
Abstract
NMDA receptor activity is involved in shaping synaptic connections throughout development and adulthood. We recently reported that brief activation of NMDA receptors on cultured ventral midbrain dopamine neurons enhanced their axon growth rate and induced axonal branching. To test whether this mechanism was relevant to axon regrowth in adult animals, we examined the reinnervation of dorsal striatum following nigral dopamine neuron loss induced by unilateral intrastriatal injections of the toxin 6-hydroxydopamine. We used a pharmacological approach to enhance NMDA receptor-dependent signaling by treatment with an inhibitor of glycine transporter-1 that elevates levels of extracellular glycine, a coagonist required for NMDA receptor activation. All mice displayed sprouting of dopaminergic axons from spared fibers in the ventral striatum to the denervated dorsal striatum at 7 weeks post-lesion, but the reinnervation in mice treated for 4 weeks with glycine uptake inhibitor was approximately twice as dense as in untreated mice. The treated mice also displayed higher levels of striatal dopamine and a complete recovery from lateralization in a test of sensorimotor behavior. We confirmed that the actions of glycine uptake inhibition on reinnervation and behavioral recovery required NMDA receptors in dopamine neurons using targeted deletion of the NR1 NMDA receptor subunit in dopamine neurons. Glycine transport inhibitors promote functionally relevant sprouting of surviving dopamine axons and could provide clinical treatment for disorders such as Parkinson's disease.
Introduction
During development and in adulthood, NMDA glutamate receptor activity is involved in synapse elimination or stabilization, and inhibition or promotion of axonal sprouting (Li et al., 1994; Katz and Shatz, 1996; Constantine-Paton and Cline, 1998; Ruthazer and Cline, 2004; Colonnese et al., 2005; Lee et al., 2005). The roles of axonal presynaptic versus somatodendritic postsynaptic NMDA receptors in these processes are not well understood. Presynaptic NMDA receptor expression on axons appears to be high during early development and drops drastically in adulthood (Herkert et al., 1998; Lien et al., 2006; Corlew et al., 2007; Wang et al., 2011). The functional relevance of presynaptic NMDA receptors is controversial (Christie and Jahr, 2008; Pugh and Jahr, 2011), although several studies suggest a modulatory effect on transmitter release (Tzingounis and Nicoll, 2004; Larsen et al., 2011).
In cultured neurons, NMDA receptors tend to be expressed in axons and axonal growth cones (Schmitz et al., 2009; Wang et al., 2011) and mediate growth cone turning in response to glutamate gradients (Zheng et al., 1996). We recently reported that a brief exposure to NMDA receptor agonists enhanced axonal growth rate and branching in cultured dopaminergic midbrain neurons (Schmitz et al., 2009) consistent with prior studies on cerebellar granule cells (Pearce et al., 1987; Rashid and Cambray-Deakin, 1992). Here, we tested the hypothesis that NMDA receptor activity promotes sprouting of dopaminergic axons in vivo by studying sprouting from spared fibers in the ventral striatum to the dorsal striatum following striatal 6-hydroxydopamine (6-OHDA)-induced lesions in mature mice. Lesions were adjusted so that most cells in the substantia nigra (SNpc) innervating the dorsal striatum were lost, but cells in the ventral tegmental area (VTA) innervating the ventral striatum were spared. This lesion model mimics the denervation pattern found in brains of patients with Parkinson's disease, where the dopaminergic innervation of the lateral putamen is lost while that of the most medial portion of the putamen, the caudate, and nucleus accumbens remains relatively intact (Miller et al., 1999).
To enhance NMDA receptor activity pharmacologically, we used an uptake inhibitor of the amino acid glycine, which is a coagonist that binds to the NR1 subunit and is required for receptor activation (Clements and Westbrook, 1991; Berger et al., 1998). Glycine transporter 1 (GlyT1) is widely expressed in the forebrain in glial as well as neuronal cells (Smith et al., 1992; Raiteri and Raiteri, 2010) and has been shown to regulate glycine occupancy of NMDA receptors in the CNS (Berger et al., 1998; Bergeron et al., 1998) leading to enhanced NMDA currents and LTP in hippocampal CA1 (Martina et al., 2004). Importantly, GlyT1 inhibitors increase glycine levels in the mouse striatum threefold (Alberati et al., 2012). GlyT1 inhibitors have been explored for potential treatment of NMDA receptor hypofunction in schizophrenia (Bridges et al., 2008; Javitt, 2008). Here we report that the GlyT1 inhibitor, ACPPB (Lindsley et al., 2006; Wolkenberg et al., 2009), promoted functional dopaminergic reinnervation of the 6-OHDA-lesioned dorsal striatum in mature mice and that this action depended on NMDA receptors expressed by dopaminergic neurons.
Materials and Methods
Mice.
Mice were kept according to National Institutes of Health guidelines under a 12 h light/dark cycle with ad libitum access to food and water. Some mice were kept under mild food restriction for up to 4 d (see below, Behavioral tests). All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at Columbia University Medical Center. C57BL/6 mice and Slc6a3Cre mice, used for the conditional inactivation of Grin1, were obtained from The Jackson Laboratory. Grin1loxP mice were kindly provided by the laboratory of Dr. Charles Inturrisi (Cornell University, New York) with the permission of Dr. Susumu Tonegawa. Mice used in this study were backcrossed with C57BL/6 mice for at least six generations. All mice were male and between 3 and 5 months old at the time of 6-OHDA injections.
