
Abstract
There is compelling evidence that oligodendrocyte apoptosis, in response to CNS inflammation, contributes significantly to the development of the demyelinating disorder multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis (EAE). Therefore, approaches designed to protect oligodendrocytes would likely have therapeutic value. Activation of pancreatic endoplasmic reticulum kinase (PERK) signaling in response to endoplasmic reticulum (ER) stress increases cell survival under various cytotoxic conditions. Moreover, there is evidence that PERK signaling is activated in oligodendrocytes within demyelinating lesions in multiple sclerosis and EAE. Our previous study demonstrated that CNS delivery of the inflammatory cytokine interferon-γ before EAE onset protected mice against EAE, and this protection was dependent on PERK signaling. In our current study, we sought to elucidate the role of PERK signaling in oligodendrocytes during EAE. We generated transgenic mice that allow for temporally controlled activation of PERK signaling, in the absence of ER stress, specifically in oligodendrocytes. We demonstrated that persistent activation of PERK signaling was not deleterious to oligodendrocyte viability or the myelin of adult animals. Importantly, we found that enhanced activation of PERK signaling specifically in oligodendrocytes significantly attenuated EAE disease severity, which was associated with reduced oligodendrocyte apoptosis, demyelination, and axonal degeneration. This effect was not the result of an altered degree of the inflammatory response in EAE mice. Our results provide direct evidence that activation of PERK signaling in oligodendrocytes is cytoprotective, protecting mice against EAE.
Introduction
Multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), are T cell-mediated autoimmune demyelinating diseases of the CNS. These disorders are characterized by CNS inflammation, demyelination, oligodendrocyte death, and axonal degeneration (Frohman et al., 2006; Trapp and Nave, 2008). Recent studies suggest that oligodendrocytes are the target of immune attack in MS and EAE and that oligodendrocyte apoptosis contributes significantly to the development of these diseases (Matute and Pérez-Cerdá, 2005; McGuire et al., 2010). Profound oligodendrocyte apoptosis has been observed in newly forming MS lesions in the absence of infiltrating lymphocytes or myelin phagocytes (Barnett and Prineas, 2004). Both oligodendrocyte-targeted expression of antiapoptotic proteins and oligodendrocyte-targeted deletion of a proapoptotic protein protect oligodendrocytes from EAE-induced apoptosis, resulting in the attenuation of demyelination and axonal degeneration in EAE lesions (Hisahara et al., 2000; McGuire et al., 2010).
Endoplasmic reticulum (ER) stress, the stress of accumulating unfolded or misfolded proteins in the ER, activates pancreatic ER kinase (PERK), which coordinates an adaptive program known as the integrated stress response (ISR) by phosphorylating translation initiation factor 2α (eIF2α) (Harding et al., 2003; Proud, 2005; Marciniak and Ron, 2006). Phosphorylation of eIF2α promotes a stress-resistant state through the global attenuation of protein biosynthesis and the induction of stress-induced cytoprotective genes. Accumulating evidence suggests that the ISR is involved in the pathogenesis of MS and EAE (Lin and Popko, 2009). Activation of the ISR was observed in multiple cell types within MS and EAE demyelinating lesions, including oligodendrocytes, T cells, microglia/macrophages, and astrocytes (Chakrabarty et al., 2004; Mháille et al., 2008; Cunnea et al., 2011). Additionally, it has been shown that the ISR is a potent regulator of inflammatory T-cell differentiation (Scheu et al., 2006; Sundrud et al., 2009) and that it also regulates the function of macrophages in inflammatory diseases (Woo et al., 2009). Our previous study demonstrated that CNS delivery of the inflammatory cytokine IFN-γ before EAE onset protected against EAE-induced oligodendrocyte death and demyelination in a PERK-dependent manner, as the protective effects of IFN-γ were associated with PERK activation in oligodendrocytes and were abrogated in PERK haploinsufficient animals (Lin et al., 2007). Nevertheless, because IFN-γ is a pleiotropic immune cytokine that regulates T-cell differentiation and microglia/macrophage activation (Boehm et al., 1997), it is not clear whether abrogation of the protective effects of IFN-γ in PERK haploinsufficient animals is the result of the impairment of ISR signaling in oligodendrocytes or in inflammatory cells. Collectively, the role of the ISR in oligodendrocytes during MS and EAE remains unclear.
In the current study, we sought to dissect the precise role of the ISR in oligodendrocytes during EAE pathogenesis. We generated transgenic mice that allow for temporally controlled activation of PERK signaling specifically in oligodendrocytes. Using this unique mouse model, we found that enhanced ISR signaling specifically in oligodendrocytes significantly alleviates EAE-induced oligodendrocyte apoptosis, resulting in the attenuation of demyelination and axonal degeneration. Our findings indicate that therapeutic approaches to activating the ISR in oligodendrocytes could be beneficial to patients with MS.
Materials and Methods
Generation of PLP/Fv2E-PERK transgenic mice.
A proteolipid protein (PLP)-based expression construct containing 2.4 kb of the 5′ flanking DNA, exon 1, and intron 1 of the mouse PLP gene, and a SV40 poly(A) signal sequence (Fuss et al., 2000, 2001) was obtained from Dr. Wendy Macklin (University of Colorado Denver, Aurora, Colorado). A 2.4 kb Fv2E-PERK cDNA segment was excised from the pBabe/Fv2E-PERK plasmid (Lu et al., 2004) with EcoRI and NsiI and was inserted into the SmaI site of a modified version of the pNEB193 plasmid (Fuss et al., 2000). The resulting pNEB193/Fv2E-PERK plasmid was further digested with AscI and PacI to release the Fv2E-PERK cDNA, which was subcloned into the multiple cloning sites region of the PLP expression construct at the same restriction sites. The pPLP/Fv2E-PERK plasmid was digested with ApaI and SacII and a linear 13.5 kb PLP/Fv2E-PERK transgene fragment was isolated for microinjection into fertilized C57BL/6J oocytes. Transgenic founder mice were identified by PCR. Tail tip DNA was amplified with the following primers: sense primer (CCAGATGAGTGTGGGTCAGA) and antisense primer (AGGCTTTAACTTCCCGCATT). The identified founders were subsequently bred with C57BL/6J mice (The Jackson Laboratory) to establish transgenic mouse lines.
To activate the transgene Fv2E-PERK in oligodendrocytes, PLP/Fv2E-PERK transgenic mice were given intraperitoneal injections of AP20187 daily at a dose of 0.5, 2, or 5 mg/kg. AP20187 was a gift from Ariad Pharmaceuticals. Lyophilized AP20187 was dissolved in 100% ethanol at a concentration of 62.5 mg/ml stock solution and stored at −20°C. Injection solutions, as recommended by Ariad Pharmaceuticals, consisted of 4% ethanol, 10% PEG-400, and 2% Tween 20 in water. The transgenic mice receiving only the vehicle (4% ethanol, 10% PEG-400, 2% Tween 20 in water) served as controls. All animal procedures were conducted in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of the University of South Alabama, the University of Chicago, and the University of Minnesota.
Mixed glial cultures and oligodendrocyte cultures.
Mixed glial cultures were prepared from neonatal PLP/Fv2E-PERK mice as described previously (Baerwald et al., 2000). The cells were cultured in DMEM (Invitrogen) with 10% FBS at 37°C with 5% CO2. After 12 d, the cells were switched to a differentiation medium containing 5 μl/ml insulin, 50 μg/ml transferrin, 30 nm selenium, 10 nm biotin, 10 nm progesterone, and 15 nm triiodothyronine, and 0.1% BSA. After 5 d of differentiation, the cells were treated with AP20187 or vehicle for 24 h. RNA was isolated from the cells using Trizol reagent (Invitrogen).
