury remain obscure. Here we test the hypothesis that thrombin, a blood-borne coagulation factor, contributes to neurovascular injury during acute focal ischemia. Stroke was induced in adult Sprague Dawley rats by occluding the middle cerebral artery. Intra-arterial thrombin infusion during ischemia significantly increased vascular disruption and cellular injury. Intravenous infusion of argatroban, a direct thrombin inhibitor, alleviated neurovascular injury. Immunostaining showed thrombin on neurons in the ischemic core. Using an activatable cell-penetrating peptide engineered to detect thrombin activity, we discovered that thrombin proteolytic activity was specifically associated with neuronal damage during ischemia. Protease activated receptor-1, the presumptive thrombin receptor, appeared to mediate ischemic neurovascular injury. Furthermore, rats receiving thrombin during ischemia showed cognitive deficit, whereas rats receiving argatroban retained intact learning and memory. These results suggest a potential role for thrombin contributing to neurovascular injury and several potential avenues for neuroprotection.
Introduction
Stroke refers to the symptoms caused by sudden loss of blood supply to the brain. Conditions that narrow or block the brain vasculature, including thrombus, might reduce blood supply causing focal tissue ischemia and stroke symptoms. Thrombin is the key regulator in clot formation and a natural target for stroke prevention and intervention.
Thrombin was generally considered to be only a clotting factor until 1991 when Coughlin et al. isolated the cellular receptor for thrombin, which is called protease activated receptor-1 (PAR-1; Vu et al., 1991). Subsequently a small family of receptors (PAR-2, PAR-3, and PAR-4) were discovered (Ishihara et al., 1997; Coughlin, 2000; Nakanishi-Matsui et al., 2000). The existence of PARs suggests that thrombin may play additional roles in normal physiology and in pathology, beyond serving as a clotting factor. The neuroprotection by PAR-1 deletion in various ischemic models also indicated toxic effects of thrombin in the CNS (Junge et al., 2003; Olson et al., 2004). Accumulating data suggest pleiotropic roles of thrombin in brain tissue, including vascular disruption (Chen et al., 2010; Liu et al., 2010), inflammatory response (Nishino et al., 1993; Xue and Del Bigio, 2001; Keep et al., 2005), oxidative stress (Choi et al., 2005; Kameda et al., 2012), and direct cellular toxicity (Donovan et al., 1997; Gingrich et al., 2000; Striggow et al., 2000); thrombin might contribute to stroke pathology through multiple mechanisms. A better understanding of thrombin toxicity should lead to new therapeutic targets in clinical application.
Previously, we developed a method to identify severe vascular disruption using a 2MDa dextran conjugated to FITC and observed severe vascular damage as early as 1 h after stroke onset in rodents (Chen et al., 2009; Friedman et al., 2009). This finding led to the hypothesis that blood–brain barrier opening during acute stroke allows blood-borne toxic factors to enter the brain and cause significant additional damage to ischemic tissue. We focused on thrombin as it is activated by endothelial injury; amplifies its own generation through positive feedback loops in the coagulation cascade; and is neurotoxic in vitro. Although numerous reports discuss the importance of thrombin activation in brain injury little is known about the effect of localized stroke-evoked thrombin activation and its effect on long-term cognitive function. Activated cell penetrating peptides (ACPPs) are probes that can be used for sensitive detection of localized protease activation in vivo. They have been used previously to monitor matrix metalloprotease (Jiang et al., 2004; Olson et al., 2009) and elastase activity (Whitney et al., 2010) within tumors and more recently adapted to detect thrombin activation in atherosclerotic plaques (Olson et al., 2012). In this report we have used a novel thrombin activated ACPP to monitor thrombin activation within a model of brain ischemia and have correlated this activation with neuronal damage and long-term loss of cognitive function.
Materials and Methods
Animal surgery.
All protocols were approved by the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center, following all national guidelines for the care of experimental animals. The procedure for middle cerebral artery occlusion (MCAo) model was performed as described previously (Longa et al., 1989; Manoonkitiwongsa et al., 2004). The subjects were male adult Sprague Dawley rats, 290 to 310 g. All animals received tail-vein injections of 0.3 ml FITC-dextran (Sigma-Aldrich, FD2000S; 2MDa, 5% solution in PBS) at the start of the surgery (Chen et al., 2009). Animals were anesthetized with 4% isoflurane mixed in oxygen and nitrous oxide (30:70) by facemask. A midline neck incision was made exposing the left common carotid artery. The external carotid and pterygopalatine arteries were ligated with 4–0 silk. An incision was made in the wall of the external carotid artery close to the bifurcation point of the external and internal carotid arteries. A 4–0 heat-blunted nylon suture (Ethicon) was then inserted and advanced ∼17.5 mm from the bifurcation point into the internal carotid arteries, thereby blocking the ostium of the middle cerebral artery for 1, 2, 3, or 4 h. A PE10 catheter connected to a syringe pump was then inserted into the external carotid artery. For argatroban treatment, the jugular vein was isolated and a PE 10 catheter inserted and secured with 6–0 silk ligatures. Indwelling catheters were tunneled under the skin to an exit incision between the scapulae. After the predetermined occlusion duration, the nylon suture was removed from carotid artery to allow the reperfusion of blood flow into the MCA. Neurological function was examined 1 h after ischemia onset and again during reperfusion using a published rodent neurological grading system (Bederson et al., 1986). At the end of the reperfusion period, animals were killed with an overdose of ketamine and xylazine, and then intracardially perfused with 250 ml of saline followed by 300 ml of 4% paraformaldehyde. Brains were removed, postfixed in 4% paraformaldehyde, cryoprotected in 30% sucrose in phosphate buffer, and then sliced into 50 μm sections on a freezing microtome (Reichert-Jung). Animals were excluded if subarachnoid hemorrhage was found postmortem.