For the conditional inactivation of Grin1, the gene that encodes the NR1 subunit that is contained in all NMDA receptors, a transgenic line, Slc6a3Cre, was used in which Cre recombinase is driven by the dopamine transporter promoter (Bäckman et al., 2006). To test for potential ectopic Cre expression, we crossed this line with a tdTomato-ROSA reporter mouse line (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J from The Jackson Laboratory). The Grin1loxP mice were bred with the Slc6a3Cre mice to obtain regional knock-out (Slc6a3Cre/wtGrin1loxP/loxP, cNR KO) and control mice (Slc6a3Cre/wtGrin1w/w, DATCre). Animals were genotyped using Grin1loxP and Slc6a3Cre primers. The successful knock-out of NR1 was confirmed by the lack of effect of NMDA on burst spiking in cell-attached whole-cell recordings in living brain slices of the SNpc.
Intrastriatal 6-OHDA injection and GlyT1 inhibitor treatment.
The 6-OHDA injections were performed as described previously (Marti et al., 1997). Mice received an intraperitoneal injection of desipramine (25 mg/kg) to block norepinephrine transporters 30 min before the 6-OHDA injection. Mice were anesthetized with ketamine/xylazine, a small hole was drilled on the left side of the skull at the site of injection, mice were placed in a stereotaxic frame (David Kopf Instruments), and 6-OHDA (Regis Technologies) solution (5 mg/ml, in 0.02% ascorbic acid in 0.9% saline) was infused at an average rate of 0.35 μl/min for 6 min (total dose: 10.5 μg) through a cannula inserted into the left striatum (coordinates relative to bregma: anteroposterior, +0.9 mm; mediolateral, +2.0 mm; dorsoventral, −0.25 mm; Franklin and Paxinos, 1997).
Beginning at 3 weeks post 6-OHDA injection, mice were treated with the GlyT1 inhibitor ACPPB (Wolkenberg et al., 2009), 30 mg/kg suspension in 0.5% methocel saline, by intraperitoneal injections three times a week for a total of 12 injections (Javitt et al., 2004). Untreated mice were injected with the vehicle alone. The plasma clearance Cl of ACPPB is 19.8 ml/min/kg, bioavailability percentage F is 32%, and t(1/2) is 4.2 h. The ancillary pharmacology profile (a Ricerca panel on radioligand binding assays) showed that ACPPB had no significant activity (no displacement >50% at 10 μm) at >200 G-protein-coupled receptors, ion channels, and transporters. Its specificity for GlyT1 versus GlyT2 was confirmed by using a [14C]glycine uptake SPA assay (Lindsley et al., 2006; Wolkenberg et al., 2009).
Immunohistology and quantification.
At 3 weeks or 7 weeks following the 6-OHDA injection mouse brains were fixed by superfusion with 4% buffered paraformaldehyde, postfixed overnight, and cryoprotected in 30% sucrose for 2 d. Coronal cryosections of the striatum and the midbrain were cut at 30 μm thickness. Cryosections were immunostained with primary antibodies against dopamine transporter and tyrosine hydroxylase (TH), respectively (Millipore). Primary antibodies were detected using Alexa Fluor 488-conjugated anti-rat and anti-mouse antibodies (Invitrogen). Images were acquired and analyzed with an Olympus IX81 microscope using MetaMorph software (Molecular Devices). Images of sections immunostained for TH from mice expressing tdTomato (cross of tdTomato-ROSA reporter mice with DATCre mice) were taken on a Leica DM6000 confocal microscope.
To estimate the lesion size in the SNpc and VTA every second section through the midbrain was collected and immunolabeled for TH. Five sections containing SNpc and three to four sections containing VTA (between bregma –2.92 and −3.16 mm; Franklin and Paxinos, 1997) were analyzed. Using MetaMorph software, a region around the area containing SNpc and VTA, respectively, was drawn and the image threshold was set above the fluorescence of the cortex. The size of the area covered by labeled cells was expressed as the percentage of the area size in the unlesioned hemisphere. All mice included in the analysis had SNpc lesions >70% and VTA lesions smaller than 50% (see Fig. 1E,F).
Fiber density in the striatum was assessed in 12–15 adjacent cryosections immunolabeled for the dopamine transporter (between bregma 1.18 and 0.82 mm). In each section, four regions of interest (0.12 mm2) arranged dorsoventrally in the center of the section were analyzed with the most dorsal directly under the corpus callosum and the most ventral region overlapping with part of the spared ventral striatum (see Fig. 2A). The threshold was set in the center of each region so that the fiber bundles that cross the striatum were excluded. The threshold area per region on the lesioned side was expressed as percentage of the threshold area per region in the respective control hemisphere and the average for each region in the 12–15 sections per mouse was calculated (Fig. 2A; see the examples of thresholding). The initial set of untreated and treated C57BL/6 mice was analyzed knowing the experimental condition, but the following ones including all DATCre and cNR KO mice were analyzed blindly.
Whole-cell patch-clamp and cell-attached recordings in brain slice.
Coronal midbrain slices were prepared from TH-eGFP mice (Matsushita et al., 2002) using a vibratome (Leica VT1200) and superfused with artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 26.2 NaHCO3, 10 glucose, 1.8 KCl, 1.2 MgCl2-6H2O, 1.0 NaH2PO4-6H2O, and 2.4 CaCl2. The recording chamber temperature was maintained at 32°C. Whole-cell patch-clamp recordings were made with pipettes (tip resistance 3–4 MOhm) pulled from borosilicate glass (G150F-4; Warner Instruments) on a P-97 Flaming-Brown micropipette puller (Sutter Instruments) and filled with internal solution containing the following (in mm): 120 cesium-methanesulfonate, 11 glucose, 10 HEPES, 5 NaCl, 5 QX314, 2 NaATP, 2 MgATP, 1.1 EGTA, 0.3 NaGTP (pH 7.3, 270–273 mOsm).
