
Abstract
Neurofibromatosis type I (NF1), caused by the mutation in the NF1 gene, is characterized by multiple pathological symptoms. Importantly, ∼50% of NF1 patients also suffer learning difficulty. Although downstream pathways are well studied, regulation of the NF1-encoded neurofibromin protein is less clear. Here, we focused on the pathophysiology of Drosophila NF1 mutants in synaptic growth at neuromuscular junctions. Our analysis suggests that the Drosophila neurofibromin protein NF1 is required to constrain synaptic growth and transmission. NF1 functions downstream of the Drosophila focal adhesion kinase (FAK) Fak56 and physically interacts with Fak56. The N-terminal region of NF1 mediates the interaction with Fak56 and is required for the signaling activity and presynaptic localization of NF1. In presynapses, NF1 acts via the cAMP pathway, but independent of its GAP activity, to restrain synaptic growth. Thus, presynaptic FAK signaling may be disrupted, causing abnormal synaptic growth and transmission in the NF1 genetic disorder.
Introduction
The hereditary disorder neurofibromatosis type I (NF1), caused by mutations in the NF1 gene, is characterized by multiple pathological symptoms, such as multiple cafe-au-lait spots and neurofibromas. Approximately 50% of NF1 patients also suffer learning disability (Allanson et al., 1991; von Deimling et al., 1995; Friedman, 1999; Costa et al., 2001; Arun and Gutmann, 2004). NF1 mutations cause reduction in the level and/or activity in the protein neurofibromin that carries a Ras GAP-related domain (GRD) (Wallace et al., 1990). In several cases, abnormality in NF1 animal models can be attributed to disregulated Ras/MAPK signaling (Gutmann et al., 1993; Weiss et al., 1999; Tong et al., 2002). In Drosophila, NF1 mutants display growth retardation primarily caused by hyperactivated Ras signaling, which can be rescued by neuronal expression of the NF1 gene (The et al., 1997; Walker et al., 2006). Circadian rhythm in NF1 mutants is also disturbed by hyperactivated Ras/MAPK signaling (Williams et al., 2001). The second pathway known to mediate neurofibromin/NF1 activity is adenylyl cyclase (AC)/cAMP signaling. Reduced AC activity contributes to the phenotypes observed in NF1−/− mouse brain (Tong et al., 2002). Several defects in NF1 mutant flies are caused by disregulated AC/cAMP signaling, such as shortened life span, diminished neuropeptide-stimulated K+ current, and defective olfactory learning and memory (Guo et al., 1997, 2000; Tong et al., 2007). Thus, Ras/MAPK and AC/cAMP mediate different cellular functions of neurofibromin/NF1 in animals. Molecularly, the GRD of neurofibromin/NF1 catalyzes turnover of Ras-GTP while the C-terminal domain is involved in AC activation (Weiss et al., 1999; Tong et al., 2002). Although downstream signaling pathways are well studied, regulation of neurofibromin/NF1 activities and upstream signaling components are not clear.
Focal adhesion kinase (FAK) functions in cerebral cortex lamination, growth cone dynamics, axon branching and pathfinding, and synapse formation (Beggs et al., 2003; Rico et al., 2004; Robles and Gomez, 2006; Endo and Yamashita, 2009; Woo et al., 2009; Myers and Gomez, 2011). FAK functions together with Src in netrin and integrin signaling to modulate axonal terminal dynamics (Li et al., 2004; Liu et al., 2004; Ren et al., 2004; Robles and Gomez, 2006). FAK also suppresses axonal arborization through a p190RhoGEF-dependent mechanism (Rico et al., 2004). In EphB-receptor signaling, FAK inhibits the activity of the F-actin-severing protein cofilin to maintain dendritic spine stability in hippocampal neurons (Shi et al., 2009). The Drosophila FAK, Fak56, confines synaptic growth and regulates basal synaptic transmission at neuromuscular junctions (NMJs) (Tsai et al., 2008).
We examined morphological and electrophysiological properties at NMJs of Drosophila NF1 mutants, and found that they displayed the same phenotypes as Fak56 mutants. Genetic and protein–protein interaction between NF1 and Fak56 suggested that NF1 functions downstream of and forms a protein complex with Fak56 through the N-terminal 400 aa region that mediates NF1 signaling activity and synaptic localization. We further showed that the GAP activity of NF1 is dispensable while the AC/cAMP pathway is required to mediate NF1 and Fak56 in presynapses to suppress NMJ growth.
Materials and Methods
Fly stocks and manipulations.
Drosophila larvae of both sexes were used in this study. Wild-type control is w1118 or an isogenic strain for NF1 mutants. Mutant alleles were backcrossed to w1118 for at least five generations. Fak56N30, Fak56K24, Fak56KG00304, rut1, dncM14, NF1E1, NF1E2, NF1P2, hs-NF1 wt, hs-NF1 K1481A, hs-NF1 R1276P, and hs-NF1 ΔGRD have been previously reported (Zhong et al., 1992; The et al., 1997; Devenport and Brown, 2004; Walker et al., 2006; Tsai et al., 2008). UAS-NF1-A, UAS-NF1-B, UAS-CFP-NF1, and UAS-CFP-NF1ΔA were constructed for this study using the Gateway System (Invitrogen and Drosophila Genomics Resource Center). GAL4 drivers elav-GAL4 and C57-GAL4 were obtained from the Bloomington Drosophila Stock Center. UAS-rut RNAi (II and III) were from the Vienna Drosophila RNAi Center. All flies were reared at 25°C.
Immunostaining of NMJs and imaging processing.
Larvae of wandering late third instar were dissected for analyzing NMJ phenotypes at A3 segments, as previously described (Tsai et al., 2008). Primary antibodies used were for synapsin (Syn) [3C11, 1:100; Developmental Studies Hybridoma Bank (DSHB)], Bruchpilot (Brp) (nc82, 1:100; DSHB), Futsch (22C10, 1:100; DSHB), Discs large (Dlg) (4F3, 1:100; DSHB), Fas2 (1D4, 1:100; DSHB), FAK [pY397] (rabbit, 1:50; Invitrogen), GFP (mouse, 1:500; Invitrogen), HRP-conjugated TRITC or Cy5 (rabbit, 1:100; Jackson ImmunoResearch). Images were acquired using confocal Zeiss LSM 510 Meta and processed by Adobe Photoshop CS. Quantification of NMJ bouton numbers were collected by projecting images from 10 z-sections within 6.5–8 μm. The NMJ 6/7 size was shown as bouton numbers divided by muscle areas revealed by phalloidin staining and measured by Zeiss LSM Image Examiner.