Oligodendrocyte progenitor cells (OPCs) were isolated from PLP/Fv2E-PERK and wild-type littermate mice using the sequential immunopanning protocol described by Dugas et al. (2006). Briefly, whole brains from 6-day-old pups were dissected and enzymatically dissociated with papain at 37°C. After enzymatic dissociation and gentle trituration, the single-cell suspension was sequentially incubated on dishes coated with antibodies against Ran-2, GalC, and O4. Ran-2−, GalC−, O4+ OPCs were trypsinized from the O4 panning plate and seeded onto poly-d-lysine coated 12 mm glass coverslips. Cells were cultured in DMEM, supplemented with human transferrin (100 μg/ml), BSA (100 μg/ml), putrescine (16 μg/ml), progesterone (60 ng/ml), sodium selenite (40 ng/ml), N-acetyl-l-cysteine (5 μg/ml), d- biotin (10 ng/ml), insulin (5 μg/ml), glutamine (2 mm), sodium pyruvate (1 mm), penicillin-streptomycin (100 U each), B-27 (1 ×; Invitrogen), Trace Elements (1 ×; Mediatech), growth factors for differentiation: forskolin (4.2 μg/ml), CNTF (10 ng/ml, PeproTech), NT-3 (1 ng/ml, PeproTech), and triiodothyronine (40 ng/ml). After 3 d of differentiation, the cells were treated with 1 nm AP20187 or vehicle for 4 h and were then fixed with cold 4% paraformaldehyde in PBS. The cells were blocked with PBS containing 10% goat serum and 0.1% Triton X-100 and incubated overnight with the primary antibody diluted in blocking solution. Appropriate fluorochrome-labeled secondary antibodies (Vector Laboratories) were used for detection. Immunostaining for phosphorylated eIF2α (p-eIF2α, 1:50; Cell Signaling Technology), 2′,3′-cyclic nucleotide phosphodiesterase (1:200; Santa Cruz Biotechnology), PLP (1:50; Santa Cruz Biotechnology), and the active form of NF-κB, p65 (1:50; Millipore) were performed. Fluorescent-stained cells were mounted with Vectashield mounting medium with 4′, 6-diamidino-2-phenylindole (DAPI, Vector Laboratories). The stained cells were visualized with the Nikon A1 confocal microscope and analyzed as previously described (Lin et al., 2005).
EAE immunization.
Seven-week-old female mice received subcutaneous injections, in the flank and at the tail base, of 200 μg of myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide emulsified in complete Freund's adjuvant (BD Biosciences), supplemented with 600 μg of Mycobacterium tuberculosis (strain H37Ra; BD Biosciences). Two intraperitoneal injections of 400 ng of pertussis toxin (List Biological Laboratories) were given 24 and 72 h later. Clinical scores (0 = healthy; 1 = flaccid tail; 2 = ataxia and/or paresis of hindlimbs; 3 = paralysis of hindlimbs and/or paresis of forelimbs; 4 = tetraparalysis; 5 = moribund or death) were recorded daily.
Western blot analysis.
Tissues harvested from mice were rinsed in ice-cold PBS and were homogenized using a motorized homogenizer as previously described (Lin et al., 2005, 2008). After incubating on ice for 15 min, the extracts were cleared by centrifugation at 14,000 rpm twice for 30 min each. The protein content of each extract was determined by DC Protein Assay (Bio-Rad Laboratories). The extracts (100 μg) were separated by SDS-PAGE and were transferred to nitrocellulose. The blots were incubated with a primary antibody, either anti-FKBP-12 (1:2000, Thermo Scientific) or anti-β-actin (1:10,000, Sigma-Aldrich), followed by an HRP-conjugated secondary antibody, and the signal was revealed by chemiluminescence.
Real-time PCR.
Deeply anesthetized mice were perfused with ice-cold PBS. RNA was isolated from the spinal cord using Trizol reagent (Invitrogen) and treated with DNaseI (Invitrogen) to eliminate genomic DNA. Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). TaqMan real-time PCR was performed with iQ Supermix (Bio-Rad Laboratories) on a Bio-Rad iQ Real-Time PCR detection system as previously described (Lin et al., 2006, 2007; Lin and Lin, 2010).
Immunohistochemistry and TUNEL staining.
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in PBS. The tissues were removed, postfixed with paraformaldehyde, cryopreserved in 30% sucrose, embedded in optimal cutting temperature compound, and frozen on dry ice. Frozen sections were cut in a cryostat at a thickness of 10 μm. For immunohistochemistry, the sections were treated with −20°C acetone, blocked with PBS containing 10% goat serum and 0.1% Triton X-100, and incubated overnight with the primary antibody diluted in blocking solution. Fluorescein, Cy3, or enzyme-labeled secondary antibodies (Vector Laboratories) were used for detection. Immunohistochemistry for CC1 (APC7, 1:50; EMD Biosciences), FKBP-12 (1:200; Thermo Scientific), GFAP (1:1000; Sternberger Monoclonals), NeuN (1:100; Millipore Bioscience Research Reagents), platelet-derived growth factor α receptor (PDGFαR, 1:100, Santa Cruz Biotechnology), p-eIF2α (1:50; Cell Signaling Technology), myelin basic protein (MBP, 1:1000; Sternberger Monoclonals), SMI-31 (1:1000; Sternberger Monoclonals), CD3 (1:50; Santa Cruz Biotechnology), CD11b (1:50; Santa Cruz Biotechnology), aspartoacylase (ASPA, 1:1000, kindly provided by Dr. M.A. Aryan Namboodiri at Uniformed Services University of the Health Sciences, Bethesda, Maryland), and the active form of NF-κB p65 (1:50; Millipore) were performed. Fluorescent-stained sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories) and visualized with the Nikon A1 confocal microscope.
TUNEL staining was performed using the ApopTag kit (Millipore Bioscience Research Reagents) according to the manufacturer's instructions. The sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories) and visualized with the Nikon A1 confocal microscope.
For quantitative analyses, cross spinal cord sections were cut at 10 μm. Every 10th section in the series from lumbar 2 to lumbar 4 spinal cord segment, a total of 20 sections per mouse, was immunostained with appropriate antibodies. We counted immunopositive cells within the anterior funiculus medially next to the anterior median fissure in the lumbar spinal cord in every section, confined to an area of 0.1 mm2 as described in our previous paper (Lin et al., 2007). For each mouse, the number of immunopositive cells from a total of 20 sections was averaged, and the average number per section was used for statistical analysis. For quantitative MBP immunohistochemistry analysis, we calculated the percentage of the demyelinated area in the lumbar spinal cord in every section by normalizing the demyelinated area against the total white matter area. The total white matter area and the demyelinated area in the lumbar spinal cord in every section were measured by NIH ImageJ software (http://rsb.info.nih.gov/ij/). For each mouse, the average percentage of the demyelinated area per section was calculated and used for statistical analysis. The histological analyses were performed by a researcher blind to the experimental groups.
Toluidine blue staining and electron microscopy analysis.
For toluidine blue staining of 1 μm sections, we anesthetized and perfused mice with PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde. The lumbar spinal cords were processed, embedded, sectioned, and analyzed as previously described (Lin et al., 2007). The total white matter area and the demyelinated area in the lumbar spinal cord were measured by NIH ImageJ software (http://rsb.info.nih.gov/ij/). The percentage of the demyelinated area was calculated by normalizing the demyelinated area against the total white matter area. For electron microscopy (EM) analysis, the demyelinated lesions within the anterior funiculus medially next to the anterior median fissure were trimmed, and then thin sections were cut, stained with uranyl acetate and lead citrate, and examined using Philips CM 100 transmission electron microscope. We counted the total number of axons and the number of degenerating axons in the demyelinated lesions, confined to an area of 100 μm2. The histological analyses were performed by a researcher blind to the experimental groups.
BrdU pulse-chase analysis.