Enhanced green fluorescent protein (EGFP)-positive SNpc cells were voltage clamped at −70 mV. EPSCs were evoked by electrical stimulation (double pulse with 50 ms interval, 100–400 μA, 100 μs duration, every 20 s) using a concentric bipolar tungsten electrode (World Precision Instruments) driven by a current-isolated stimulator (Iso-Flex) placed within 200 μm of the recording site. Baseline EPSCs were recorded (Axopatch 200B amplifier; Molecular Devices) for 10 min in picrotoxin (50 μm), then the NMDA current was isolated pharmacologically by bath addition of NBQX (10 μm) in external magnesium concentration of 0.1 mm. The glycine transport inhibitor ALX5407-HCl (0.5 μm; Tocris Bioscience) was coapplied for 10 min, followed by addition of the NMDA receptor antagonist d-APV (10 μm). Peak EPSC amplitudes were measured and sweeps were averaged for the last 5 min of each drug condition. Data were analyzed in Clampfit (Molecular Devices) after a baseline correction and EPSC values are reported as mean ± SEM. A paired Welsh t test showed significance for p < 0.5.
To test for efficient NMDA receptor knock-out, coronal midbrain slices (250 μm) were prepared from cNR KO and DATCre mice (5–15 weeks old). Cell-attached patch recordings were obtained from individual neurons (digitization at 10 kHz, Bessel filter at 5 kHz cutoff). Spontaneous firing frequency was recorded in voltage-clamp mode after a gigaohm seal was established from a command potential of −60 mV. Only cells with a stable baseline activity for 5 min were analyzed for tonic firing. NMDA (20 μm) was bath applied for 100 s. The coefficient of variation (CV) for interspike intervals (ISIs) was calculated as CV = (SD of ISIs)/mean ISI. At the end of each experiment, the seal was ruptured to obtain whole-cell intracellular recordings (internal solution containing (in mm): 100 potassium-gluconate, 20 KCl, 10 HEPES, 10 Na2phosphocreatine, 10 glucose, 4 MgATP, 0.3 NaGTP, pH 7.3, 276 mOsm) for measurements of Ih sag depolarization in current-clamp mode to identify dopaminergic neurons. All drugs were from Sigma Aldrich unless otherwise specified.
Cyclic voltammetry recordings in brain slice.
At the 7 week time point, evoked dopamine release was recorded in coronal brain slices (Schmitz et al., 2001). Striatal brain slices were cut on a vibratome at 250 μm thickness. Recordings were obtained from slices between bregma +1.54 mm to + 0.62 mm, in ACSF containing (in mm): 119 NaCl, 3.0 KCl, 26 NaHCO3, 2.0 CaCl2, 1.2 MgCl2·H2O, 1.0 KH2PO4, and 10 glucose, at 31°C. Cylinder carbon fiber electrodes (5 μm diameter) were used for recordings at three sites in the dorsal, central, and ventral striatum in the lesioned and control hemisphere. For cyclic voltammetry, a triangular voltage wave (−450 to +825 mV at 293 V/s vs Ag/AgCl) was applied to the electrode every 100 ms. Current was recorded with an Axopatch 200B amplifier (Molecular Devices), with a low-pass Bessel filter setting at 10 kHz, digitized at 40 kHz (ITC-18 board; InstruTech) and analyzed with IGOR software (WaveMetrics). Striatal slices were electrically stimulated (1 ms) with a bipolar stimulating electrode placed ∼100 μm from the recording electrode using an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I.). Stimulation magnitude was selected by plotting a current–response curve and selecting the minimum value that reached the plateau. Background-subtracted cyclic voltammograms obtained in dopamine solutions of known concentration served to calibrate the electrodes and to identify the released catecholamine.
HPLC measurements of striatal dopamine content.
Brain blocks (cut between bregma 2 and 0 mm) from untreated and treated mice at 7 weeks were divided into left and right hemispheres and processed for HPLC analysis. Tissue was homogenized in 500 μl of 0.2 m perchloric acid using a tissue dismembrator. Samples were spun in a cooled microcentrifuge at 14,000 × g for 10 min. The supernatant was removed and stored at –80°C for at most 2 d before HPLC-ED of norepinephrine, dopamine, DOPAC, and serotonin content. The mobile phase (adjusted to pH 3.2) contained 5% methanol and (in mm): 45 sodium dihydrogen phosphate, 0.2 EDTA, and 1.4 heptanesulfonic acid. The HPLC system consisted of an ESA Coulochem II with a 5011 analytical cell and a BAS Biophase ODS column. Catecholamine levels were normalized to amount of protein measured using the Pierce BCA protein assay kit (Pierce Biotechnology).
Behavioral tests.
Lateralization of motor behavior in mice was tested 3 and 7 weeks following the 6-OHDA lesion with the “cylinder” and “corridor” tests (Dowd et al., 2005; Grealish et al., 2010). Forelimb use was assessed using the cylinder test in which mice were placed in a glass beaker and were videotaped for 5 min. The number of left and right paw touches while rearing was determined frame by frame. Data were expressed as the number of right paw touches as percentage of the number of all touches.
The corridor test was conducted as detailed in Grealish et al. (2010). Briefly, mice were kept on a restricted diet (1.5 g of regular chow per 2 5 g of mouse per day plus a few sugar pellets) for a total of 4 d. They were tested on mornings of days 3, 4, and 5. Mice were placed in a narrow corridor with a total of 10 pairs of small cups along the left and right walls containing three sugar pellets each. The number of sugar pellets retrieved on the left and right sides were counted during a 10 min interval and expressed as the number of right retrievals (each counted as a single retrieval regardless of the number of pellets retrieved) divided by the number of all retrievals.
Statistics.
Statistical analysis was performed in Prism 5 (GraphPad Software) using one-way ANOVA followed by Tukey–Kramer's post hoc test for comparisons across multiple groups or two-tailed Welch t test for comparison of two groups. The significance level was p < 0.05.