Drug preparation.
The infusion of drugs was started immediately after the onset of MCA occlusion and continued using a syringe pump at 0.2 ml/h for the duration of MCA occlusion. Rat thrombin (Sigma-Aldrich, T5772) was dissolved in saline at 1U/ml and infused through a catheter placed in external carotid artery; common carotid artery flow carried the infusate into the internal carotid artery and then into the middle cerebral artery. Argatroban (Enzo Life Sciences, BML-PI146) was dissolved in saline at 0.56 mg/ml and was infused through the jugular vein. PAR-1 antagonist SCH79797 (Tocris Bioscience, Catalog #1592) was dissolved in DMSO at 8.3 mg/ml as stock solution, and was diluted in saline and infused into the jugular vein at the final concentration of 8.3 μg/ml, following the optimal dose as previously reported (Strande et al., 2007). PAR-1 agonist peptide TFLLR (Tocris Bioscience, Cat#1464) was dissolved in double distilled water at 450 μm and infused into the external carotid artery catheter (de Garavilla et al., 2001; Kawao et al., 2003). The control peptide RLLFT (Tocris Bioscience, Catalog #3393) was prepared in the same diluents at the same concentration.
For the experiment using ACPP to localize thrombin activation, 30 nmol of ACPP was infused in a bolus through catheterization of external carotid artery immediately after de-occlusion.
Behavioral assessment.
Behavioral tests were conducted to assess the motor function as well as memory and learning ability of the experimental animals. For this experiment, animals were subjected to 2 h ischemia and randomly assigned to receive one of the three treatments during ischemia: thrombin, argatroban, and saline infusion. One week after stroke onset, these animals were tested using the rotarod and cylinder test. For the rotarod test, the animals first went through a training session and learned to stay on a rotating spindle that accelerates from 4 to 40 rpm in 2 min. Each trial ended when the animal fell off the spindle. Following the training, three testing sessions were conducted and the average time the animal stayed on the spindle was measured to evaluate the performance. For the cylinder test, the animals were placed in a 20 cm wide transparent glass cylinder and a video camera was used to record a minimum of 20 contacts of each forepaw with the wall of the glass cylinder. Data were recorded as left, right or both paws simultaneously. Each placement was counted and divided by 20 to obtain percentage of paw usage.
Two weeks after stroke onset, these animals were tested with the neurological screen and open field followed by the Barnes Maze test. The neurological screen included tests of neurological reflexes, righting/startle response, and visual/tactile placing. For open field testing, study animals were placed in an open topped, clear Plexiglas box, surrounded by a series of photobeams, connected to a computer to record locomotor behavior. Data were recorded over a 60 min test session. Both number of horizontal beam breaks (general activity) and vertical beam breaks (rearing) were recorded. In the Barnes Maze test, the animals were placed on a flat circular platform with a bright spotlight on the center as the aversive stimulus. The platform contained 20 holes evenly spaced around the perimeter; only one of the holes led to the true escape box. The animals were given 3 min to locate the escape hole and the trial ended when they entered the box. The animals underwent three trials a day for 4 consecutive days, followed by a 2 day break. The position of the escape box remained consistent and thus maintained its relative location with respect to external spatial cues. On the 7th day, the animals were tested for their ability to locate the escape box at the same position. On the 8th and 9th day, the position of the escape box was moved to a new position (spatial probe and retention). The latency to enter the box was recorded. The learning curve for each animal was determined by calculation of the reciprocal of the average latency for each day in the learning period (Days 1–4). The slope of this curve is an indicator of learning speed with a higher slope characterizing animals that learned faster (Lyden et al., 1992). In addition, primary errors were calculated as the sum of errors made before the first encounter of the escape box.
Immunohistochemistry.
Brain sections were immunostained with primary antibodies goat anti-thrombin (Santa Cruz Biotechnology, sc-23335, 1:100), goat anti-plasmin (Santa Cruz Biotechnology, sc-15036, 1:100), rabbit anti-Factor-X (Santa Cruz Biotechnology, sc-20673, 1:100), rabbit anti-PAR-1 (Santa Cruz Biotechnology, sc-5605, 1:200), mouse anti-NeuN (Millipore, MAB377, 1:1000), mouse anti-GFAP (Millipore, MAB360, 1:5000), rabbit anti-Iba1 (Wako, 019-19741, 1:500), and Lycopersicon esculentum Lectin (Vector Laboratories, FL-1171, 1:1000). Selective Cy3- or Cy5-conjugated secondary antibodies were obtained from Millipore. The protocol for immunostaining is described in brief: sections were permeabilized with 5% bovine serum albumin and 0.1% Triton X-100 for 1 h, incubated with primary antibody at 4°C for 2 d, and followed by incubation with secondary antibody at room temperature for 4 h. Sections were washed in PBS, mounted on slides, and coverslipped with ProLong Gold antifade reagent (Invitrogen).