PLP/Fv2E-PERK mice immunized with MOG 35–55 peptide were given intraperitoneal injections of 0.5 mg/kg AP20187 or vehicle daily starting on PID10. All mice received additional injections of 50 mg/kg BrdU (Sigma-Aldrich) dissolved in H2O on PID11, PID13, PID15, and PID17. Mice were then anesthetized and transcardically perfused, with lumbar spinal cord isolated and fixed as described above. To colabel with ASPA and BrdU, immunofluorescent staining was conducted in two cycles: sections were first treated as described above using ASPA (1:1000) with positive staining confirmed after fluorescent secondary antibody labeling. Slides were then reintroduced to −20°C acetone for 10 min, washed with PBS, incubated with 2N HCl for 30 min at 37°C to denature DNA, and neutralized with 0.1 m sodium borate for 10 min at room temperature. Sections were then blocked and incubated with antibody to BrdU (1:250, Abcam), secondary antibody, and finally dyed with Hoechst 33258 (Invitrogen) before mounting and coverslipping. Tissue images were visualized with a CRi Pannoramic Scan Whole Slide Scanner (Cambridge Research and Instrumentation) and captured using a Zeiss AxioCam MRm digital camera. The quantitative analysis of ASPA and BrdU double-positive cells in the lumber spinal cord was performed as described above.
T-cell proliferation and viability assay and cytokine assay.
Single-spleen cell suspensions were generated from mice with EAE at postimmunization day (PID) 10. Red blood cells were lysed with ACK lysis buffer (Invitrogen), washed, and plated at 5 × 105 splenocytes per well in 96-well microtiter plates. Samples were plated in triplicate. The cells were simultaneously treated with AP20187 (0, 0.1, or 1 nm) and MOG35–55 peptide (0, 1, 10, or 100 μg/ml), and were incubated at 37°C and 5% CO2. After 48 h, 20 μl of BrdU labeling solution (Millipore) was added to the culture media for 24 h. Cell proliferation was determined using the Colorimetric BrdU Cell Proliferation kit (Millipore) according to the manufacturer's instructions. We quantified the cytokines in the culture supernatants of the triplicate wells using the ELISA kits (Thermo Scientific) according to the manufacturer's instructions. For the T-cell viability assay, the spleen cells treated with AP20187 and MOG35–55 peptide were incubated at 37°C and 5% CO2 for 72 h. Cell viability was determined by the MTT assay kit (Progema) according to the manufacturer's instructions.
Statistics.
Data are mean ± SD. For quantitative histology analyses, the average number for each mouse was used for statistical analysis. Multiple comparisons were statistically evaluated by the one-way ANOVA test using SigmaStat 3.1 software (Hearne Scientific Software). A p value <0.05 was considered significant.
Results
Generation of transgenic mice that express Fv2E-PERK specifically in oligodendrocytes
The PERK protein contains three functional domains: a stress-sensing ER luminal domain, an ER transmembrane domain, and a cytosolic eIF2α kinase domain. Activation of PERK signaling is initiated by PERK dimerization and autophosphorylation, which is driven by the stress-sensing ER lumenal domain of PERK (Harding et al., 1999). Fv2E-PERK is an artificial PERK derivative, which was generated by fusing the eIF2α kinase effector domain of PERK to a polypeptide containing two modified FK506 binding domains (Fv2E) (Lu et al., 2004). The activity of this artificial kinase is subordinate to the FK506 dimerizing agent AP20187 and is uncoupled from upstream ER stress signaling (Lu et al., 2004). To generate transgenic mice that specifically express Fv2E-PERK in oligodendrocytes, we assembled a transgenic construct that expresses Fv2E-PERK under the control of a PLP-based construct (Fig. 1A). A number of studies have shown that this construct, containing 2.4 kb of the 5′ flanking DNA, exon 1, and intron 1 of the mouse PLP gene, drives strong and specific transgene expression in oligodendrocytes (Fuss et al., 2000, 2001; Doerflinger et al., 2003; Balabanov et al., 2006). The linearized PLP/Fv2E-PERK transgene was injected into fertilized C57BL/6J mouse oocytes, and seven distinct lines of transgenic mice carrying the PLP/Fv2E-PERK transgene were established. Mice of all seven lines were fertile, transmitted the transgene in a Mendelian fashion, and appeared healthy. RT-PCR analysis amplifying a region spanning the modified FK506 binding domains and the eIF2α kinase domain showed that PLP/Fv2E-PERK transgenic line 18 expressed Fv2E-PERK mRNA in the CNS (data not shown) and was used for all subsequent studies.

Structure of the transgene and characterization of the transgenic mice. A, Partial structure of the PLP/Fv2E-PERK transgene. Transcription initiates within the PLP portion of the transgene and drives expression of the Fv2E-PERK open reading frame. B, Western blotting showed that the Fv2E-PERK protein was expressed in the brains of PLP/Fv2E-PERK mice (Tg), but not in littermate wild-type mice (WT). Moreover, Fv2E-PERK was not expressed in heart, lung, liver, kidney, or spleen in the transgenic mice. C, CC1 and FKBP12 double immunostaining showed that wild-type mice did not express Fv2E-PERK in the brain and that Fv2E-PERK was expressed specifically in oligodendrocytes in the brains of PLP/Fv2E-PERK mice. D, GFAP and FKBP12 double immunostaining showed that Fv2E-PERK was not expressed by astrocytes in the brains of PLP/Fv2E-PERK mice. E, NeuN and FKBP12 double immunostaining showed that Fv2E-PERK was not expressed by neurons in the brains of PLP/Fv2E-PERK mice. F, CD11b and FKBP12 double immunostaining showed that Fv2E-PERK was not expressed by microglia in the brains of PLP/Fv2E-PERK mice. G, PDGFαR and FKBP12 double immunostaining showed that Fv2E-PERK was not expressed by OPCs in the brains of PLP/Fv2E-PERK mice; N = 3 animals. Scale bar: C, 23 μm; D–G, 31 μm.
We further determined the expression pattern of Fv2E-PERK in this line of transgenic mice using Western blot and immunohistochemisty analyses using the anti-FKBP-12 antibody, which recognizes the modified FK506 binding domain and has been successfully used to detect the expression of Fv2E-PERK by Western blot analysis (Ranganathan et al., 2008). Moreover, its immunoreactivity is specific to the modified domain, as no staining is observed in the CNS of wild-type mice (Fig. 1C). Western blots probed with anti-FKBP-12 showed that Fv2E-PERK was expressed in the CNS of the transgenic mice, but not in other organs, such as heart, lung, liver, kidney, or spleen (Fig. 1B). Double immune labeling for CC1 (a marker for mature oligodendrocytes) and FKBP-12 in the CNS of PLP/Fv2E-PERK mice revealed that the majority of CC1-positive oligodendrocytes were also FKBP-12-positive. Importantly, we found that all FKBP-12-positive cells were CC1-positive oligodendrocytes (Fig. 1C). Additionally, double immunolabeling showed that the FKBP-12 immunoreactivity was undetectable in other types of cells in the CNS of the transgenic mice, such as GFAP-positive astrocytic cells (Fig. 1D), NeuN-positive neurons (Fig. 1E), CD11b-positive microglial cells (Fig. 1F), and PDGFαR-positive OPCs (Fig. 1G). Therefore, the PLP/Fv2E-PERK transgenic mice specifically express Fv2E-PERK in mature oligodendrocytes.
Activation of PERK signaling is not detrimental to oligodendrocytes in adult animals
We first determined whether AP20187, a synthetic small chemical compound that induces homodimerization of fusion proteins containing the modified FK506 binding domains, could successfully control the activity of Fv2E-PERK in oligodendrocytes by using mixed glial cell cultures. Mixed glial cells cultured from neonatal PLP/Fv2E-PERK mice were allowed to differentiate for 5 d, a time point when the expression of Fv2E-PERK was detectable by RT-PCR (data not shown), and then were treated with AP20187 for 24 h. Real-time PCR analysis showed that AP20187 treatment increased the expression of PERK-responsive genes encoding CAATT enhancer binding protein homologous protein (CHOP) and growth arrest and DNA damage 34 (GADD34) in a dose-dependent manner (Fig. 2A), but did not significantly affect the expression of mRNA encoding binding Ig protein (BIP). These results were consistent with previous reports showing that CHOP induction is completely dependent on the activity of PERK in ER-stressed cells, and the effect of PERK signaling on BIP expression is minimal (Harding et al., 2000).