Results
Effects of GlyT1 inhibition on NMDA currents in dopaminergic neurons
We tested whether Glyt1 inhibitors enhanced NMDA receptor currents in dopaminergic SNpc neurons in a manner similar to hippocampal neurons (Bergeron et al., 1998). Whole-cell patch-clamp recordings were made in brain slices prepared from TH-eGFP mice. GFP-positive cells in the SNpc were voltage-clamped at −70 mV and EPSCs were evoked by local electrical stimulation (double pulse with 50 ms interval, applied every 20 s) in the presence of the GABA-A receptor antagonist picrotoxin, 50 μm. After stable EPSC amplitudes were recorded for 10 min (mean peak amplitude: −100 ± 19 pA), the NMDA current was isolated pharmacologically by bath application of the AMPA receptor antagonist NBQX. We then coapplied the GlyT1 inhibitor ALX5407-HCl for 10 min followed by the NMDA receptor antagonist d-APV for another 10 min. In Figure 1A an example of EPSCs recorded in NBQX (gray) and in ALX5407-HCl (black) is shown (average of the last four traces recorded in each drug). The maximal amplitude in response to the first pulse (the response to the second pulse was more variable) is plotted for the entire time course of an experiment in Figure 1B. In all four cells recorded, ALX5407-HCl enhanced the stimulus-evoked NMDA currents (Fig. 1C). The average peak amplitude of the NMDA-mediated current before ALX5407-HCl was −33 ± 8 pA and with ALX5407-HCl −57 ± 11 pA. The average increase in current was 92 ± 29% (Welsh t test, p < 0.05). d-APV application eliminated the evoked currents (mean peak amplitude of −8 ± 1 pA), confirming that they were mediated by NMDA receptors. Thus, application of Glyt1 inhibitors in the SNpc enhanced NMDA receptor activity to a similar extent as in hippocampal neurons (Bergeron et al., 1998).

GlyT1 inhibitor effect on NMDA receptor currents in SNpc neurons and the lesion model. A, Brain slices (from TH-eGFP mice) were perfused first with NBQX (10 μm) in low Mg2+ medium (0.1 mm) to isolate the NMDA-mediated component of the EPSCs, followed by addition of the GlyT1 inhibitor ALX5407 (0.5 μm), and finally with the NMDA receptor antagonist d-APV (50 μm) to confirm the nature of the recorded current. EPSCs (average of the last 4 traces in each condition) recorded in an SNpc neuron (identified by GFP expression) in response to a double stimulus pulse are shown for NBQX incubation (gray) and ALX5407 incubation (black). B, EPSC amplitudes for the first pulse are plotted over the time course of the experiment. C, The average EPSC amplitudes over the last 5 min of the respective drug superfusion are plotted for NBQX and ALX5407 for four cells. D, Time course of the recovery experiments: 6-OHDA was injected intrastriatally, behavior was tested at 3 weeks following the injection, when treatment with the GlyT1 inhibitor ACPPB was initiated. Mice received three intraperitoneal (i.p.) injections per week for the following 4 weeks. At 7 weeks post-injection, behavior was tested again and mice were perfusion-fixed and striatal and midbrain sections were stained for DAT and TH, respectively. E, Fluorescence micrographs of coronal brain sections immunolabeled for DAT in rostrocaudal order through the control striatum (left) and the lesioned striatum (right) 3 weeks following 6-OHDA injections. The dorsal striatum is mostly devoid of DAT-stained fibers, whereas the innervation of the ventral striatum is well preserved. Scale bar, 1 mm. F, Micrographs of coronal brain sections of the SNpc and VTA, immunolabeled for TH, show that on the lesioned side (right) most cell bodies in the SNpc were lost, whereas most cells in the VTA were spared. Scale bar, 100 μm. G, Lesion size, assessed at 7 weeks post-lesion, is expressed as area covered by immunolabel in the lesioned hemisphere in percentage of the control hemisphere (average ± SEM). Only mice that displayed <30% remaining TH label in the SNpc and retained >50% in the VTA (dotted lines) were included in the study. There was no difference in lesion size between the control (n = 6) and the ACPPB-treated group (n = 7, two-tailed Welch t test, p > 0.05).
Intrastriatal 6-OHDA lesions sparing the VTA
Our model for studying striatal dopaminergic reinnervation was to induce dopamine neuron loss in most of the SNpc and to spare the VTA so that remaining axons from the VTA projection to the ventral striatum could be induced to innervate the depleted dorsal striatum. Fig. 1D shows the time course of the experiments and the chemical structure of the Glyt1 inhibitor, ACPPB, a highly selective GlyT1 inhibitor that exhibited no effects on GlyT2 or on >200 receptors, ion channels, and transporters assayed (Lindsley et al., 2006; Wolkenberg et al., 2009). At 3 weeks following intrastriatal 6-OHDA injections into the dorsal striatum, the dorsal striatum was devoid of dopaminergic fibers, while the ventral striatum was relatively spared as seen in Fig. 1E, which shows a series of rostrocaudal sections immunolabeled for the dopamine transporter (DAT) with the intact hemisphere on the left and the lesioned hemisphere on the right. For the reinnervation experiments, lesion size in the SNpc and VTA was determined in midbrain sections immunolabeled for TH at 7 weeks post-lesion (Fig. 1F). Lesion size was expressed as percentage of area covered by TH label compared with the corresponding unlesioned hemisphere. Only mice that displayed a lesion size of at least 70% in the SNpc and <50% in the VTA were included in the study to ensure that there was no significant difference in lesion sizes between the treated and control groups (Fig. 1G).
ACPPB effects on the dopaminergic reinnervation of the dorsal striatum
Mice in the treatment group received intraperitoneal injections of ACPPB (30 mg/kg) three times a week from week 3 through week 7 following the lesion, while control mice received only vehicle. In contrast to 3 weeks post-lesion (Fig. 2A, first row, second section), at 7 weeks DAT-labeled fibers in the dorsal striatum were clearly visible in control mice (Fig. 2A, first row, third section). The fibers appeared to grow from the spared ventral area, as there was an increasing dorsoventral density gradient of label. In mice treated for 4 weeks with ACPPB, the fiber density of the central area in the striatum was clearly enhanced as compared with untreated mice (Fig. 2A, first row, fourth section).