Electronic microscopy of boutons.
The procedure has been described (Tsai et al., 2008). Briefly, dissected larval body walls with attached ventral nerve cords and motor axons were fixed in modified Trump's fixative and postfixed in 2% osmium tetroxide in 0.1 m sodium cacodylate buffer. The dissected muscle 6/7s of A3 segments were stained en bloc in 2% aqueous uranyl acetate, dehydrated in a graded ethanol series, and embedded in the Spurr's medium. Thin sections (90 nm) were stained with uranyl acetate and lead citrate, and imaged from a Tecnai G2 Spirit TWIN electron microscope (FEI) and a Gatan CCD Camera (794.10.BP2 MultiScan). TEM data were quantified by MetaMorph V6.3r7 (Molecular Devices).
Preparation of dibutyryl-cAMP-supplemented or forskolin-supplemented larval food.
The stock solutions of dibutyryl-cAMP (db-cAMP) and forskolin (Sigma-Aldrich) were prepared in distilled water and DMSO, respectively. The stock solutions or solvents (mock controls) were added into food vials to reach final concentrations of 10 μm, which was repeated every 2 d while larvae were growing to reach late third instar.
Immunoprecipitation, immunoblotting, and GST pull-down assay.
Embryos of 0–24 h or transfected S2 cells were homogenized in lysis buffer (100 mm NaCl, 10 mm Tris, pH 7.6, 1 mm EDTA, 1% Triton X-100, 10 mm glycerol phosphate, 10 mm NaF, 1 mm Na3VO4, 1 mm PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). Lysates of 1.5 mg of total protein were used for immunoprecipitation by anti-NF1 (guinea pig; Walker et al., 2006) or anti-Flag (1:20,000 dilution; M2; Sigma-Aldrich) antibodies. For Western blots, anti-Fak56 (rabbit, 1:5000; Palmer et al., 1999), anti-NF1 (Mab11, 1:1; Walker et al., 2006), anti-phosphotyrosine (1:1000; 4G10; Millipore) and anti-Myc (1:3000; 9E10; Santa Cruz Biotechnology) antibodies were used. Seven NF1 cDNA fragments (see Fig. 4A for the range of each fragment) were subcloned into pGEX4T-3. GST-NF1 fusion proteins were expressed, purified, and immobilized on glutathione beads and incubated with 500 μg of total protein from S2 cell lysates made in radioimmunoprecipitation assay lysis buffer (150 mm NaCl, 50 mm Tris, pH 7.5, 1% sodium deoxycholate, 1% NP40, 0.1% SDS, 1 mm PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). The GST pull-down complexes were analyzed by Western blot using anti-Fak56 antibodies.
Electrophysiological recording.
The basal transmission properties were measured in muscle 6 of A3 segments as previously described (Tsai et al., 2008). Briefly, larval body walls were dissected in cold (4°C) HL3.1 Ca2+-free saline (Feng et al., 2004) and recorded in HL3.1 containing 0.2–0.4 mm CaCl2 at 22°C. Only samples with resting potential from −80 to −65 mV and membrane resistance of >5 MΩ were measured. The measured excitatory junction potential (EJP) amplitudes were corrected for nonlinear summation by using two different correction factors (Stevens, 1976; McLachlan and Martin, 1981). Paired-pulse facilitation (PPF) is the ratios of second EJPs to first EJPs, averaged from responses to 3–5 consecutive stimulations (separated by 10 s rest) at 20–500 ms interpulse intervals. Signals were digitized at 50 kHz by a DigiData 1440 interface (Molecular Devices), low-pass filtered at 10 kHz, and saved on IBM-compatible PC for analysis.
Results
Presynaptic NF1 suppresses NMJ growth
To examine whether NF1 has a role in NMJ growth regulation, we examined NMJ morphology of late third instar homozygous or transheterozygous for NF1E1 and NF1E2, compared with isogenic wild-type control. NMJ morphology is revealed by immunostaining for HRP labeling axonal processes and Syn labeling presynaptic vesicles (Fig. 1A). Also, muscle fibers were labeled by phalloidin (data not shown). Both NF1E1/+ and NF1E2/+ heterozygotes show no difference to wild-type control, which is an isogenic line used to generate these two alleles. However, both NF1E2/E2 and NF1E1/E2 displayed larger NMJ size (Fig. 1B,C), suggesting that NF1E2 is a stronger allele than NF1E1 in NMJ growth control. The overgrowth phenotype is not limited to NMJ 6/7s as the sizes of NMJ 4s and NMJ 12/13s were also increased in NF1E2/E2 (Fig. 1D–H). NF1 mutants also display smaller body and muscle size (Walker et al., 2006). However, the increase in NMJ size was not attributed to the reduction in muscle size, as muscle expression of UAS-CFP-NF1 by the C57-GAL4 driver restored the muscle size but failed to suppress the overgrowth of NMJs in NF1E2/E2 (Fig. 1I). In contrast, neuronal expression of UAS-CFP-NF1 suppressed NMJ overgrowth, indicating that NF1 is required in presynapses in NMJ growth control (Fig. 1I).