Persistent activation of PERK signaling specifically in oligodendrocytes was not detrimental to these cells or to myelin in adult animals. A, Real-time PCR analysis showed that AP20187-induced PERK signaling increased the expression of CHOP and GADD34 in the mixed glial cultures generated from neonatal PLP/Fv2E-PERK mice in a dose-dependent manner but did not significantly affect the expression of BIP. B, Real-time PCR analysis showed that AP20187 treatment increased the expression of CHOP and GADD34 in the spinal cords of PLP/Fv2E-PERK mice in a dose-dependent manner but did not significantly affect the expression of BIP. C, D, CC1 and p-eIF2α double immunostaining showed that p-eIF2α was undetectable in oligodendrocytes in the spinal cords of PLP/Fv2E-PERK mice treated with vehicle but became detectable in the oligodendrocytes of mice treated with AP20187. E, Cell counting revealed that the numbers of CC1-positive oligodendrocytes in the white matter of the spinal cords and the cerebellums of PLP/Fv2E-PERK mice treated with AP20187 were comparable with those of mice treated with vehicle. F, G, MBP immunostaining showed that AP20187 treatment did not alter myelin integrity in the spinal cords of adult PLP/Fv2E-PERK mice; N = 3 animals. Error bars indicate SD. ★p < 0.05. Scale bar: C, D, 12 μm; F, G, 200 μm.
To determine whether AP20187 could also control Fv2E-PERK activity in oligodendrocytes in vivo, 21-day-old PLP/Fv2E-PERK transgenic mice were given intraperitoneal injections of the compound at a dose of 0.5, 2, or 5 mg/kg or were injected with only the vehicle (4% ethanol, 10% PEG-400, 2% Tween 20 in water) once daily for 2 d. Real-time PCR analysis showed that AP20187 treatment enhanced the expression of the mRNAs encoding CHOP and GADD34 in the CNS of the transgenic mice in a dose-dependent manner but did not alter the expression of BIP (Fig. 2B). Together, these data indicate that the activity of PERK signaling in oligodendrocytes in PLP/Fv2E-PERK mice can be temporally manipulated by the administration of AP20187.
Next, we determined the effects of the activation of PERK signaling on oligodendrocytes in adult mice. Six-week-old PLP/Fv2E-PERK mice were treated with 2 mg/kg AP20187 or vehicle daily for 4 weeks. All AP20187-treated mice were indistinguishable from vehicle-treated mice and littermate wild-type mice. In accordance with previous studies (Lin et al., 2005, 2006, 2007), CC1 and p-eIF2α double immunostaining showed that p-eIF2α was undetectable in oligodendrocytes in the CNS of PLP/Fv2E-PERK mice treated with vehicle. Importantly, we found that AP20187 treatment markedly increased the level of p-eIF2α specifically in the oligodendrocytes of these mice (Fig. 2C,D). Cell counting showed that AP20187 treatment did not significantly alter CC1-positive oligodendrocyte numbers in the CNS of PLP/Fv2E-PERK mice (Fig. 2E). Moreover, MBP immuohistochemistry showed that AP20187 treatment had no noticeable effect on myelin integrity in the CNS of PLP/Fv2E-PERK mice (Fig. 2F,G). Additionally, PDGFαR immunostaining showed that AP20187 treatment did not significantly change the number of PDGFαR-positive OPCs in the CNS of PLP/Fv2E-PERK mice (data not shown). Collectively, these data demonstrate that persistent activation of PERK signaling specifically in oligodendrocytes is not detrimental to the cells or myelin of adult animals.
Enhancing the ISR specifically in oligodendrocytes attenuates EAE-induced demyelination
A number of studies have shown that AP20187 treatment alone has no noticeable effect on mice under physiological or pathological conditions (Mallet et al., 2002; Neff et al., 2002; Steel et al., 2010). In agreement with these studies, we found that AP20187 treatment alone did not influence the development of EAE in wild-type C57BL/6J mice (data not shown). To assess the effects of the enhanced ISR specifically in oligodendrocytes on the development of EAE, one group of 7-week-old female PLP/Fv2E-PERK mice immunized with MOG35–55 peptide was given intraperitoneal injections of 0.5 mg/kg AP20187 daily starting on PID10. A second group of immunized PLP/Fv2E-PERK mice was treated with vehicle to serve as a control. Real-time PCR analysis showed that AP20187 treatment significantly increased the levels of CHOP mRNA in the CNS of PLP/Fv2E-PERK mice at PID12 (Fig. 3B). CC1 and p-eIF2α double immunostaining also showed that AP20187 treatment noticeably increased the level of p-eIF2α specifically in oligodendrocytes at PID12 (Fig. 3C,D). All mice immunized with MOG35–55 peptide developed neurological signs of disease starting around PID14. Nevertheless, PLP/Fv2E-PERK mice treated with AP20187 demonstrated significantly milder EAE clinical symptoms at the peak of disease (PID18–PID23) compared with control mice (Fig. 3A). The majority of control mice (22 of 24) developed hind-limb paralysis, a severe symptom of EAE. In contrast, none of the 18 PLP/Fv2E-PERK mice treated with AP20187 displayed hind-limb paralysis. Additionally, we found that neither increasing the dose of AP20187 (2 or 5 mg/kg) nor starting the treatment at earlier time points (PID1 or PID5) attenuated EAE disease severity in PLP/Fv2E-PERK mice to a greater extent than treatment with 0.5 mg/kg AP20187 starting on PID10 (data not shown).

Enhancing the PERK-mediated ISR specifically in oligodendrocytes attenuated EAE disease severity. A, Mean clinical score were recorded daily: 0 = healthy; 1 = flaccid tail; 2 = ataxia and/or paresis of hindlimbs; 3 = paralysis of hindlimbs and/or paresis of forelimbs; 4 = tetraparalysis; 5 = moribund or death. N = 12 animals. Error bars indicate SD. ★p < 0.05. B, Real-time PCR analysis showed that AP20187 treatment increased the expression of CHOP in the spinal cords of PLP/Fv2E-PERK mice at PID12; N = 4 animals. Error bars indicate SD. ★p < 0.05. C, D, CC1 and p-eIF2α double immunostaining showed that the level of p-eIF2α was markedly increased in oligodendrocytes in the lumbar spinal cords of PLP/Fv2E-PERK mice treated with AP20187 at PID12; N = 3 animals. Scale bar: C, D, 23 μm.