Striatal dopaminergic reinnervation. A, Top row, Micrographs of coronal striatal sections immunolabeled for DAT: on the left a noninjected control hemisphere, on the right examples of lesioned hemispheres. At 3 weeks post-lesion (left), very few stained fibers were left in the dorsal striatum. At 7 weeks post-lesion (middle), fibers had reinnervated some of the dorsal striatum. This reinnervation was clearly denser in ACPPB-treated mice (right). Scale bar, 200 μm. The ROIs that were analyzed for labeled fiber density are indicated in the control hemisphere (left; white ovals). Below the same sections are shown at higher magnification including the ROIs and the threshold mask in yellow used for quantification of the area covered with labeled fibers. The threshold was chosen so that the fiber bundles crossing the striatum were excluded. Scale bar, 200 μm. B, DAT-labeled area (in percentage of control side, average ± SEM) for 3 weeks post-lesion (red), 7 weeks post-lesion in untreated (light blue), and 7 weeks post-lesion in ACPPB-treated mice (blue). Very little reinnervation was present in the dorsal-most region (1). In ROIs 3–4, significantly more area was covered with DAT-positive fibers at 7 weeks (n = 6) than at 3 weeks (n = 4), and treated mice (n = 7) showed a significantly greater DAT-labeled area than untreated mice (one-way ANOVA with Tukey post hoc test, **p < 0.01, ***p < 0.001, only significantly different, and meaningful comparisons are marked). C, Micrographs of putative growth cones found in DAT-labeled sections in the lesioned dorsal striatum. Scale bar, 20 μm.
To quantify the reinnervation of the dorsal striatum, brain sections were analyzed for fiber density in four dorsoventrally arranged regions of interest (ROIs) in the middle of the striatum, as indicated by the ovals in the first panel (control hemisphere) in Fig. 2A. Higher magnifications of the ROIs with the threshold mask for detection of DAT-labeled fibers in yellow are shown below (Fig. 2A, second to fifth row). The resulting area of DAT immunolabel was normalized to the corresponding ROIs of the control hemisphere in the same section (12–15 sections per mouse brain, bregma 1.18–0.82 mm). In the most dorsal region (ROI 1), located just below the corpus callosum, very little reinnervation was present; the same was found for the very medial and lateral edges of the striatum. In the adjacent ROI 2, axon density was not statistically higher, while in the two ventral-most regions (ROIs 3 and 4), axon density was approximately twice as high in treated than in control mice (Fig. 2B). Structures that resembled axonal growth cones, suggesting an innate capacity to sprout, were frequently seen in sections from control and treated mice at 7 weeks post-lesion (Fig. 2C).
ACPPB effects on behavioral recovery
Axonal sprouting does not necessarily provide functional recovery, and we therefore tested the effects of ACPPB treatment on behavior. Two tests that measure lateralized motor behavior caused by unilateral nigrostriatal lesions were administered. In the cylinder test, a mouse was placed into a glass beaker and left and right paw touches while rearing up on the glass wall were counted (Fig. 3A). Mice used the contralateral paw (right paw) less than the one ipsilateral to the lesion (left paw). Forelimb use was expressed as right paw touches divided by the sum of left and right paw touches. Only mice that displayed a right paw preference of <40% at 3 weeks were included in the analysis. The initial average paw preference at 3 weeks was 32 ± 1.4% in the control group and 33 ± 1.3% in the treated group. There was no significant improvement in both groups at 7 weeks (35 ± 1% in the control group and 36 ± 1.6% in the treated group). This result confirms earlier reports (Grealish et al., 2010) that behavior data from the cylinder test correlate relatively weakly with striatal lesion size (R2 = 0.138). Furthermore, the lack of effect of ACPPB treatment in this test might reflect the minimal level of reinnervation in the most dorsal and lateral areas of the striatum that encompass the forepaw motor region (West et al., 1990).

Lateralized behavior. A, Mice were tested at 3 and 7 weeks following 6-OHDA lesions in the cylinder test in which left and right paw touches were counted while rearing (photo). Only mice that used the right paw <40% at 3 weeks post-lesion were included. The box-and-whisker plot (with minimum, maximum, mean, and 25 and 75% quartile) shows that there was no significant difference between 3 (light red and blue) and 7 weeks (dark red and blue) and no difference between control (light and dark blue, n = 12) and ACPPB-treated (light and dark red, n = 9) animals. The plot on the right shows the differences between 3 and 7 weeks for individual mice. There was little improvement and no significant difference (t test > 0.05) between control (blue) and treated mice (red). B, Mice were tested at 3 and 7 weeks following 6-OHDA lesions in the corridor test (photo) in which the number of retrieval of sugar pellets from the left or right side of the corridor were counted. Only mice that retrieved from the right side by <33% at 3 weeks post-lesion were included. There was significant improvement between 3 and 7 weeks in control mice (blue, n = 10) and a more pronounced recovery in ACPPB-treated mice (red, n = 12), which was significantly larger than in controls (one-way ANOVA with post hoc Tukey test, ***p < 0.001). The plot on the right shows the percentage recovery between 3 and 7 weeks for individual mice. There was a significant difference (two-tailed Welsh t test, ***p < 0.001) between control (blue) and ACPPB-treated mice (red).