Presynaptic function of NF1 in NMJ growth regulation. A, B, D, E, G, H, NMJ 6/7s (A, B), NMJ 4s (D, E), and NMJ 12/13s (G, H) are shown by labeling for HRP (green) and Syn (red) in wild type (A, D, G) and NF1E2/E2 (B, E, H). Scale bars: A, D, G, 20 μm. C, F, NMJ sizes quantified as bouton numbers divided by muscle areas for NMJ 6/7s, shown as bouton number times 10−4/muscle area (μm2), which were then normalized to wild-type control 100% (C) or bouton numbers for NMJ 4s (F). Average NMJ sizes (mean ± SEM) in this and all following figures were compared by Student's t test for statistical significance, with asterisk indicating p < 0.05, and no significance (n.s.) for p > 0.05. Sample number (n) is shown within bar. I, NMJ overgrowth in NF1E2 was rescued by presynaptic expression of UAS-CFP-NF1 (elav>CFP-NF1 NF1E2/E2) but not postsynaptic expression (C57>CFP-NF1 NF1E2/E2). J, Expressions of Syn in presynapses, Brp at active zones, Futsch for microtubules, and Dlg in postsynapses are shown for wild-type control (left) and NF1E2/E2 (right) at NMJ 4s that were costained with HRP (magenta). All images are shown as single sections. Scale bar, 5 μm. K, L, Electron micrographs of cross sections through a type I bouton of muscle 6/7s of wild type (K) and NF1E2/E2 (L) larvae reared at 22°C. Squares show active zones with a T-bar, which are enlarged in lower left corner. Subsynaptic reticula (SSR), active zones (arrows), and mitochondria (Mt) are indicated. Scale bar, 0.2 μm.
The overgrown NMJs in NF1E2/E2 showed normal distribution of synaptic proteins analyzed by immunostaining. These proteins include synaptic vesicle-localized Syn, active zone-localized Brp, microtubule-associated Futsch, and postsynaptic PDZ-protein Dlg (Fig. 1J). In addition, analysis by transmission electron microscopy revealed normal morphology and size of boutons and subsynaptic reticula, and distribution of ultrastructures, such as active zones and T-bars (Fig. 1K,L, Table 1). These results suggest that NF1 functions in presynapses to confine normal NMJ growth during larval development.
Features of synaptic ultrastructures were quantified, and statistical significances (p value) between wild type and NF1E2/E2 calculated by Student's t test are shown in right column
Synaptic transmission is increased in NF1 mutants
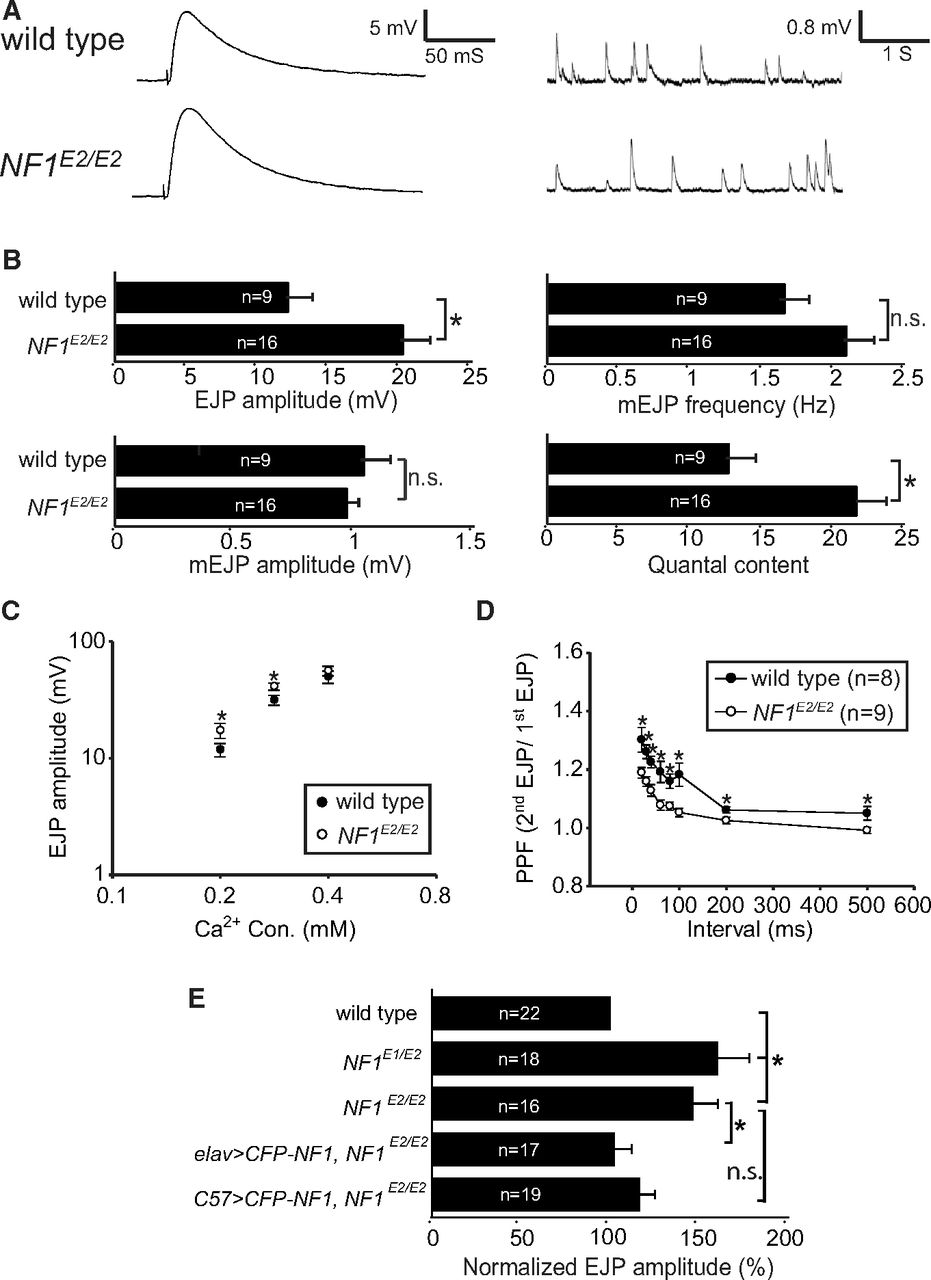
To examine whether NF1 contributes to the transmitter release, postsynaptic responses were recorded at NF1 mutant NMJs (Fig. 2A,B). When compared with the isogenic wild-type control, the amplitude of evoked junctional potential (EJP) was increased by 67% in NF1E2/E2 (wild type: 12.04 ± 1.75 mV; NF1E2/E2: 20.13 ± 1.66 mV; p = 0.003, Student's t test). To determine whether the increase in the EJP amplitude in NF1E2/E2 was caused by greater response from postsynaptic glutamate receptors or by increased release probability of presynaptic vesicles, we compared spontaneous release [or miniature EJP (mEJP)] between wild type and NF1 mutants. Whereas the amplitude of mEJP remained constant in NF1E2/E2 (wild type: 1.04 ± 0.11 mV; NF1E2/E2: 0.97 ± 0.06 mV; p = 0.592, Student's t test), a slight increase, although not statistically significant, was detected in the frequency of mEJP (wild type: 1.65 ± 0.19 Hz; NF1E2/E2: 2.08 ± 0.17 Hz; p = 0.104, Student's t test). The similarity in mEJP amplitudes suggests that the postsynaptic response was largely normal in NF1E2/E2 mutants. Instead, the quantal content, calculated by dividing EJP with mEJP, was significantly increased in NF1E2/E2 mutants (wild type: 12.61 ± 2.39; NF1E2/E2: 21.52 ± 2.02; p = 0.009, Student's t test). These changes in basal transmission properties suggest that NF1 likely regulates presynaptic release probability.