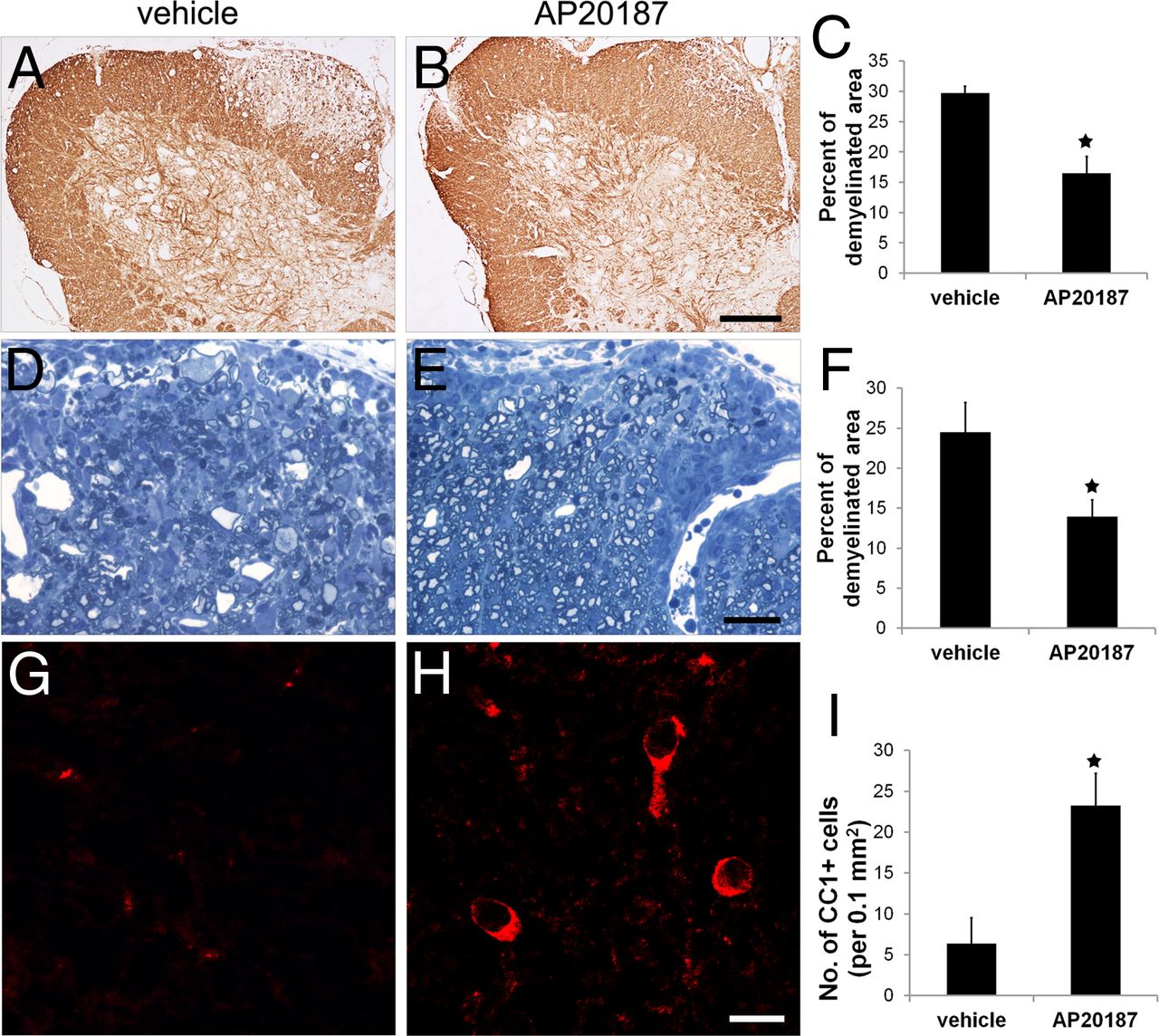
CNS tissues were prepared at the peak of disease (PID19) and analyzed for tissue damage. MBP immunostaining revealed severe myelin damage in the lumbar spinal cord of PLP/Fv2E-PERK mice treated with vehicle at PID19 (Fig. 4A,C). Interestingly, AP20187 treatment significantly alleviated EAE-induced myelin damage in these mice (Fig. 4B,C). We further verified the reduction of myelin damage in the lumbar spinal cord of PLP/Fv2E-PERK mice treated with AP20187 via toluidine blue staining. Quantitative analysis of toluidine blue staining showed that the percentage of area in the white matter of the lumbar spinal cord that was demyelinated in PLP/Fv2E-PERK mice treated with AP20187 was significantly reduced compared with that of mice treated with vehicle (Fig. 4D–F). CC1 immunostaining showed that oligodendrocytes in the demyelinated lesions of PLP/Fv2E-PERK mice treated with vehicle were largely lost, whereas there were significantly more oligodendrocytes remaining in the demyelinated lesions of the mice treated with AP20187 (Fig. 4G–I). Moreover, whereas phosphorylated neurofilament-H (SMI-31) immunostaining of lumbar spinal cord longitudinal sections showed severe axon loss and axon degeneration in the demyelinating lesions of the mice treated with vehicle, there were markedly less axon loss and axon degeneration in the mice treated with AP20187 (Fig. 5A,B). As expected, EM analysis showed severe axon degeneration in the demyelinating lesions of the mice treated with vehicle (Fig. 5C). Degenerating axons displayed all forms of degeneration, including swelling, shrinkage, and accumulation of electron-dense deposits and membranous structures. Importantly, EM analysis verified that AP20187 treatment significantly reduced the number of degenerating axons and increased the density of axons in the demyelinating lesions in the lumbar spinal cord of PLP/Fv2E-PERK mice (Fig. 5C–F). Together, these data demonstrate that enhancing the ISR specifically in oligodendrocytes protects mice against EAE.

Enhancing the PERK-mediated ISR specifically in oligodendrocytes ameliorated EAE-induced demyelination and oligodendrocyte loss. A–C, At PID19, MBP immunostaining revealed large demyelinating lesions in the lumbar spinal cords of PLP/Fv2E-PERK mice treated with vehicle. Interestingly, AP20187 treatment significantly reduced the size of demyelinating lesions in PLP/Fv2E-PERK mice; 20 sections per mouse, N = 4 animals. D–F, Toluidine blue staining showed that AP20187 treatment significantly reduced the percentage of the white matter area of the lumbar spinal cord that was demyelinated in PLP/Fv2E-PERK mice at PID19; 5 serial sections per mouse, N = 4 animals. G–I, CC1 immunostaining showed that AP20187 treatment significantly attenuated the oligodendrocyte loss observed at PID19 in the demyelinating lesions in the lumbar spinal cord of PLP/Fv2E-PERK mice; 20 sections per mouse, N = 4 animals. Error bars indicate SD. ★p < 0.05. Scale bar: A, B, 200 μm; D, E, 30 μm; G, H, 15 μm.

Enhancing the PERK-mediated ISR specifically in oligodendrocytes ameliorated EAE-induced axonal degeneration. A, B, SMI-31 immunostaining of lumbar spinal cord longitudinal sections showed that AP20187 treatment markedly attenuated axon loss and axon degeneration in the demyelinating lesions in PLP/Fv2E-PERK mice at PID 19. C–F, EM analysis showed that AP20187 treatment significantly reduced the number of degenerating axons (*, arrow) and increased the number of total axons in the demyelinating lesions in PLP/Fv2E-PERK mice at PID19; N = 4 animals. Error bars indicate SD. ★p < 0.05. Scale bar: A, B, 10 μm; C, D, 2 μm.
Treatment with the dimerizer AP20187 has no significant effect on the inflammatory response in PLP/Fv2E-PERK mice undergoing EAE
During EAE, T cells are primed in the mouse peripheral immune system before PID10 (Stromnes and Goverman, 2006), suggesting that treatment with AP20187 starting on PID10 would have no impact on T-cell priming. Therefore, we investigated whether AP20187 treatment had a direct affect on the activity of primed T cells in PLP/Fv2E-PERK mice during EAE. Spleen leukocytes were generated from PLP/Fv2E-PERK mice undergoing EAE at PID10. As expected, RT-PCR analysis showed that the spleen leukocytes did not express the Fv2E-PERK transgene (data not shown), which was consistent with the Western blot data (Fig. 1B). The spleen leukocytes were then treated with AP20187 or vehicle, and their in vitro response to a secondary exposure to MOG35–55 peptide was analyzed. Importantly, in vitro recall assays showed that AP20187 treatment did not alter T-cell proliferation, viability, or cytokine production (Fig. 6). Collectively, these data rule out the possibility that AP20187 directly acts on T cells to influence their activity in PLP/Fv2E-PERK mice subjected to EAE.

AP20187 treatment did not directly act on T cells to influence their activity in PLP/Fv2E-PERK mice undergoing EAE. A, The BrdU cell proliferation assay showed that AP20187 treatment did not affect T-cell proliferation in response to MOG35–55 peptide. B, The MTT-cell viability assay showed that AP20187 treatment did not alter T-cell viability in response to MOG35–55 peptide. C, D, ELISA analyses showed that AP20187 treatment did not significantly affect the ability of T cells to produce the Th1 cytokine IFN-γ or the Th2 cytokine IL-4 in response to MOG35–55 peptide. N = 4 animals. Error bars indicate SD.