Cell death assay.
To detect cellular death associated with ischemic vascular disruption, a TUNEL staining protocol was performed using the In Situ Cell Death Detection Kit (Roche, #12156792) with minor modification. Sections prepared above were pretreated with fresh 0.1% Triton X-100 and 0.1% sodium citrate buffer for 10 min on ice. Then the sections were rinsed twice with PBS, and incubated with TUNEL reaction mixture prepared according to the manufacturer's instruction for 3 h at 37°C. Finally, the sections were rinsed in PBS, mounted with ProLong Gold antifade reagent. Fluorojade staining was used to detect general neuronal degeneration following the protocol of manufacturer (Millipore, AG325).
Western blot.
Brain samples from ischemic and nonischemic tissue were homogenized with 1× RIPA buffer (Thermo Fisher). Protein concentrations were normalized with MicroBCA kit (Thermo Fisher). Samples were loaded onto 4–12% NuPage Bis-Tris gels (Invitrogen) and transferred onto PVDF membranes. Blots were incubated with goat polyclonal antibody against thrombin (Santa Cruz Biotechnology, sc-23335, 1:200) or mouse monoclonal antibody against GAPDH (Santa Cruz Biotechnology, sc-32233, 1:5000), followed by horseradish peroxidase-conjugated secondary antibody (Sigma-Aldrich). Protein signals were visualized by Amersham ECL Substrate (GE Healthcare, RPN2106) and detected by ChemiDoc XRS imaging system (Bio-Rad). Band intensities were quantified using Image Lab software (ImageLabs).
Imaging and quantitative analysis.
Vascular disruption and cellular injury of ischemic brain sections were imaged at low power using epifluorescence microscopy with a highly sensitive CCD camera (Apogee, Alta U32). The signal of FITC-dextran, TUNEL, and Fluorojade staining were quantified using Image-Pro Plus (Media Cybernetics) as described previously (Chen et al., 2009). In brief, an operator blind to the subject's group or duration of ischemia examined each section and adjusted the brightness and contrast levels to optimize the appearance of the fluorescence. For FITC-dextran and Fluorojade quantification, the operator outlined the region of interest (striatum and cortex), and applied semiautomatic thresholding and segmentation to measure the total area of fluorescence. For TUNEL staining, the operator set a threshold that differentiated the fluorescence from the background and the software automatically counted the number of cells. For cellular colocalization studies, brain sections were imaged by confocal microscopy (Leica TCS SP), and the colocalization information were manually examined and counted in Image-Pro Plus as well.
Statistics.
All data were summarized for analysis using Excel 2010 (Microsoft). Means were tested using the t test or ANOVA as appropriate. All variances presented are SEs. Multivariate analysis of variation (MANOVA) was conducted by statistical software JMP (SAS). All values were corrected for multiple comparisons using the Bonferroni approach.
Results
Thrombin level increased in the ischemic brain region
We caused focal cerebral ischemia in Sprague Dawley rats by occluding the middle cerebral artery (MCA) and labeled severe vascular disruption with high molecular weight FITC-dextran (Fig. 1a). To investigate the involvement of thrombin in ischemic cytotoxicity during stroke, we labeled the brain sections with thrombin antibody (Fig. 1b). Significant thrombin antigen was found in regions of severe vascular disruption, compared with regions with less or no vascular disruption contralaterally (Fig. 1c). Western blotting of tissue samples from ischemic core (high FITC-dextran leakage) confirmed the significantly elevated level of thrombin (Fig. 1d,e). Double staining with cellular markers (NeuN for neurons, GFAP for astrocytes, and Iba-1 for microglia) revealed that 65% of thrombin was associated with neurons, 15% with microvessels and 10% with glial cells (Fig. 1f–i).

Extravasated thrombin associated with ischemic neurons. Focal cerebral ischemia was induced by occluding the middle cerebral artery for 4 h and 2MDa FITC-dextran was infused to label severe vascular disruption. a, Subregions of brain section with high, low, and no FITC fluorescence were selected for immunostaining and confocal microscopy. The no-leakage section comes from a contralateral brain region homologous to the ischemic zone. Scale bar: 2 mm. b, Representative images of brain subregions visualized with FITC-dextran and Cy5-labeled thrombin antibody. Scale bar: 50 μm. c, Significant amounts of thrombin were present in areas with severe vascular disruption (n = 6–9; ANOVA, p < 0.05; post hoc Bonferroni test, *p < 0.05). d, e, Western blots showed a threefold increase of thrombin level in the ischemic regions with high levels of leakage, compared with regions with low levels of leakage or contralateral regions with no leakage (n = 7; ANOVA, p < 0.01; post hoc Bonferroni test, *p < 0.05, **p < 0.01). f–i, Immunohistochemistry with thrombin and cellular marker revealed the association of thrombin antigen with ischemic neurons (n = 9–17; ANOVA, p < 0.001; post hoc Bonferroni test, ***p < 0.001). Arrows indicated the colocalization of thrombin antigen and neuronal marker NeuN. Scale bars: f–h, 50 μm.