Synaptic transmission properties in NF1E2/E2. A, Electrophysiological recordings at muscle 6s of wild type and NF1E2/E2, showing a representative EJP or mEJP trace for each. Resting potentials: wild type, −75.69 ± 2.10 mV; NF1E2/E2, −73.23 ± 1.60 mV. B, Average EJP amplitude, mEJP amplitude, mEJP frequency, and quantal content in wild type and NF1E2/E2 are shown. C, EJP amplitudes in 0.2, 0.3, and 0.4 mm Ca2+ concentrations in wild type (close symbols) and NF1E2/E2 (open symbols) with at least 10 samples in each average EJP. D, Measurement of PPFs for wild type and NF1E2/E2 with interstimulus intervals between 20 and 500 ms. E, Comparison of EJPs in NF1E1/E2 and NF1E2/E2 mutants and rescue of EJPs in NF1E2/E2 by elav-GAL4 or C57-GAL4 drivers. The EJP is shown as percentages to wild type (100%). In all comparisons in this figure, significant difference is indicated by asterisk with p < 0.05 by Student's t test. Not significant (n.s.), p > 0.05. The p values are 0.016 between NF1E2 and elav>CFP-NF1 NF1E2/E2 and 0.074 between NF1E2 and elav>CFP-NF1 NF1E2/E2. Resting potentials: wild type, −73.10 ± 1.37 mV; NF1E1/E2, −72.29 ± 1.36 mV; NF1E2/E2, −73.23 ± 1.60 mV; elav>CFP-NF1 NF1E2, −69.42 ± 1.26 mV; C57>CFP-NF1 NF1E2, −69.94 ± 1.40 mV.
To further study the increase in the release probability, we measured EJPs in different Ca2+ concentrations. The EJP difference between wild type and NF1E2/E2 was largest at 0.2 mm, decreased at 0.3 mm, and diminished at 0.4 mm (Fig. 2C). Thus, NF1E2/E2 mutant EJP amplitudes are higher when the Ca2+ concentration is low. We chose 0.3 mm Ca2+ for performing PPF. At wild-type NMJ 6/7s, the second pulse was consistently larger than the first pulse at all interstimulus intervals. In NF1E2/E2 mutants, while the PPF was still detected, the second pulse was significantly reduced at all interstimulus intervals (Fig. 2D), consistent with the idea that the release probability at NF1E2/E2 NMJs is increased.
The increase in the EJP amplitude was also detected in NF1E1/E2 (Fig. 2E). To address whether NF1 functions in presynapses or postsynapses to regulate the evoked release, we performed rescue experiments in NF1E2/E2. When UAS-CFP-NF1 was driven by elav-GAL4, the EJP amplitude was restored to almost wild-type levels (102 ± 10% of wild type, p = 0.016). While postsynaptic expression also suppressed the enlarged EJP amplitude, it was not statistically significant (116 ± 8% of wild type, p = 0.074). Thus, NF1 mainly functions in presynapses to regulate synaptic transmission.
NF1 physically interacts with Fak56
Similar to NF1, Fak56 functions in presynapses to regulate NMJ growth and synaptic transmission properties (Tsai et al., 2008). We then tested the genetic interaction between Fak56 and NF1. The normal NMJ size in hypomorphic Fak56N30/KG (Fak56hypo) was enhanced by the NF1E2 allele (Fig. 3A). The strong genetic interaction prompted us to test whether Fak56 and NF1 function in the same pathway in NMJ growth regulation. Importantly, while presynaptic expression of NF1-CFP had no effect on the normal NMJ growth in the heterozygous Fak56N30/+ mutant, it fully suppressed the overgrown NMJ in the Fak56-null mutant (Fak56N30/K24, Fig. 3B), suggesting that NF1 functions downstream of or in parallel to Fak56 in NMJ growth regulation. As presynaptic overexpression of Fak56 by several neuronal GAL4 drivers caused embryonic lethality, we were unable to perform the reverse experiment of rescuing NF1E2/E2 NMJ overgrowth by Fak56. In line with the notion that Fak56 acts upstream of or in parallel to NF1, the level of Fak56 tyrosine phosphorylation, an indication for the activation of integrin signaling, remained normal at NF1E2 NMJs (Fig. 3C). Together, these results are most consistent with the notion that NF1 functions downstream of Fak56 in regulating NMJ growth.