During EAE pathogenesis, primed T cells enter the CNS and induce tissue damage well before the onset of clinical disease (Hickey et al., 1991). We found that AP20187 treatment had no noticeable effect on the number of CD3-positive T cells, the number of CD11b-positive microglia/macrophages, or the expression of immune cytokines in the spinal cords of PLP/Fv2E-PERK mice before the onset of clinical disease (PID12, Fig. 7A–F). Moreover, we found that AP20187 treatment did not significantly alter the number of CD3-positive T cells or the number of CD11b-positive microglia/macrophages in the lumbar spinal cords of PLP/Fv2E-PERK mice at the peak of disease (PID19, Fig. 7G–K). Nevertheless, double immunostaining showed that both T cells and microglia/macrophages were confined to the small demyelinating lesions with high cell density in AP20187-treated mice but were diffuse across the big demyelinating lesions in vehicle-treated mice. Importantly, real-time PCR analysis showed that AP20187 treatment did not significantly change the expression of immune cytokines in the spinal cords of PLP/Fv2E-PERK mice at PID19 (Fig. 7L). Together, our data suggest that AP20187 treatment does not significantly affect the degree of the inflammatory response in the peripheral immune system or the CNS of PLP/Fv2E-PERK mice during EAE. Thus, it is unlikely that AP20187 treatment attenuates EAE-induced oligodendrocyte loss, demyelination, and axonal degeneration in PLP/Fv2E-PERK mice by altering the degree of the inflammatory response.

AP20187 treatment did not alter the degree of the inflammatory response in the CNS of PLP/Fv2E-PERK mice undergoing EAE. A, B, E, CD3 immunostaining showed that AP20187 treatment did not affect T-cell infiltration into the white matter of the lumbar spinal cord in PLP/Fv2E-PERK mice at PID12; 20 sections per mouse, N = 4 animals. C–E, CD11b immunostaining showed that AP20187 treatment did not change the number of microglia/macrophages present in the white matter of the lumbar spinal cord in PLP/Fv2E-PERK mice at PID12; 20 sections per mouse, N = 4 animals. F, Real-time PCR analysis showed that AP20187 treatment did not significantly alter the levels of the mRNAs encoding iNOS, TNF-α, IFN-γ, IL-4, IL-5, IL-12, IL-17, and IL-23 in the spinal cord of PLP/Fv2E-PERK mice at PID12; N = 4 animals. G, H, K, CD3 and MBP double immunostaining showed that AP20187 treatment did not change the number of T cells present in the white matter of the lumbar spinal cord in PLP/Fv2E-PERK mice at PID19; 20 sections per mouse, N = 4 animals. I–K, CD11b and MBP double immunostaining showed that AP20187 treatment did not change the number of microglia/macrophages present in the white matter of the lumbar spinal cord in PLP/Fv2E-PERK mice at PID19; 20 sections per mouse, N = 4 animals. L, Real-time PCR analysis showed that AP20187 treatment did not significantly alter the levels of the mRNAs encoding iNOS, TNF-α, IFN-γ, IL-4, IL-5, IL-12, IL-17, and IL-23 in the spinal cord of PLP/Fv2E-PERK mice at PID19; N = 4 animals. Error bars indicate SD. Scale bar: A, B, 20 μm; C, D, 60 μm; G, H, 25 μm; I, J, 25 μm.
An enhanced ISR cell-autonomously protects oligodendrocytes against EAE-induced apoptosis
Several lines of evidence have indicated that immune cytokine cytotoxicity, reactive oxidative and nitrative stress, and glutamate excitoxicity contribute significantly to oligodendrocyte apoptosis in MS and EAE (Buntinx et al., 2002; Bradl and Lassmann, 2010). It has been demonstrated that the ISR protects cells against all of these factors, as well as against ER stress (Harding et al., 2003; Lu et al., 2004; Lin et al., 2008). Therefore, we next sought to determine whether the enhanced ISR protects oligodendrocytes against immune attacks. Several studies have shown that oligodendrocyte apoptosis is the earliest structural change in newly forming demyelinating lesions in both MS and EAE (Barnett and Prineas, 2004; Henderson et al., 2009). In accordance with these studies, CC1, TUNEL, and DAPI triple labeling revealed, before the onset of clinical disease, a number of apoptotic oligodendrocytes, which were CC1 and TUNEL positive with nuclear fragmentation, in the lumbar spinal cord of PLP/Fv2E-PERK mice treated with vehicle (PID12, Fig. 8E–H,M). Importantly, we found that, at PID12, the number of apoptotic oligodendrocytes was significantly reduced in the lumbar spinal cord of PLP/Fv2E-PERK mice treated with AP20187 (Fig. 8I–M). As expected, we did not find any apoptotic oligodendrocytes in the CNS of age-matched naive PLP/Fv2E-PERK mice (Fig. 8A–D,M). CC1 and activated caspase-3 double immunostaining also showed that AP20187 treatment noticeably reduced the number of CC1 and activated caspase-3 double-positive cells in the lumbar spinal cord of PLP/Fv2E-PERK mice at PID12 (data not shown). Moreover, we found minimal number of apoptotic PDGFαR-positive OPCs in the lumbar spinal cord of PLP/Fv2E-PERK mice treated with either AP20187 or vehicle at PID12 (data not shown). Thus, these results demonstrate that enhancing the ISR specifically in oligodendrocytes protects these cells against immune attacks in EAE mice. Additionally, BrdU pulse-chase experiments showed that AP20187 treatment did not significantly change the number of BrdU and ASPA double-positive cells in the lumbar spinal cord of PLP/Fv2E-PERK mice at PID 19 (data not shown). Together, these data indicate that the beneficial effects of AP20187 treatment on the development of EAE are the result of its cytoprotective effects on mature oligodendrocytes instead of newly generated oligodendrocytes.

Enhancing the PERK-mediated ISR specifically in oligodendrocytes protected these cells against immune attack. A–D, M, CC1, TUNEL, and DAPI triple labeling showed that there is no apoptotic oligodendrocytes in the lumbar spinal cords of 10-week-old naive PLP/Fv2E-PERK mice. E–H, M, At PID12, CC1, TUNEL, and DAPI triple labeling revealed a number of apoptotic oligodendrocytes (arrow) in the lumbar spinal cords of PLP/Fv2E-PERK mice treated with vehicle. Inset, Apoptotic oligodendrocytes were CC1- and TUNEL-positive with nuclear fragmentation. I–M. Nevertheless, the number of apoptotic oligodendrocytes was dramatically reduced in the lumbar spinal cords of PLP/Fv2E-PERK mice treated with AP20187; 20 sections per mouse, N = 4 animals. Error bars indicate SD. ★p < 0.05. Scale bar: A–L, 20 μm.
It has been shown that the ISR activates the NF-κB pathway by repressing the translation of the NF-κB inhibitor IκBα during ER stress (Deng et al., 2004). Importantly, our recent study demonstrates that IFN-γ activates PERK signaling as well as NF-κB signaling in oligodendrocytes and that PERK is required for IFN-γ-induced NF-κB activation in oligodendrocytes (Lin et al., 2012). NF-κB is increasingly recognized as an antiapoptotic transcription factor (Karin and Lin, 2002). Several studies have suggested that NF-κB activation promotes oligodendrocyte survival during inflammatory insults (Vollgraf et al., 1999; Nicholas et al., 2001; Hamanoue et al., 2004). Therefore, we were interested in the potential role of the NF-κB pathway in the cytoprotective effects of ISR signaling on oligodendrocytes. Double immunostaining for ASPA, a marker for oligodendrocytes (Madhavarao et al., 2004; Locatelli et al., 2012), and the active form of NF-κB p65 showed that immunoreactivity for the active form of NF-κB p65 was undetectable in the CNS of adult naive mice (Fig. 9A). In accordance with a previous study (Bonetti et al., 1999), double immunostaining at PID12 revealed that a number of ASPA-positive oligodendrocytes were positive for the active form of NF-κB p65 in the white matter of the lumbar spinal cord in PLP/Fv2E-PERK mice treated with vehicle (Fig. 9B,D). Importantly, we found that AP20187 treatment significantly increased the number of oligodendrocytes that stained positive for the active form NF-κB p65 in the lumbar spinal cord of PLP/Fv2E-PERK mice at PID12 (Fig. 9C,D). Moreover, as expected, we found that AP20187 treatment had no effect on eIF2α phosphorylation or NF-κB activation in differentiated oligodendrocytes cultured from wild-type mice (Fig. 10A,B). Interestingly, AP20187 treatment resulted in eIF2α phosphorylation and NF-κB activation in differentiated oligodendrocytes cultured from PLP/Fv2E-PERK mice (Fig. 10A,B). Collectively, these data suggest the possibility that activation of NF-κB signaling contributes to the protective effects of the ISR in oligodendrocytes during EAE.