PPRSFL-ACPP specifically detected thrombin proteolytic activity induced by stroke
Thrombin antibody staining localizes the antigen but not necessarily the proteolytic activity of the enzyme. To assay thrombin activity in vivo, we applied a novel imaging probe, an activated cell penetrating peptide (ACPP). The probe consists of 9 d-glutamates and 9 d-arginines, connected by a linker sequence (initially DPRSFL) which was based on an amino acid sequence in the exosite of the PAR1 receptor (Olson et al., 2012). A new optimized ACPP substituted PPRSFL in place of DPRSFL within the activation region. The PPRSFL substrate was identified using a substitution mutagenesis screen to identify substrates with increased specificity for cleavage by thrombin with reduced cleavage by the related enzymes plasmin and factor X (Fig. 2a). The thrombin-activated PPRSFL-ACPP was infused into the ischemic brain through a catheter implanted in the external carotid artery. In this model the infusate flows from the catheter tip into the common carotid artery where anterograde flow carries the infused probe into the internal carotid and ultimately the middle cerebral artery (Fig. 2b). Thrombin cleavage of the ACPP jettisons the polyglutamate, allowing the Cy5-labeled polyarginine to bind and internalize into nearby cells. Uncleaved probes wash out of the tissue during reperfusion. To confirm the selectivity of PPRSFL-ACPP for thrombin in this model, argatroban was administered intravenously during the intra-arterial infusion of ACPP probe to temporarily block thrombin activity. Argatroban reduced fluorescence uptake of PPRSFL-ACPP, confirming that thrombin activity was required for in vivo labeling (Fig. 2c–e). To further confirm the selectivity of PPRSFL-ACPP for detecting thrombin activity in stroke, brain sections from rats treated with PPRSFL-ACPP were immunostained with antibodies for thrombin, plasmin, and factor X, respectively (Fig. 2f–h). As shown in Figure 2i, 80% of the PPRSFL-ACPP labeling colocalized with thrombin immunostaining while significantly less ACPP labeling colocalized with the immunoreactivity of plasmin or factor X (p < 0.01, χ2).

Thrombin activity was specifically labeled with ACPP in vivo. a, In vitro assay of ACPP after 15 min incubation with different serine proteases. The upper panel shows that the original thrombin cleavage sequence, DPRSFL, was susceptible to multiple proteases including thrombin, plasmin, and factor X; the lower panel demonstrates the higher specificity of a modified sequence, PPRSFL, to thrombin cleavage. PPRSFL-ACPP was used for the subsequent in vivo experiments. b, Scheme of experimental design. Animals underwent 2 h occlusion followed by 6 h reperfusion. FITC-conjugated 2MDa dextran was infused at occlusion onset to label vascular disruption; Cy5-conjugated ACPP probe was infused intra-arterially at de-occlusion followed by 6 h recirculation to allow clearance of uncleaved ACPP. c–e, ACPP labeling was reduced after intravenous argatroban, a direct thrombin inhibitor, suggesting that the ACPP labeling is thrombin dependent (n = 10; independent samples t test, **p < 0.01). The area of Cy5-ACPP signal and FITC fluorescence was plotted as in d. In e, the ratio of ACPP area to FITC area was calculated to adjust for the reduced vascular disruption by argatroban. Scale bar: c, 100 μm. f–i, Immunostaining with an antibody directed against thrombin colocalized with ACPP-positive cells (yellow arrow), confirming the selectivity of ACPP. Colocalization of ACPP signal with the other two proteases, plasmin and factor X, was significantly lower (n = 4; ANOVA, p < 0.01; post hoc Bonferroni test, **p < 0.01). Scale bars: f–h, 50 μm.