Genetic and protein–protein interaction between Fak56 and NF1. A, B, Quantification of NMJ 6/7 size was done as for Figure 1C. A, Enhancement of Fak56hypo NMJ 6/7 growth by NF1E2 in Fak56hypo;NF1E2/+. B, Rescue of Fak56null NMJ overgrowth by elav-GAL4 expression of NF1 by comparing NMJ 6/7 sizes between elav-GAL4;Fak56null and elav>CFP-NF1;Fak56null, and elav-GAL4;Fak56N30/+ and elav>NF1-CFP;Fak56N30/+ as controls. C, pFAK signals shown in green (left) or white (right) are indistinguishable between wild-type and NF1E2/E2 NMJs costained with HRP in magenta. Scale bar, 5 μm. D, Coimmunoprecipitation of Fak56 and NF1. NF1 immunoprecipitates from embryo extracts were blotted by NF1 and Fak56 antibodies (left). Input control, 2% of wild-type extracts. As control for antibody specificity, wild type, NF1P2/P2, and Fak56null were blotted by Fak56, NF1, or tubulin (Tub) antibodies (right). E–G, Phosphorylation-independent complex formation between Fak56 and NF1. E, S2 cells transfected with Myc-Fak56 and Flag-NF1 were immunoprecipitated by Myc antibody and treated with or without the alkaline calf intestine phosphatase (CIP). Top, Detection of NF1 levels showed no difference between wild type and NF1E2/E2 mutant. F, G, S2 cells transfected with Myc, Myc-Fak56, or Myc-Fak56Y430F with Flag-NF1 (F, G) or Flag (F) were immunoprecipitated by Myc antibodies, and immunoblotted by Myc or Flag antibodies to reveal NF1 and Fak56 levels (F, top), or immunoprecipitated by Flag antibody, and immunoblotted with pY or Flag antibodies (G, top two panels). As control, S2 cell extracts were blotted by Flag, Myc, or Tub antibodies without immunoprecipitation. H, S2 cells transfected with Flag-NF1 and Myc-Fak56 and treated with 6 mm ionomycin for 0, 1, and 5 min. The Myc immunoprecipitates were blotted with Flag, pY, and Myc antibodies (top). Total cell extracts without immunoprecipitation were also blotted with Flag, Myc, and Tub antibodies (bottom).
We next examined whether NF1 physically interacts with Fak56. In NF1 immunoprecipitates from embryonic extracts, Fak56 was detected by anti-FAK antibodies in Western blot analysis. As controls, NF1-immunoprecipitates from Fak56 or NF1 mutant extracts failed to reveal any Fak56 signal (Fig. 3D, right). In addition, Flag-tagged NF1 and Myc-tagged Fak56 proteins in S2 cells also existed in the same immunocomplex (Fig. 3E–H). Thus, NF1 and Fak56 either interact with each other directly or exist within the same protein complex.
Tyrosine phosphorylation of FAK mediates FAK interaction with diverse proteins for signaling activities (Mitra et al., 2005). However, the Flag-NF1/Myc-Fak56 complex was not dissociated upon the treatment with calf intestine phosphatases (Fig. 3E, lane 4). Furthermore, Myc-Fak56Y430F with mutation in the autophosphorylation site associated with Flag-NF1 as efficiently as Myc-Fak56wt (Fig. 3F). Furthermore, overexpression of Myc-Fak56wt or Myc-Fak56Y430F failed to alter the level of tyrosine phosphorylation on NF1 (Fig. 3G). These results suggest that the Fak56-NF1 interaction is independent of Fak56 tyrosine phosphorylation activity.
We next tested whether Ca2+ influx regulates the interaction between Fak56 and NF1. Elevation of the Ca2+ influx by treatment of S2 cells with ionomycin diminished the interaction between NF1 and Fak56 (Fig. 3H), suggesting that the interaction between NF1 and Fak56 is Ca2+-sensitive. Consistent with a previous report (Ueda et al., 2008), the level of tyrosine-phosphorylated Fak56 was also reduced in response to ionomycin (Fig. 3H). Because the interaction between NF1 and Fak56 was independent of tyrosine phosphorylation, intracellular Ca2+ concentration likely affects the Fak56-NF1 association and Fak56 tyrosine phosphorylation independently.
The N-terminal A region of NF1 is required for Fak56 interaction and presynaptic localization
The Fak56–NF1 interaction may function to recruit or stabilize NF1 to the presynaptic sites. To test this, we first defined the interacting region of NF1 by the GST pull-down assay. Seven GST fusion proteins (NF1-A–NF1-G) with overlapping NF1 sequence were generated to test interaction with Fak56. Only NF1-A (amino acids 1–408) was able to pull down Fak56 (Fig. 4A). Further deletion analysis within the NF1-A region failed to reveal smaller Fak56-interacting regions, suggesting that the intact A region or >1 motifs within NF1-A are required for Fak56 interaction.