For the corridor test (Dowd et al., 2005) a mouse was placed in a narrow corridor containing pairs of containers with sugar pellets placed adjacently along the left and right walls. Unilaterally lesioned mice tended to collect sugar pellets mostly from the ipsilateral side (Fig. 3B). Mice were tested in the corridor test at 3 and 7 weeks following the lesion. Only mice that exhibited a lateralized response of <33% retrievals on the right side at 3 weeks were included. Both groups improved between the 3 and 7 week time points and there was a statistically significant difference between untreated and treated mice. At 3 weeks post-lesion, mice exhibited average right-side retrievals of only 16%. At 7 weeks, the untreated mice improved to 32%, while treated mice completely recovered and exhibited equal side preference (50%). Thus, ACPPB-treated mice exhibited both greater dopaminergic reinnervation and a more pronounced behavioral recovery in the corridor test than untreated mice. Although reinnervation of the dorsal striatum at 7 weeks was far from complete, the treated mice exhibited full recovery of behavior, indicating that dopaminergic innervation in the more ventral part of the dorsal striatum is sufficient for decreasing the 6-OHDA-induced lateralization in this test.
Striatal dopamine release, uptake, and tissue content
To assess ACPPB effects on dopaminergic transmission, we recorded dopamine release evoked by a single pulse stimulus in acutely prepared striatal brain slices using cyclic voltammetry. The peak of the dopamine signals chiefly reflects the amount of dopamine released and the half-life of the signal mostly reflects dopamine uptake (Schmitz et al., 2001, 2003). Recordings were obtained from a dorsal, central, and ventral area in the lesioned and the intact hemisphere as indicated in Fig. 4A. As expected from the pattern of reinnervation seen with DAT immunolabel, dopamine release in the dorsal-most region could rarely be detected whereas release in the ventral-most region was similar to the intact hemisphere (data not shown). We measured dopamine release and reuptake in slices obtained from mice that exhibited a sufficient response to the lesion in the behavioral tests (some mice were only tested in the cylinder test (Fig. 4B, black symbols). The data for mice in the corridor test at 3 and 7 weeks post-lesion are shown in Fig. 4C. The amount of evoked dopamine release varied greatly, especially in the ACPPB-treated mice (Fig. 4B). Although there was a slight trend toward higher dopamine release in ACPPB-treated slices, the difference was not statistically significant, nor was the half-life of the signals (which could only be determined for signals with a sufficient peak height). We attributed the release variability to the “patchiness” of the reinnervation, as cyclic voltammetry recordings with carbon fiber electrodes sample an area of only a few square micrometers (Schmitz et al., 2001; Rice and Cragg, 2008). While recordings could typically be acquired from a single slice per animal in the intact and lesioned hemispheres at all three sites with the same electrode, obtaining recordings from more sites in a slice was not possible because of the instability of the electrode kinetics following tissue exposure and the time-dependent decline of tissue health. We therefore measured overall tissue catecholamine levels using HPLC analysis. All mice for this analysis were tested for behavioral lateralization in the corridor test at 3 and 7 weeks following 6-OHDA lesion (Fig. 4D). The tissue blocks for HPLC analysis of catecholamine content comprised both ventral and dorsal striatum (from bregma 1.18 to 0.82 mm). There was a relatively small, but statistically significantly increase in dopamine and DOPAC levels in the striatal tissue of ACPPB-treated compared with vehicle-treated mice (Fig. 4E). The correlation between dopamine tissue levels and behavioral recovery was relatively weak with R2 = 0.115 (Fig. 4F). In comparison, the correlation of the corridor test behavior with lesion size had an R2 of 0.46 (Grealish et al., 2010). Together the cyclic voltammetry and HPLC data suggest that relatively local effects on sprouting (patchiness) rather than a more uniform increase in innervation and dopamine levels may account for the robust behavioral recovery of sensorimotor function observed in ACPPB-treated mice.

Cyclic voltammetry recordings of evoked dopamine release and dopamine tissue content. A, Cyclic voltammetry recordings of electrically evoked dopamine release in living coronal brain slices were performed in control and ACPPB-treated mice at 7 weeks following 6-OHDA lesions. Only mice that showed <40% right paw touches in the cylinder test or <33% right retrievals in the corridor test (C) at 3 weeks post-lesion were included. Recordings were acquired alternately from the unlesioned and lesioned hemisphere in three different locations, dorsal, central, and ventral, as indicated. In almost all lesioned hemispheres, evoked dopamine release could not be detected in the most dorsal location (upper traces), whereas evoked release in the most ventral location was indistinguishable between lesioned and control sides (lower traces). B, Peak amplitudes of evoked dopamine release measured in the central area normalized to the corresponding nonlesioned side from control (triangles n = 7) and ACPPB-treated (circles n = 10) mice are shown in the left scatter plot. Although data were more widely spread for treated mice, the difference was not statistically significant (Welch t test, p > 0.05). The right scatter plot shows the half-life of the same dopamine signals (note: half-life of very small signals could not be determined), which was not significantly different between control and treated mice. C, Behavior data from the mice that were tested in the corridor test before recordings were performed: there was a clear difference in behavioral recovery from 3 to 7 weeks between the three control (gray) and the four ACPPB-treated mice (black, box-and-whisker plot with minimum, maximum, mean, and 25 and 75% quartile, Welsh t test, *p < 0.05). D, Mice used for HPLC measurements of catecholamine content were tested for behavioral recovery in the corridor test (Welsh t test, *p < 0.05). E, Overall tissue content of norepinephrine (NE), 3,4-dihydroxyphenylacetic acid (DOPAC), dopamine (DA), and serotonin (5-HT) in brain blocks containing dorsal and ventral striatum from the lesioned hemisphere was normalized to values obtained from the intact hemisphere. There was an increase (average ± SEM) (one-tailed Welch t test, *p < 0.05) in DOPAC and DA content in brains from ACPPB-treated mice (black columns, n = 6) compared with controls (gray columns, n = 6). F, Behavioral recovery is plotted versus dopamine tissue content for control (gray triangles) and treated (black dots) mice. The correlation coefficient was R2 = 0.115.