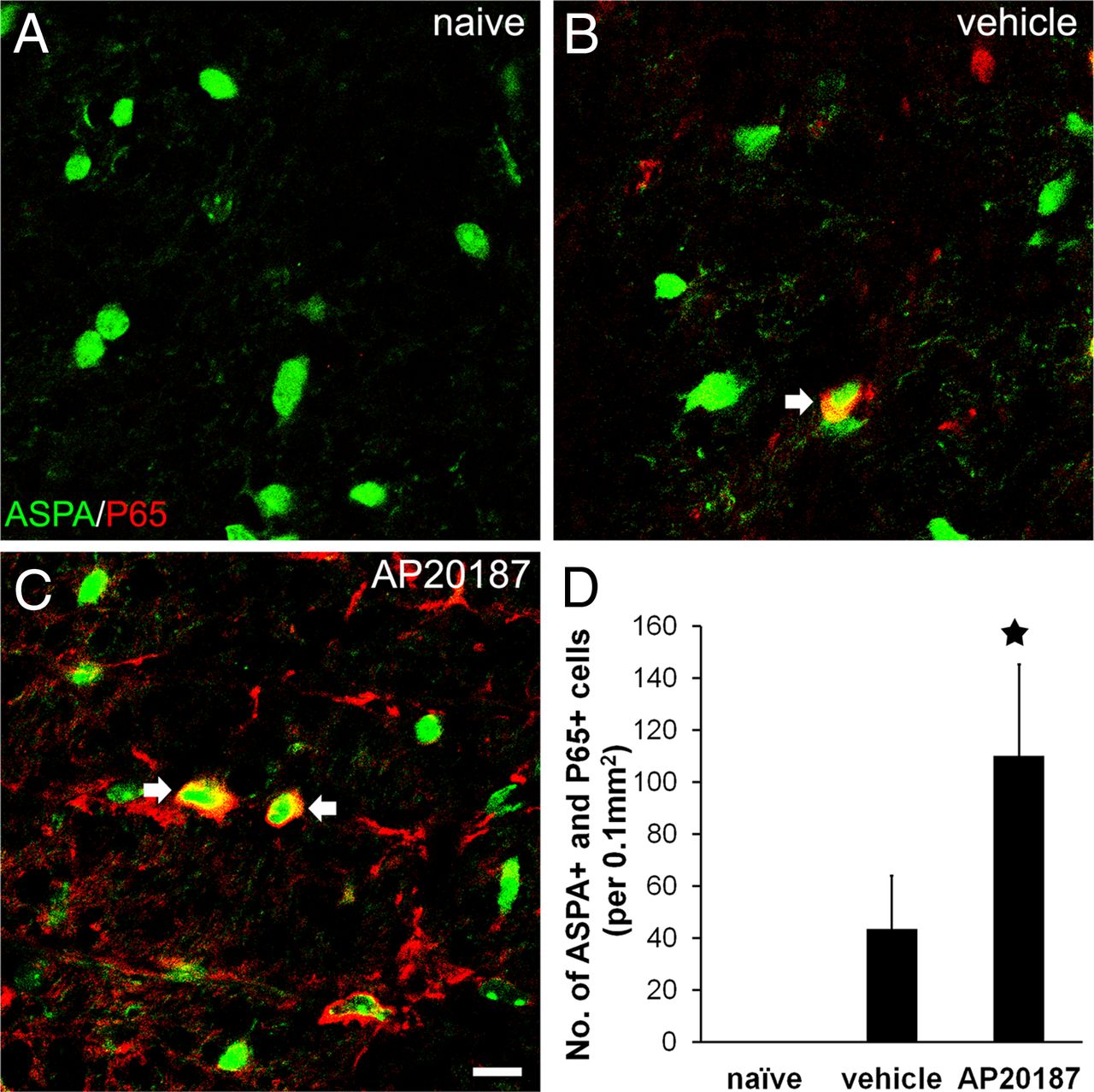
Enhancing the PERK-mediated ISR specifically in oligodendrocytes increased activation of the NF-κB pathway in the CNS of EAE mice. A, Double immunostaining for ASPA and the active form of NF-κB p65 showed that the active form of NF-κB p65 was undetectable in oligodendrocytes in the CNS of adult naive mice. B, However, the active form of NF-κB p65 was detectable at PID12 in a number of oligodendrocytes in the lumbar spinal cord white matter of PLP/Fv2E-PERK mice treated with vehicle. C, Interestingly, the number of oligodendrocytes positive for the active form of NF-κB p65 was further increased in the lumbar spinal cords of AP20187-treated mice. D, Quantitative analysis showed that the number of oligodendrocytes positive for the active form of NF-κB p65 in the lumbar spinal cord white matter of PLP/Fv2E-PERK mice treated with AP20187 was significantly increased compared with vehicle-treated mice; 20 sections per mouse, N = 4 animals. Error bars indicate SD. ★p < 0.05. Scale bar: A–C, 20 μm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The PERK-mediated ISR activated the NF-κB pathway in oligodendrocytes. A, PLP and p-eIF2α double immunostaining showed that AP20187 treatment had no effect on eIF2α phosphorylation in differentiated oligodendrocytes cultured from wild-type mice. In contrast, AP20187 treatment resulted in eIF2α phosphorylation in the cells cultured from PLP/Fv2E-PERK mice. B, 2′,3′-Cyclic nucleotide phosphodiesterase and the active form of NF-κB p65 double immunostaining showed that AP20187 treatment had no effect on NF-κB p65 activation in differentiated oligodendrocytes cultured from wild-type mice. Interestingly, AP20187 treatment resulted in NF-κB p65 activation in the cells cultured from PLP/Fv2E-PERK mice. Scale bar: A, B, 20 μm.
Discussion
Oligodendrocytes produce the myelin sheath that wraps around axons and facilitates action potential conduction along axons and maintains axonal integrity (Simons and Trotter, 2007). The mean surface area of myelin per oligodendrocyte in the CNS is thousands of times greater than the surface area of a typical mammalian cell; thus, each oligodendrocyte must synthesize an enormous amount of myelin membrane proteins and membrane lipids to assemble myelin and maintain its integrity (Pfeiffer et al., 1993). The ER is responsible for the modification and folding of myelin membrane proteins and the production of membrane lipids. Therefore, it is not surprising that oligodendrocytes are highly sensitive to the disruption of ER homeostasis and that ER stress in oligodendrocytes plays an important role in the pathogenesis of a number of myelin disorders (Gow and Wrabetz, 2009; Lin and Popko, 2009).