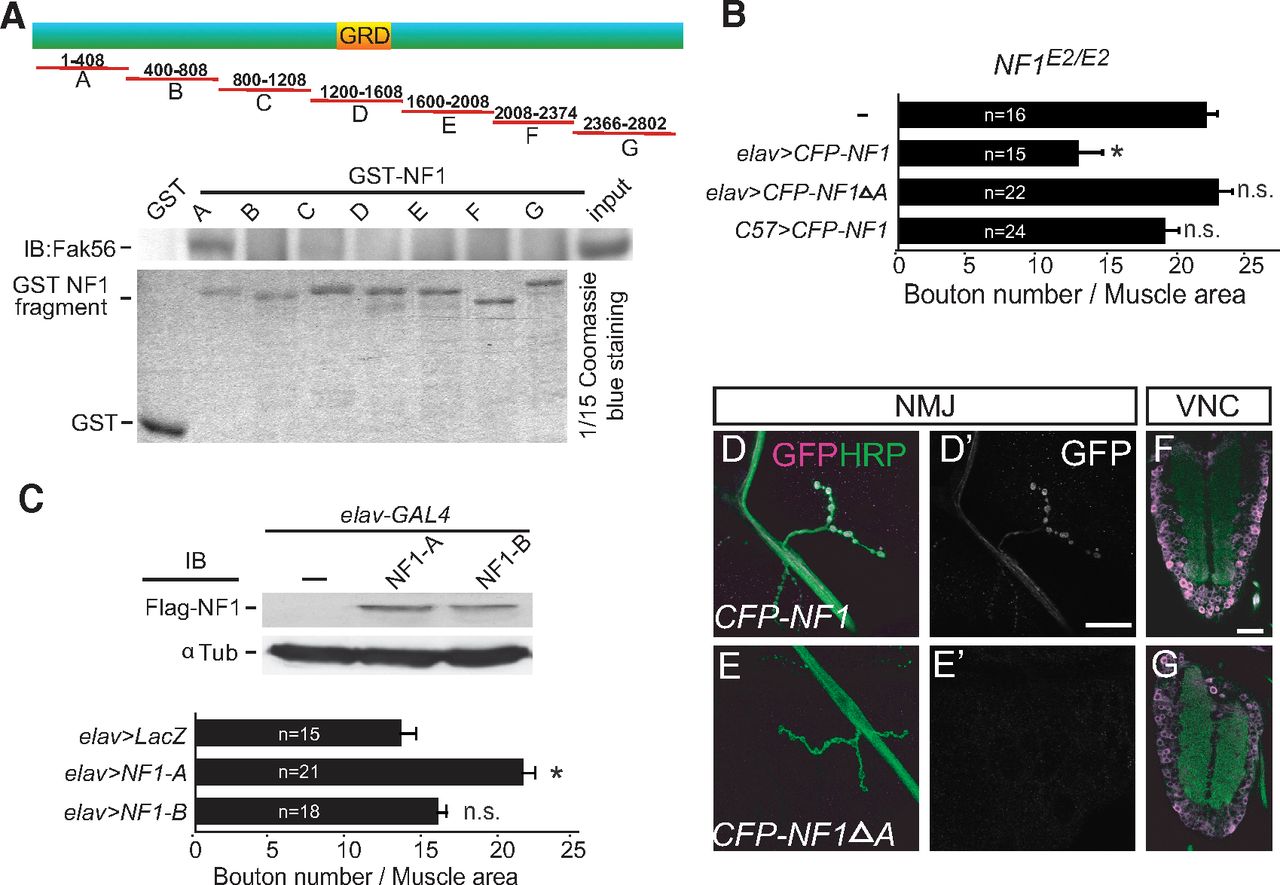
The N-terminal A region mediates Fak56 interaction and NF1 synaptic localization. A, The A region of NF1 precipitates with Fak56. NF1 was divided into seven fragments to generate GST fusions (GST-NF1A to NF1G, top diagram). The GRD and the size of each fragment are indicated. GST-NF1 fusions were purified (bottom) for pull-down assays in S2 cell extracts, and the precipitates were blotted with Fak56 antibodies (middle). B, The A region is required for NF1 activity in regulating NMJ size in NF1E2/E2. Quantification of NMJ 6/7 size, as done in Figure 1C, showing neuronal expression of NF1 (NF1E2/E2; elav>CFP-NF1) or A region-truncated NF1 (NF1E2/E2; elav>CFP-NF1ΔA), or muscle expression of NF1 (NF1E2/E2; C57>CFP-NF1). C, Dominant-negative effect of the A region in causing NMJ overgrowth. Quantification of NMJ 6/7 size in elav>LacZ, elav>NF1-A, and elav>NF1-B (bottom). Expression levels of Flag-NF1-A and Flag-NF1-B by elav-GAL4 in adult brain are shown in Flag Western blot (top). The elav>LacZ serves as control. *p < 0.05 by Student's t test. D–G, NF1 expressions detected by the GFP antibody (D', E', magenta or white) at axons and NMJs of elav>CFP-NF1 (D) but not elav>CFP-NF1ΔA (E) costained with HRP (green). Both were detected in ventral nerve cords (F, G). Scale bars: D', 20 μm; F, 50 μm.
We further tested whether the A region is required for NF1 to regulate NMJ growth. While the full-length NF1 (CFP-NF1) transgene restored NMJ growth in NF1E2/E2 mutants, the A region-truncated NF1 (CFP-NF1ΔA) failed to suppress NMJ overgrowth (Fig. 4B). If the A region mediates a protein–protein interaction between Fak56 and NF1, overexpression of this region could disrupt the interaction and induce dominant-negative effects. Indeed, when overexpressed in presynapses, the NF1-A region significantly enhanced NMJ growth (Fig. 4C, bottom). As a control, overexpression of the adjacent NF1-B region (Fig. 4A, amino acids 400–808) had no effect on NMJ growth (Fig. 4C), although both proteins were expressed at equivalent levels (Fig. 4C, top). These results support the notion that the NF1-A region, when overexpressed, blocks the Fak56 and NF1 interaction crucial for NMJ growth regulation.
We found that the NF1 fusion protein CFP-NF1 was localized at NMJs when expressed by neuronal elav-GAL4 (Fig. 4D). Interestingly, the A region-deleted NF1, CFP-NF1ΔA, failed to reveal NMJ localization (Fig. 4E). In addition, only CFP-NF1 signal was detected in axonal tracts. However, both proteins were expressed at similar levels as they were detected in the soma of ventral nerve cords (Fig. 4F,G). These results suggest that the A region is required for NF1 presynaptic localization.
NMJ growth regulation is independent of the GAP activity of NF1
The NF1 activity is known to modulate two signaling pathways, suppression of the Ras pathway and activation of the cAMP pathway (Shilyansky et al., 2010). We first investigated whether the RasGAP activity of NF1 that downregulates the Ras pathway contributes to NMJ growth regulation. NF1 transgenes with mutations in the GRD were tested for their ability to rescue NMJ overgrowth in NF1E2/E2 mutants. The arginine to proline (RP) mutation (hs-NF1 RP) specifically affects RasGAP activity while the lysine to alanine (KA) mutation (hs-NF1 KA) affects the electrostatic interaction with Ras (Walker et al., 2006). Interestingly, both transgenes, as well as the one with complete removal of GRD (hs-NF1 ΔGRD), rescued the NMJ overgrowth defect in NF1E2/E2, and their rescuing activities were comparable to wild-type hs-NF1 (Fig. 5A). In contrast, transgenes with defect in GAP activity (RP and ΔGRD) did not rescue the body size of NF1E2/E2 pupae (Fig. 5B), which was also reported (Walker et al., 2006). Thus, the GAP activity of NF1 is not essential in NMJ growth regulation.