ACPPB effects in conditional NMDA receptor knock-out mice
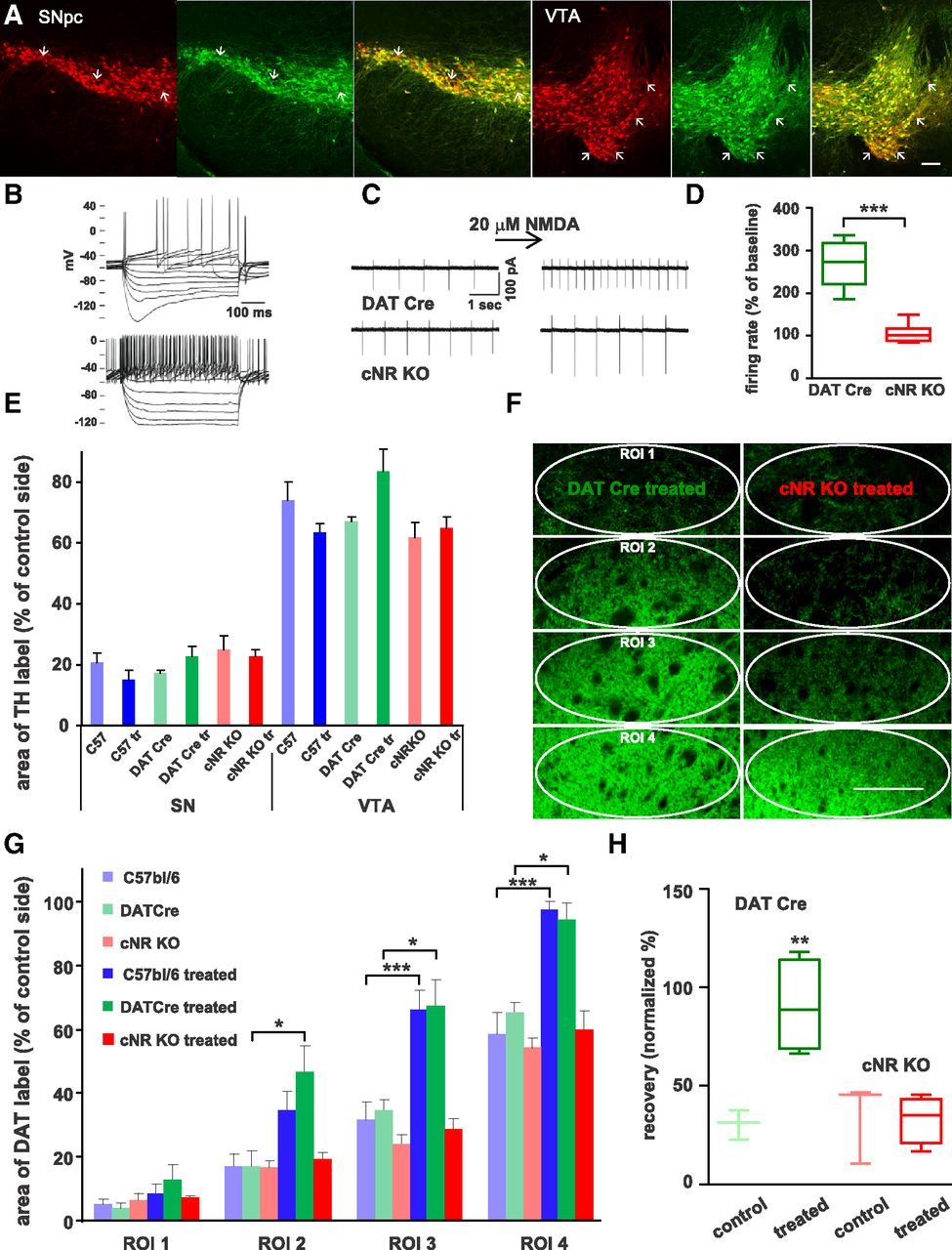
To test whether ACPPB effects on axonal sprouting and behavioral recovery in 6-OHDA-lesioned mice were mediated by NMDA receptors in dopamine neurons as postulated, we bred a mouse line lacking NMDA receptors selectively in dopaminergic neurons. Slc6a3Cre mice (Bäckman et al., 2006) were used for the conditional inactivation of Grin1. As a control comparison for the conditional knock-outs (Slc6a3Cre/wtGrin1loxP/loxP, cNR KO) DATCre mice (Slc6a3Cre/wtGrin1w/w) were used, as heterozygous Slc6a3Cre mice have been reported to express slightly reduced levels (83%) of the dopamine transporter (Bäckman et al., 2006), which could interfere with 6-OHDA toxicity. The Slc6a3Cre mouse line was reported to show little ectopic Cre expression (Bäckman et al., 2006; Tritsch et al., 2012). Similarly, we found little ectopic expression in the SNpc and only slightly higher ectopic expression in the VTA in a cross of the Slc6a3Cre mouse line with a tdTomato-ROSA reporter mouse line (Fig. 5A). Thus, although a small population of other cell types may show Cre expression in crosses with this mouse line, the majority of Cre recombination was found in dopaminergic cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Conditional NR KO mice. A, Sections of SNpc and VTA (3 sections each) from a mouse derived from crossing tdTomato-ROSA reporter mice with the DATCre mouse line (red) were immunolabeled for TH (green) to check for ectopic expression of Cre (merge: right sections). There was some ectopic expression of Cre in both SNpc and VTA (arrows), but the percentage was relatively low (average of 8% for SNpc and 9% for VTA, n = 7 sections in each group). Scale bar, 100 μm. B, Electrophysiological recordings in cNR KO mice derived from crossings of Grin1loxP mice with Slc6a3Cre mice: whole-cell recordings were obtained from neurons in brain slices containing SNpc; the presence of Ih currents characteristic for dopamine neurons (upper traces) in contrast to putative GABA cells (lower traces) was used to identify dopamine neurons. C, Spontaneous firing was recorded in cell-attached mode: in DATCre mice, superfusion with 20 μm NMDA elicited an increase in firing rate (upper traces), in contrast, no such increase was seen in cNR KO mice (lower traces). D, Firing rate increased in DATCre mice (n = 5, green) in response to NMDA by ∼160%, while there was no change in cNR KO mice (red, n = 6, box-and-whisker plot with maximum, minimum, mean, and 25 and 75% quartile, Welsh t test, ***p < 0.001). E, To test effects of ACPPB on striatal reinnervation, only mice with lesion sizes in the SNpc >70% and <50% in the VTA were included in the analysis. There was no significant difference in the lesion sizes in the SNpc or VTA between the groups (average ± SEM, ANOVA, p > 0.05). F, Fluorescence micrographs of coronal brain sections through the striatum immunolabeled for DAT from a DATCre mouse and a cNR KO mouse both treated with ACPPB with the ROIs analyzed indicated (ovals). Scale bar, 200 μm. Striatal reinnervation in treated cNR KO mice was far less dense than in treated DATCre mice. G, Bar graph (average ± SEM) of the DAT-labeled area (in percentage of control side) for DATCre mice (controls: light green, n = 5; treated: dark green, n = 4), cNR KO mice (controls: light red, n = 5; treated: red, n = 7), and also for C57BL/6 mice (light blue, n = 6 and dark blue, n = 7; data from Fig. 2B) for comparison. ACPPB treatment had no effect on the innervation density in cNR KO mice (one-way ANOVA with post hoc Tukey test, *p < 0.05, ***p < 0.001; note: only meaningful and statistically significant comparisons are labeled). H, ACPPB-treated DATCre mice showed pronounced behavioral recovery in the corridor test (dark green, n = 4), whereas the untreated DATCre (light green, n = 3) and cNR KO mice (light red, n = 3), as well as the treated cNR KO mice (red, n = 5), showed little recovery (box-and-whisker plot, ANOVA with post hoc Tukey test, **p < 0.01).