Thrombin activity was associated with neuronal damage
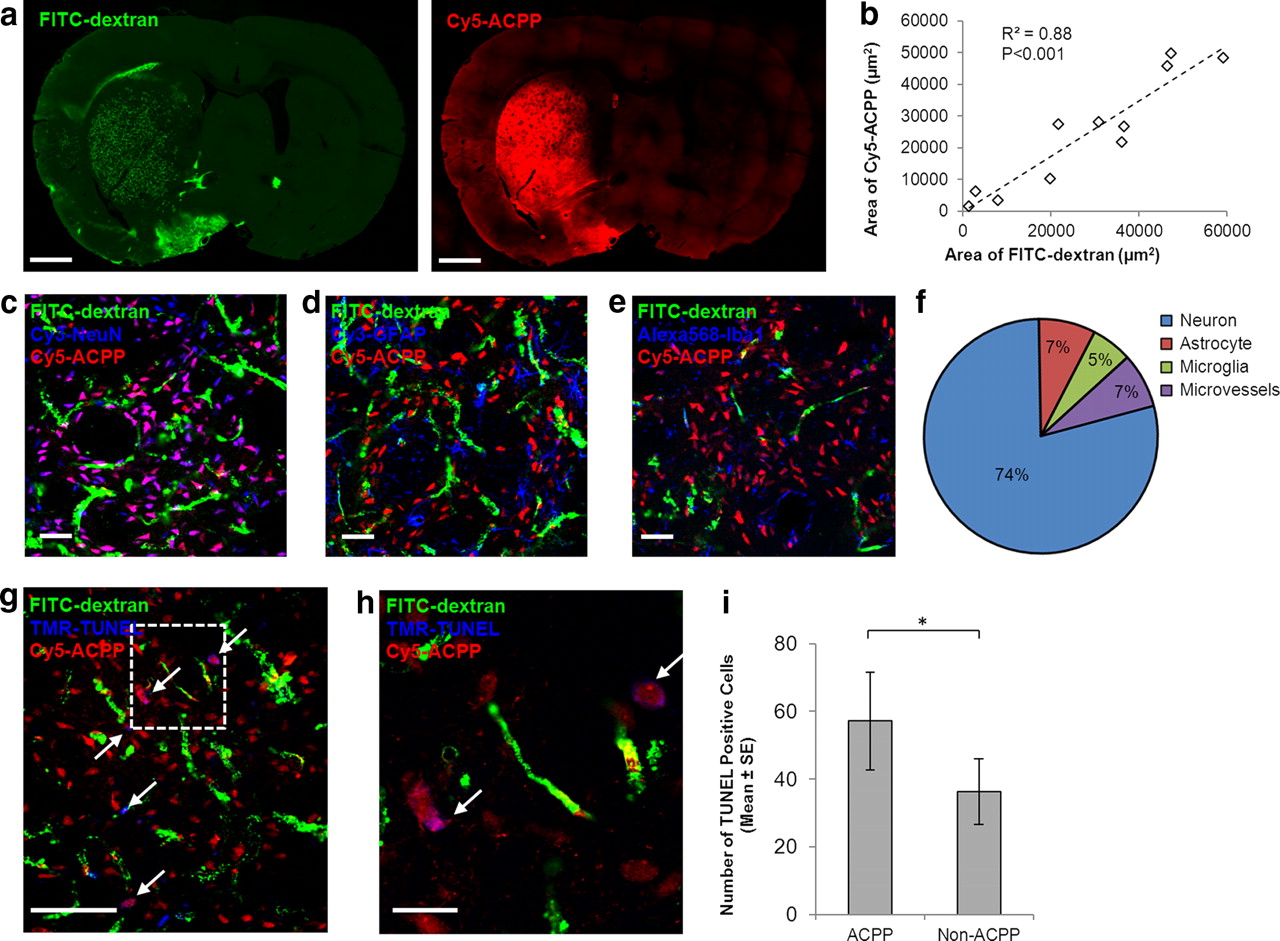
We then sought to characterize the topographic distribution of thrombin activity during ischemia. PPRSFL-ACPP accumulated in regions of severe vascular disruption (Fig. 3a). Random micrographs across the brain were generated using confocal microscopy, and a close correlation between PPRSFL-ACPP labeling and vascular disruption was found (Fig. 3a,b). ACPP-positive cells (as detected by Cy5 fluorescence) were immunostained with cellular markers for neurons (NeuN), astrocytes (GFAP), microglia and macrophages (Iba1), and microvessels (Lycopersicon Esculentum Lectin). Among all PPRSFL-ACPP-positive brain cells, 74% also stained with NeuN, confirming that thrombin activity localized mostly in neurons (Fig. 3c–f). In addition, ischemic cell death was detected with TUNEL staining. Among the sections examined (N = 6), 61% of the TUNEL-positive cells were associated with PPRSFL-ACPP probe, confirming that thrombin activity was associated with neurovascular injury (Fig. 3g–i).

Thrombin activity was associated with neurovascular damage. a, b, Thrombin ACPP was deposited in parenchymal tissues containing severe vascular disruption (FITC-dextran leakage). The area of Cy5-ACPP signal correlated with the area of FITC-dextran fluorescence (n = 11; correlation coefficient r = 0.939; R2 = 0.88; p < 0.001). Scale bar: a, 3 mm. c–f, ACPP-positive cells were identified with cellular markers for neurons (NeuN), astrocytes (GFAP), microglia and macrophages (Iba1), and microvessels (Lycopersicon Esculentum Lectin, data not shown). Of the ACPP-positive cells, 74% were associated with neurons, and significantly fewer with glia and microvessels (n = 7–11; ANOVA, p < 0.001; post hoc Bonferroni test, p < 0.001 between neurons and all other cell types). Scale bars: c–e, 50 μm. g, h, Cell death was reported by TUNEL staining. The inbox was magnified in h. Scale bars: g, 100 μm; h, 25 μm. i, TUNEL signal localized more in ACPP-positive cells (n = 6; paired sample t test, *p < 0.05).