Suppression of NMJ growth by NF1 is independent of RasGAP activity. A, B, Quantification of the NMJ 6/7 size (A), as done in Figure 1C, and the length of pupal cases (B), measured at 48 h after puparium formation, are compared between wild type, and NF1E2/E2, NF1E2/E2 hs-NF1 wt, NF1E2/E2 hs-NF1 KA, NF1E2/E2 hs-NF1 RP, and NF1E2/E2 hs-NF1 ΔGRD. All transgenics were treated identically for 30 min at 37°C each day before assay. *p < 0.05 by Student's t test compared with wild type. C, D, Expression of Fas2 (C, D, green; C', D', white) at NMJ 4s was shown in wild type (C) and not affected in NF1E2/E2 (D) with costained HRP in magenta. E, Quantification of Fas2 levels at NMJ 4s shows no difference between wild type and NF1E2/E2. Fas2 intensity within boutons (outlined by costained HRP) was normalized to HRP intensity quantified from the same area.
To test whether NF1 regulates Ras signaling at NMJs, we examined the protein level of the cell adhesion molecule Fas2. The gene dosage of fas2 modulates the NMJ size and the Fas2 protein level at NMJs is a sensitive readout of Ras signaling (Koh et al., 2002). We found that the Fas2 levels were indistinguishable between NF1E2/E2 and wild-type control (Fig. 5C–E). Together, these results suggest that NF1 functions independently of the Ras pathway in regulating NMJ growth.
NMJ overgrowth in NF1 and Fak56 mutants is suppressed by elevating cAMP levels
NF1 functions as a regulator of ACs to modulate cellular cAMP levels (Guo et al., 1997, 2000). To test the involvement of cAMP in NF1-regulated NMJ growth, larvae were fed with food supplemented with db-cAMP or the AC activator forskolin (Tong et al., 2007). We found that the NMJ size in wild-type larvae remained the same when fed with either chemical (Fig. 6A). However, NMJ overgrowth in NF1E2/E2 mutants was suppressed in both feeding conditions, compared with mock controls (Fig. 6A). Thus, cAMP is likely insufficient to suppress NMJ growth in NF1 mutants.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The cAMP pathway downstream of Fak56/NF1 in NMJ growth suppression. Quantification of NMJ 6/7 size was done as for Figure 1C. A, Higher cAMP levels suppress NMJ overgrowth in Fak56null and NF1E2/E2. Larvae were reared in food supplemented with 10 mm db-cAMP (left) or 10 mm forskolin (right) and mock controls (black bars) are water for db-cAMP and DMSO for forskolin. *p < 0.05 by Student's t test. B, rut enhances Fak56hypo NMJ growth phenotype by comparing NMJ 6/7 size of Fak56hypo, rut1/+;Fak56hypo, rut1/+, Fak56null, rut1/Y;Fak56null, and rut1/Y. C–E, Quantification of NMJ sizes in the depletion of rut by two RNAi lines (II, III) driven by da-GAL4 (C), elav-GAL4 (D), and C57-GAL4 (E). Significant difference (*p < 0.05 by Student's t test) was compared with LacZ control. F, dnc suppresses NMJ phenotype in neuronal expression of Fak56RNAi (elav>Fak56RNAi) by comparing with or without carrying the dncM14 allele (elav>Fak56RNAi; dncM14/+). G, Suppression of NMJ 6/7 overgrowth in neuronal expression of the A region (elav>NF1-A) by 10 mm db-cAMP (top) or 10 mm forskolin (bottom) compared with respective mock controls (as done for Fig. 6A). H, Schematic drawing of presynaptic integrin signaling mediated by the AC/cAMP and Ras/MAPK pathways. See Discussion for details.
Similar to NF1E2/E2, feeding Fak56null larvae with db-cAMP or forskolin significantly suppressed NMJ overgrowth compared with the respective mock control (Fig. 6A). To genetically link the cAMP pathway to Fak56 signaling in NMJ growth control, rutabaga (rut) encoding the cAMP-synthesizing AC was tested for genetic interaction with Fak56. We found that the NMJ size in hypomorphic Fak56hypo was enhanced by the rut1-null allele (Fig. 6B), suggesting that rut is also involved in suppressing NMJ growth. In addition, the rut1;Fak56null double mutant displayed the same NMJ size as the Fak56null single mutant, consistent with the idea that they function in the same pathway. Although the rut1 mutant shows normal NMJ growth, this might be explained by different requirements in presynaptic and postsynaptic compartments (Zhong and Wu, 1991; Zhong et al., 1992; Cheung et al., 1999). To test this, we used two independent rut-RNAi lines to deplete rut expression(Pan et al., 2009). Consistent with rut mutant phenotypes, depleting rut expression ubiquitously by da-GAL4 showed no alteration of NMJ size (Fig. 6C). However, depleting rut expression in presynaptic neurons by elav-GAL4 induced NMJ overgrowth, and depleting rut expression in muscles caused NMJ size reduction (Fig. 6D,E). We also tested the role of dnc encoding the cAMP-hydrolyzing phosphodiesterase in NMJ growth regulation. The NMJ overgrowth induced by presynaptic Fak56-RNAi knockdown (Tsai et al., 2008) was suppressed by the null dncM14 allele (Fig. 6F). In summary, Fak56 functions in presynapses to maintain higher levels of cAMP, which is necessary to suppress NMJ growth.
Finally, we tested whether the interaction between Fak56 and NF1 is required in maintaining higher cAMP levels to suppress NMJ growth. The NMJ overgrowth caused by NF1-A overexpression was suppressed by the addition of either db-cAMP or forskolin to larval food (Fig. 6G), similar to the suppression effect on Fak56 and NF1 mutants. Thus, the dominant-negative effect of NF1-A, presumably disrupting the interaction between Fak56 and NF1, could be suppressed by elevating the cAMP levels during NMJ growth regulation.