Our previous study showed that the presence of IFN-γ in the CNS before EAE onset almost completely blocked oligodendrocyte death, demyelination, and axonal degeneration in the CNS of EAE mice. We also demonstrated that the protective effects of IFN-γ in EAE were completely abrogated in PERK-deficient animals (Lin et al., 2007). Because of the complexity of immune-mediated demyelinating diseases, which involve the interaction of the CNS and the immune system, the mouse models used in our previous study did not allow us to delineate the specific contribution of PERK signaling in individual CNS cell types (oligodendrocytes, astroctytes, microglia, and neurons) or blood-borne immune cell types (T cells and macrophages) to the development of EAE. A number of studies have suggested that oligodendrocyte apoptosis is the earliest structural change in MS and EAE demyelinating lesions, and oligodendrocyte apoptosis is, at least in part, responsible for myelin destruction and subsequent axonal degeneration in these diseases (Matute and Pérez-Cerdá, 2005; McGuire et al., 2010). Therefore, in this study, we focused on dissecting the precise role of PERK signaling in oligodendrocytes during EAE. We generated a novel transgenic mouse model, PLP/Fv2E-PERK, which allows for temporally controlled activation of PERK signaling, in the absence of ER stress, specifically in oligodendrocytes. Using this unique mouse model, we demonstrated that persistent activation of PERK signaling specifically in oligodendrocytes of adult animals had no effect on oligodendrocyte viability or myelin integrity. These results likely reflect the reduced metabolic requirements of adult oligodendrocytes, which are only responsible for the slow replenishment of myelin components. Importantly, we found that, when enhanced specifically in oligodendrocytes, the PERK-mediated ISR greatly reduced oligodendrocyte apoptosis before the onset of clinical disease and attenuated EAE-induced demyelination and axonal degeneration at the peak of disease. Moreover, we did not find any evidence suggesting that treatment with the dimerizer AP20187, which activates the Fv2E-PERK transgene specifically in oligodendroctes, significantly altered the degree of the immune response in the peripheral immune system or the CNS of PLP/Fv2E-PERK mice undergoing EAE. Additionally, BrdU pulse-chase experiments showed that AP20187 treatment did not significantly change the number of newly generated oligodendrocytes in the lumbar spinal cord of PLP/Fv2E-PERK mice at the peak of disease. Collectively, these results provide direct evidence that the activation of PERK signaling in oligodendrocytes protects these cells from immune attacks, resulting in the attenuation of demyelination and axonal degeneration in immune-mediated demyelinating diseases. Nevertheless, compared with the almost complete protection provided by IFN-γ, the degree of protection provided by enhancing PERK signaling specifically in oligodendrocytes is moderate in EAE mice. Because PERK deficiency completely abrogated the protective effects of IFN-γ in EAE (Lin et al., 2007), these findings likely reflect that activation of PERK signaling in other CNS cell types (astroctytes or microglia) or blood-borne immune cell types is also required for the full protective effects of IFN-γ during EAE.
Recent studies have suggested that axonal degeneration occurs early in MS and EAE and is the principal cause of chronic disability in MS (Trapp and Nave, 2008; Siffrin et al., 2010). Both the inflammatory response and demyelination are thought to contribute to axonal degeneration in these diseases (Herz et al., 2010; Siffrin et al., 2010). We demonstrated here that enhancing PERK signaling specifically in oligodendrocytes attenuated axonal degeneration but did not attenuate the degree of the inflammatory response in EAE demyelinating lesions. Therefore, it is unlikely that enhanced PERK signaling, when induced specifically in oligodendrocytes, attenuates EAE-induced axonal degeneration through downregulation of the inflammatory response. It is more likely that enhanced PERK signaling in oligodendrocytes protects axons in EAE by maintaining myelin integrity. Although the specific protective mechanisms of oligodendrocytes remain to be fully understood, our findings suggest that therapeutic approaches to enhancing ISR signaling in oligodendrocytes will have a positive impact on preserving the neurological functions of MS patients.
Also remaining to be elucidated are the precise molecular mechanisms responsible for the cytoprotective effects of PERK signaling. It is thought that PERK signaling contributes to cytoprotection by blocking new protein synthesis, which conserves energy, and by activating a transcriptional program that promotes the expression of numerous cytoprotective genes (Marciniak and Ron, 2006). Recent studies have shown that PERK signaling activates NF-κB, an antiapoptotic transcription factor, by repressing the translation of IκBα, an inhibitor of NF-κB (Deng et al., 2004). Indeed, we see an increase in the activation of the NF-κB pathway in the CNS of EAE mice when PERK signaling is enhanced specifically in oligodendrocytes. Therefore, it is possible that activation of the NF-κB pathway contributes to the protective effects of PERK signaling in oligodendrocytes during EAE.
In this study, attenuation of EAE symptoms was performed without affecting the degree of the inflammatory response. It is known that the dimerizer AP20187 has no effect on immunomodulation (Alfa et al., 2009). Additionally, we demonstrated that leukocytes in the peripheral immune system, as well as microglia and astrocytes in the CNS, did not express the PLP/Fv2E-PERK transgene. We also found that AP20187 treatment did not directly affect the activity of T cells in PLP/Fv2E-PERK mice undergoing EAE. Moreover, we showed that AP20187 treatment did not significantly alter the number of inflammatory cells and the expression of immune cytokines in the CNS of PLP/Fv2E-PERK mice during the course of EAE. Interestingly, we found that enhancing PERK signaling specifically in oligodendrocytes noticeably affected the distribution pattern of inflammatory cells in the CNS of EAE mice. Both T cells and microglia/macrophages were confined to the small demyelinating lesions with high cell density in AP20187-treated PLP/Fv2E-PERK mice. In contrast, previous studies showed that the oligodendrocyte-targeted expression of antiapoptotic proteins and the oligodendrocyte-targeted deletion of proapoptotic proteins attenuated EAE-induced oligodendrocyte apoptosis and demyelination and subsequently resulted in the reduction of the inflammatory response in EAE demyelinating lesions (Hisahara et al., 2000; McGuire et al., 2010). There is evidence that oligodendrocytes, through the production of a number of chemokines, actively participate in the inflammatory response in the CNS in MS and EAE (Balabanov et al., 2007; Zeis et al., 2008). It is possible that the degree of inflammatory response was not altered in the AP20187-treated PLP/Fv2E-PERK mice because treatment with the dimerizer was not initiated until PID 10, which might be too late to alter the number of CNS infiltrating inflammatory cells. The studies from Hisahara et al. (2000) and McGuire et al. (2010) were performed with mice carrying genetic alterations present at the time of EAE induction, which might explain the decreased inflammation in these models. Clearly, the role that oligodendrocytes play in the CNS inflammatory response is a critical area of study, such that these issues are deserving of additional study.
A previous report has shown that AP20187, the synthetic small chemical compound used in this study that induces homodimerization of the modified FK506 binding domain-containing fusion proteins, can penetrate the blood–brain barrier; however, its blood–brain barrier permeability is moderate (Burnett et al., 2004). In this study, we showed that the treatment with AP20187 at the dose of 0.1 nm significantly increased the expression of PERK-responsive genes CHOP and GADD34 in mixed glial cells cultured from neonatal PLP/Fv2E-PERK mice. In agreement with the in vitro study, we found that the treatment with AP20187 at a dose of 0.5 mg/kg daily was sufficient to activate Fv2E-PERK in oligodendrocytes in the CNS of PLP/Fv2E-PERK mice. Together, our results verify the blood–brain barrier permeability of AP20187 and suggest that the transgene-derived Fv2E-PERK is very sensitive to this compound in oligodendrocytes in cell culture and in the CNS of PLP/Fv2E-PERK mice.
In conclusion, this study provides direct evidence that the activation of PERK signaling in oligodendrocytes protects these cells against immune attack and subsequently results in the attenuation of demyelination and axonal degeneration in an immune-mediated demyelinating disease. These data demonstrate the importance of oligodendrocytes in the pathogenesis of MS and EAE, particularly through their role in axonal degeneration. Currently, there is no effective therapy for MS that promotes the restoration and protection of neurological functions. The results of the present study suggest that therapeutic strategies that activate PERK signaling in oligodendrocytes may have beneficial effects on neurological function and delay disability progression in MS patients.
Footnotes
- Received April 3, 2012.
- Revision received February 14, 2013.
- Accepted February 22, 2013.
This work was supported by National Multiple Sclerosis Society Grants TA 3026-A-1 and RG-4813-A-2 to W.L., National Institutes of Health Grant NS34939 to B.P., and the Myelin Repair Foundation to B.P. This work used the Nikon A1 confocal microscope funded by National Institutes of Health Grant S10RR027535. We thank Dr. Wendy Macklin (University of Colorado Denver, Aurora, Colorado) for providing the proteolipid protein-based expression construct and Dr. M.A. Aryan Namboodiri (Uniformed Services University of the Health Sciences, Bethesda, Maryland) for providing the antibody against aspartoacylase.
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: Dr. Wensheng Lin, Department of Neuroscience, University of Minnesota, 2101 6th Street SE, WMBB4-410, Minneapolis, MN 55455, linw{at}umn.edu; or Dr. Brian Popko, Center for Peripheral Neuropathy, Department of Neurology, University of Chicago, 5841 South Maryland Avenue, MC2030, Chicago, IL 60637, bpopko{at}neurology.bsd.uchicago.edu
- Copyright © 2013 the authors 0270-6474/13/335980-12$15.00/0