Thrombin caused neurovascular damage during stroke
To investigate whether thrombin caused neurovascular damage during ischemia, we induced MCA occlusion in animals for 1, 2, 3, and 4 h and infused intra-arterial thrombin during ischemia. Animals receiving thrombin infusion showed increased vascular permeability to IgG (Fig. 4a,b), severe vascular disruption (Fig. 4c,d), and cellular injury (Fig. 4e,f) compared with animals receiving saline infusion. 24 h after ischemia, the thrombin-treated animals also developed more apoptotic neuronal cells in the ischemic brain compared with saline-treated animals. Intravenous argatroban instead of thrombin decreased the progression of vascular disruption as well as cellular injury (Fig. 4a–h). In addition to cellular injury, argatroban infusion significantly ameliorated the metabolic compromise detected at 48 h with TTC staining (Fig. 4i,j).

Thrombin promoted neurovascular damage during acute ischemia. Intra-arterial infusion of thrombin significantly increased vascular permeability to IgG (a, b), severe vascular disruption labeled by FITC-dextran (c, d), and neuronal damage labeled by Fluoro-Jade staining (e, f). Intravenous infusion of argatroban protected the brain from ischemic neurovascular damage (n = 4–8 for each time point; MANOVA, p < 0.01). g, h, The number of TUNEL-positive cells was significantly increased with intra-arterial thrombin and reduced with intravenous argatroban treatment (n = 5–7 for each group; ANOVA, p < 0.001; post hoc Bonferroni test, *p < 0.05, **p < 0.01). i, j, 48 h TTC exclusion volume was significantly smaller with argatroban treatment compared with saline control (n = 6; independent samples t test, *p < 0.05). Scale bars: a, 1 mm; c, e, 200 μm; g, 100 μm.
Thrombin toxicity leads to cognitive deficit in stroke animals
To further demonstrate the translational relevance of thrombin toxicity in stroke, we assessed behavior after intra-arterial thrombin, intravenous argatroban, or vehicle. Animals were subjected to 2 h MCA occlusion and received thrombin, argatroban, or vehicle infusion during ischemia. One week after stroke, there were no significant differences among treatment groups in unilateral motor function (cylinder test, Fig. 5a), or locomotor and balance function (rotarod test, Fig. 5b). Two weeks after stroke no significant differences were found in neurological screen or spontaneous motor behavior (open field test, Fig. 5c). However, significant differences in learning and memory were shown using a Barnes maze. We used a standard learning paradigm (4 d of three trials per day) followed by 2 d off; a memory test (day 7); and a learning test using a reversal of the escape hole (day 8 and 9). We measured the time required for each subject to locate the correct escape hole (latency). Thrombin treatment worsened all measures while argatroban-treated animals showed a similar learning curve and latency time compared with the control group (Fig. 5d–f). Reversing the hole on day 8 differentiated the behavior of the animals to the maximum extent (Fig. 5d).
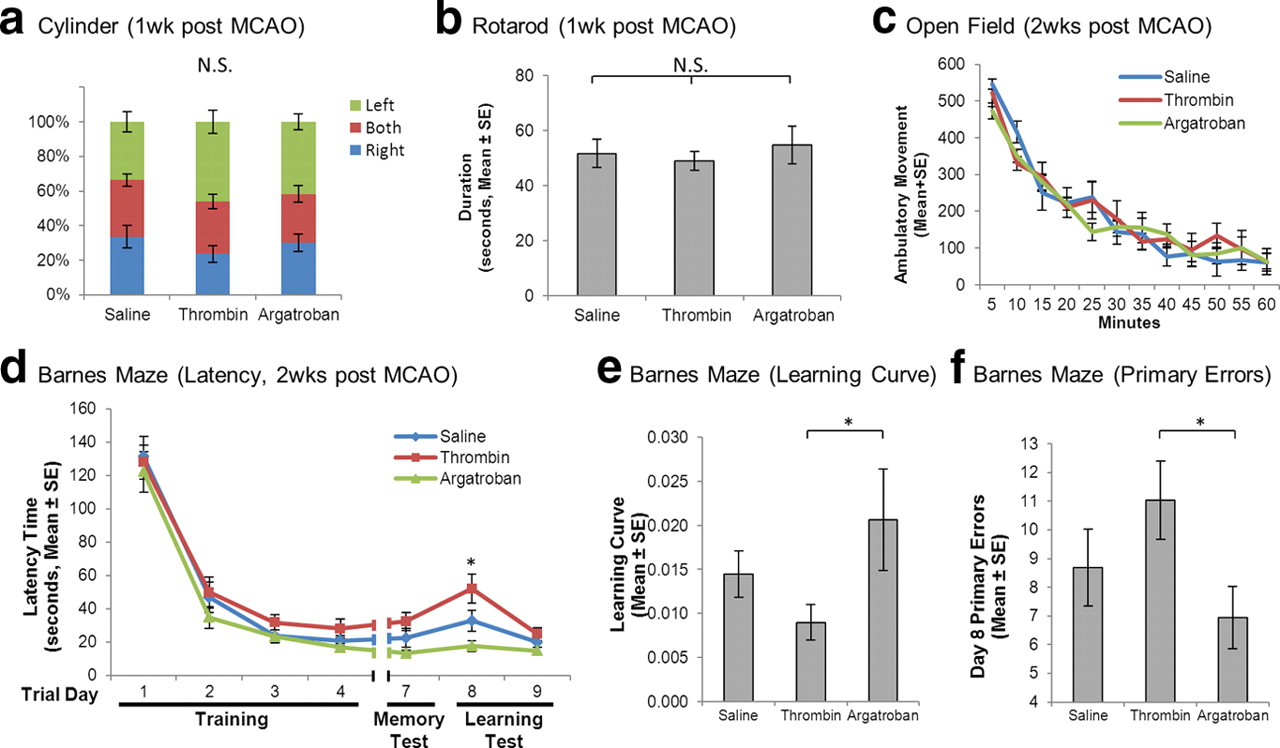
Thrombin promoted cognitive deficit. Animals underwent 2 h ischemia, treated with thrombin, argatroban, or vehicles during occlusion period, and survived for 3 weeks. No significant differences among treatment groups were found in unilateral motor function (cylinder test) (a), locomotor and balance function (rotarod; n = 10; ANOVA, p > 0.05) (b), or spontaneous motor behavior (open field; n = 10; MANOVA, p > 0.05) (c). Cognitive functions of the animals were further assessed by Barnes Maze test. Animals receiving thrombin treatment appeared to have longer latency time (d), slower learning curve (e), and higher primary errors (f) during the test. Argatroban-treated animals showed higher performance in all measures (n = 10; MANOVA, p < 0.05). The behavioral differences among treatment groups were maximal on day 8 when the escape hole was relocated (n = 10; ANOVA, p < 0.05; post hoc Bonferroni test, *p < 0.05).