Discussion
We studied NF1 mutant phenotypes at Drosophila larval NMJs and found that both Fak56 and NF1 showed very similar phenotypes. We suggest that NF1 mediates Fak56 signaling activity through a protein–protein interaction with Fak56. The interaction with Fak56 is mediated through the N-terminal 400 aa region of NF1 that is important for NF1 function and localization. The NF1 activity in NMJ growth regulation is independent of its GAP activity but mediated through the cAMP pathway. Together with our other results (Tsai et al., 2008, 2012), these findings indicate that presynaptic Fak56 mediates integrin signaling to transduce through the NF1/AC/cAMP pathway (Fig. 6H). Our study reveals the role of NF1 in synapse growth modulation and its relationship with integrin/FAK signaling, which might contribute to the understanding of pathogenesis in the NF1 genetic disorder.
Integrins promote maturation of central hippocampal synapses and peripheral NMJs, mainly through the postsynaptic activity of integrin signaling (Chavis and Westbrook, 2001; Beumer et al., 2002; Schwander et al., 2004; Cingolani and Goda, 2008). The nonconventional integrin/FAK/NF1 signaling pathway identified in this study functions in the presynaptic compartment, as shown by neuronal but not muscular expression of βν (Tsai et al., 2012), Fak56 (Tsai et al., 2008), and NF1 (Fig. 1I) in rescuing NMJ growth phenotypes in respective mutants. In addition to NMJ overgrowth, presynaptic release probabilities at Fak56 and NF1 mutant NMJs were increased, displaying enlarged EJPs and quantal contents, likely reflecting their enlarged NMJ sizes. The link between Fak56 and NF1 is mediated through the N-terminal A region of NF1 of ∼400 aa. Overexpression of the A region mimicked NF1 and Fak56 loss-of-function mutants in inducing NMJ overgrowth suppressed by cAMP elevation. In addition, the A region is important for NF1 function in NMJ growth regulation and for NF1 localization at NMJs (Fig. 4). Previous studies define several functional domains of NF1, including the Ras GRD (Xu et al., 1990), the Leucine-rich domain (Wang et al., 2011), and the C-terminal domain (Patrakitkomjorn et al., 2008), in recruiting interacting partners, such as Ras (Xu et al., 1990), AC, syndecans (Lin et al., 2007), valosin-containing protein (Wang et al., 2011), and collapsin response mediator protein 2/4 (Lin and Hsueh, 2008). The identification of the A region in the interaction with Fak56 extends our understanding to the complex nature of NF1 regulation and activity.
Modulations of two signaling activities by NF1, the cAMP pathway and the Ras pathway, have been well studied. We showed that the GAP activity of NF1 that causes the downregulation of Ras signaling is not needed in NMJ growth regulation. Instead, the GAP activity participates in body size control and circadian rhythm in Drosophila (Williams et al., 2001; Walker et al., 2006). On the other hand, the cAMP regulation of NF1 is important in learning and memory (Guo et al., 2000) and life-span control (Tong et al., 2007), in addition to NMJ growth regulation. Together, our genetic analysis and feeding of cAMP analogs suggest that Fak56/NF1 signaling maintains sufficient cAMP activity in suppressing NMJ growth. The cAMP activity through activation of PKA regulates local reorganization of actin cytoskeleton (Lin et al., 2007). It could also function through the transcription factor CREB to regulate gene expression (Davis et al., 1996; Yukawa et al., 1999). Our results suggest that a threshold of cAMP levels is maintained by the Fak56/NF1 in presynapses to confine NMJ growth. The different requirements of the cAMP pathway in presynapses and postsynapses in regulating NMJ size may indicate that presynaptic and postsynaptic interaction leads to the normal size of NMJs in ubiquitous knockdown (Fig. 6C–E).
Fak56-mediated presynaptic integrin signaling that regulates NMJ growth is mediated through both AC/cAMP activation and Ras/MAPK inhibition (Tsai et al., 2008). NF1 is known to regulate both pathways through distinct domains (Guo et al., 1997; The et al., 1997; Hannan et al., 2006). Whereas the Ras-GAP activity of NF1 is necessary to regulate pupal size (Walker et al., 2006), this activity is dispensable in regulating NMJ growth. Furthermore, NF1 does not affect MAPK activation at NMJs, as we have detected normal phospho-ERK (data not shown) and Fas2 levels at NMJs in NF1 mutants (Fig. 5C–E). How does Fak56 inhibit ERK activity at NMJs? In addition to NF1, we found that the RasGAP protein vacuolar peduncle (Vap) (Botella et al., 2003) regulates NMJ growth and genetically interacts with Fak56 in NMJ growth regulation (Wang et al., 2011; data not shown). Therefore, Fak56-mediated integrin signaling may function through both NF1 and Vap to modulate AC/cAMP and Ras/MAPK pathways, respectively, leading to the regulation of NMJ growth.
Loss of neurofibromin is the cause of the common human disorder NF1. Children with NF1, in addition to a predisposition to benign and malignant tumors, also suffer from learning and behavioral disadvantages (North et al., 1994; Cichowski and Jacks, 2001). We have shown that the N-terminal A region of NF1 is required for NF1 to interact with Fak56 at synapses. At least 18 missense mutations within the 400 aa of the A region have been identified within the corresponding region of human NF1 (according to Human Gene Mutation Database), raising the possibility that disregulation of the FAK–NF1 interaction and hence presynaptic integrin activity, if conserved in humans, may contribute to NF1 pathogenesis.
Footnotes
- Received April 11, 2012.
- Revision received August 20, 2012.
- Accepted September 17, 2012.
This work was supported by grants from Academia Sinica and the National Science Council to C.-T.C. We thank N.H. Brown, R. Palmer, Y. Zhong, the Developmental Studies Hybridoma Bank, the Drosophila Genomics Resource Center, the Bloomington Drosophila Stock Center, and the Vienna Drosophila RNAi Center for fly stocks and antibodies. We are grateful to S.-P. Lee and S.-P. Tsai at the Institute of Molecular Biology Image Facility for assistance with confocal and electron microscopy. We also thank laboratory members in the laboratories of Drs. Chien and Chen for suggestions.
The authors declare no competing financial interests.
- Correspondence should be addressed to Cheng-Ting Chien, Institute of Molecular Biology, Academia Sinica, 128 Academia Road, Section 2, Nankang, Taipei 115, Taiwan. ctchien{at}gate.sinica.edu.tw
- Copyright © 2012 the authors 0270-6474/12/3216971-11$15.00/0