PAR1-mediated thrombin toxicity during stroke
Protease activated receptor-1 (PAR-1) is the presumptive cellular receptor for thrombin. We labeled ischemic brain sections with PAR-1 antibody targeting thrombin binding sites on the N-terminal extracellular domain (Cheng et al., 2003; Dömötör et al., 2003). Upon activation, PAR-1 is rapidly internalized and degraded. Therefore the activation of PAR-1 is evident by the loss of PAR-1 antigen signal (Hoxie et al., 1993; Giacaman et al., 2009). In subregions of severe vascular disruption, PAR-1 signal was significantly reduced compared with subregions with less or no vascular disruption (Fig. 6a,b). Next, we infused a PAR-1 agonist peptide, TFLLR, through the carotid artery into the brain and found a 50% increase in severe vascular disruption compared with a scrambled peptide RLLFT (Independent sample t test, p < 0.05, Fig. 6c). The number of TUNEL-positive cells in the agonist group was also greater (Fig. 6d). To confirm, we infused SCH79797, a PAR-1 antagonist, through the jugular vein. The average area of severe vascular disruption in the treatment group was significantly smaller than in the control group (Independent sample t test, p < 0.05, Fig. 6e). The number of TUNEL-positive cells was also significantly reduced (p < 0.05, Fig. 6f).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PAR-1-mediated thrombin toxicity on the neurovascular unit. Focal ischemia was induced for 4 h and immunohistochemistry was used to examine the involvement of PAR-1, the presumed thrombin receptor, in mediating thrombin toxicity. a, b, Activation of PAR-1 was evident by the loss of antigen signal in the subregions of severe vascular disruption, compared with subregions with less or no FITC-dextran (n = 9; ANOVA, p < 0.001; post hoc Bonferroni test, *p < 0.05, ***p < 0.001). Scale bar: a, 100 μm. c, d, Intra-arterial infusion of TFLLR, a PAR-1 agonist that mimics the tethered N-terminal of the receptor, increased vascular disruption and cell death. e, f, Intravenous infusion of SCH79797, a small molecule PAR-1 inhibitor, significantly decreased the level of neurovascular injury. (n = 8–12; independent samples t test, *p < 0.05).
Discussion
In this study we found that in vivo thrombin activity mediates neuronal damage during acute focal cerebral ischemia. Using immunohistochemistry and pharmacological agonist/antagonist studies, we showed a strong association between thrombin and neurovascular damage in the ischemic core. Using a novel ACPP imaging probe to detect proteolytic activity of thrombin, we could relate early thrombin activation to cell death. We also found an association between thrombin toxicity and long-term cognitive deficit after stroke. Additional studies suggested that thrombin mediates cytotoxicity via the PAR-1 receptor. These data support an important role for thrombin toxicity during acute stroke and suggested thrombin antagonism as a neuroprotection treatment for acute ischemic stroke.
Although our data strongly suggest a role for PAR-1, the exact mechanism of thrombin-mediated vascular and tissue injury during acute ischemia requires further investigation. Our data suggest that thrombin has different effects on different cell types (Figs. 1, 3) and tissue damage after stroke could be the combinatorial result of several cellular responses. For example, thrombin effects on vasculature might be mediated by its direct effect on blood–brain barrier tight junctions (Vandenbroucke et al., 2008; Kondo et al., 2009), or by affecting neurons and glial cells which disrupt the integrity of neurovascular unit (Donovan et al., 1997; Gingrich et al., 2000), or by inducing an inflammatory response that indirectly mediates further damage (Nishino et al., 1993; Keep et al., 2005). As most thrombin-related cellular responses are mediated by the PAR-1 receptor, for future studies cell-specific PAR-1 knock-out animals would be very helpful to dissect the roles of thrombin in different cell types. Alternatively, virus-based shRNA could be infused to knock down the expression of PAR-1 or other downstream genes.
Our data show that activated thrombin accumulated in regions of severe vascular disruption but we cannot infer whether thrombin originated in the blood stream or from brain parenchyma. The fact that thrombin is induced by vascular damage and amplifies itself near the injured sites might suggest a vascular origin. However, we cannot rule out the possibility that thrombin was produced in situ during ischemia. Previous reports suggest that thrombin mRNA could be detected in neural tissues (Dihanich et al., 1991) and increased 24 h after global ischemia (Riek-Burchardt et al., 2002). In our study, we did detect thrombin protein in the nonischemic brain (Fig. 1d), consistent with the inference that thrombin could originate in the parenchyma. Further experiments will be needed to clarify the origin of thrombin and how it is related to neurovascular unit effects. Nevertheless, while the source of thrombin itself is an interesting question, the answer does not affect the pathological link between thrombin protease activity and ischemic damage, as observed in our experiments.
Our data further demonstrate the interesting role of the thrombin receptor in mediating damage to the neurovascular unit (Fig. 6). The presence of the PAR-1 receptor in brain leads us to speculate on the endogenous function of thrombin and its receptor in the brain. It is possible that thrombin contributes to injury during acute stroke but participates in brain repair later. The angiogenic effect of thrombin has been explored in several studies in vitro (Haralabopoulos et al., 1997; Tsopanoglou and Maragoudakis, 1999). Thrombin induces capillary tube formation in a dose-dependent manner. Low dose thrombin promotes angiogenesis. Several mechanisms have been implicated, including the activation of protein C and upregulation of the vascular endothelial growth factor receptor. Whether or not this is a physiologically relevant event in vivo remains unknown. It will be highly interesting if thrombin actually participates in the angiogenic response in injured tissue after stroke. In addition to angiogenesis, thrombin might play an important role in vascular development (Griffin et al., 2001). Mice deficient in thrombin receptor PAR-1 have abnormal bleeding and half of them die during embryonic development. Furthermore, thrombin might also participate in neurogenesis. Thrombin directly injected into brain causes increased expression of doublecortin, a marker for immature neurons (Yang et al., 2008). Experiments using other markers for neurogenesis (e.g., BrdU) would be necessary to further examine the concept of thrombin-mediated neurogenesis. It would be of great interest to investigate the potential engagement of thrombin in neurogenesis in postischemic brain as well.
Our data suggest therapeutic implications. The association of thrombin activity with neuronal damage and long-term cognitive deficit may be clinically significant and suggests a role for early thrombin inhibitor therapy for stroke patients. A pilot trial of argatroban has been completed and further trials are planned (Sugg et al., 2006; Barreto et al., 2012). However, current clinical trials are designed using argatroban for its anti-thrombotic property and the dose is adjusted to elevate activated partial thromboplastin time. Our results suggest that these trials may need to be altered to find a neuroprotective dose that may be quite different from an anti-thrombotic dose. In addition, thrombin effects on learning and memory impairment have been reported in other neurodegenerative diseases. Thrombin expression in neuron and glia was detected in the Alzheimer disease brains (Arai et al., 2006). Accumulation and leakage of thrombin in the microvessels of degenerative brain were also reported (Grammas et al., 2004; Bell et al., 2010). Direct intraventricular injection of thrombin to the rodent brain significantly impaired reference memory (Mhatre et al., 2004). These studies provide additional evidence to confirm the neurotoxicity of thrombin as a pathological mechanism in brain degeneration. In the future, new imaging agents such as ACPPs detectable by magnetic resonance imaging could improve diagnosis of thrombin related diseases and provide insight into molecular events in patients (Olson et al., 2010).
Footnotes
- Received January 25, 2012.
- Revision received April 6, 2012.
- Accepted April 12, 2012.
Author contributions: B.C., R.Y.T., and P.D.L. designed research; B.C., B.F., M.A.W., J.A.V.W., I.-F.L., E.S.O., Q.C., B.P., and L.Z. performed research; M.A.W. and R.Y.T. contributed unpublished reagents/analytic tools; B.C. and P.D.L. analyzed data; B.C., B.F., M.A.W., R.Y.T., and P.D.L. wrote the paper.
This study was supported by the NIH Grant R01 NS075930 to P.D.L., NIH Grant NS27177, Department of Defense W81XWH-05-1-0183 to R.Y.T., and American Heart Association Grant 09PRE2280276 to B.C. We thank lab members for helpful discussion and the Biobehavioral Core at Cedars-Sinai Medical Center for technical assistance.
M.A.W., E.S.O., and R.Y.T. have consulting agreements with Avelas Biosciences. The authors have no additional competing financial interests.
- Correspondence should be addressed to Dr. Bo Chen, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, Davis 2094D, Los Angeles, CA 90048. bochen.neuro{at}gmail.com
- Copyright © 2012 the authors 0270-6474/12/327622-10$15.00/0
This article is freely available online through the J Neurosci Open Choice option.