Cell-attached patch-clamp recordings of firing frequency in SNpc neurons in ventral midbrain slices confirmed that NR1 was deleted in cNR KO mice, as superfusion with NMDA had no effect on firing rates, in contrast to DATCre control mice in which firing rates increased on average to 280% in response to NMDA (Fig. 5B–D). The mice did not show any gross histological abnormalities with respect to dopaminergic innervation of the ventral or dorsal striatum: DAT-stained fiber density assessed as area of coverage in dorsal striatum and nucleus accumbens shell and core appeared normal (data not shown).
cNR KO and DATCre mice received unilateral, intrastriatal 6-OHDA injections and were injected intraperitoneally for 4 weeks with ACPPB or vehicle. Mice were tested in the corridor test at 3 and 7 weeks post-lesion and lesion size was determined in the SNpc and VTA (Fig. 5E). The dopaminergic fiber density in the striatum in untreated and treated DATCre mice, respectively, was similar to the density reported in untreated and treated C57BL/6 mice. In contrast, ACPPB treatment failed to increase axonal area of coverage in cNR KO mice, whereas the area of coverage in untreated cNR KO mice was comparable to untreated C57BL/6 and DATCre mice (Figs. 5F,G). Thus, although there was some regrowth of dopamine fibers into the dorsal striatum in cNR KO mice, ACPPB treatment did not increase the density of the axon network as it did in DATCre and C57BL/6 mice.
The amount of behavioral recovery could not be determined in all lesioned cNR KO mice, as 5 of 16 mice (∼30%) mice did not perform the test, i.e., they either remained in one corner of the corridor or ran up and down the corridor while rearing extensively and ignoring the sugar pellets. This behavior may reflect a relatively high level of anxiety as displayed by another cNR KO mouse line (Zweifel et al., 2011). In addition, 2 of 9 (∼20%) DATCre mice failed to perform the test, while for C57BL/6 mice the failure rate was ∼2%. In mice that performed the test, ACPPB treatment caused almost complete behavioral recovery in DATCre mice, but had no effect in cNR KO mice (Fig. 5H), indicating that NMDA receptors in dopamine neurons mediate the behavioral recovery caused by ACPPB treatment.
Discussion
We found that following unilateral 6-OHDA-induced lesions that spared the mesoaccumbens projection, an inhibitor of GlyT1 promoted dopaminergic reinnervation of the dorsal striatum and normalized 6-OHDA-induced lateralization of sensorimotor behavior. Both effects were dependent on the presence of NMDA receptors in dopamine neurons as revealed by cNR KO mice. If functional sprouting could be induced in dopamine axons in areas that are spared from denervation in Parkinson's disease (i.e., the caudate nucleus, medial portions of the putamen, and the nucleus accumbens), GlyT1 inhibitors might point toward future therapeutic treatments for patients.
The 6-OHDA unilateral lesion model and regenerative sprouting
A potential for recovery from partial lesions by dopaminergic sprouting was suggested in early studies using 6-OHDA-injections (Dravid et al., 1984; Onn et al., 1986). It is known that toxins can cause loss of TH expression rather than actual cell loss, and thus a recovery revealed by TH staining may simply constitute re-expression (Tatton et al., 1990). While we cannot completely rule out such a possibility, we think this is unlikely, because we used immunolabel for DAT in the striatum, which is more stable than TH following toxin injections (Przedborski et al., 1995), and second, because only mice with a lesion size in the SNpc of >70% (assessed by TH staining) were included. To explain our data with a loss and subsequent reinstatement of TH/DAT expression, one would have to assume that dopaminergic markers remained downregulated in cell bodies, but were upregulated in the distal axons by 7 weeks post-lesion.
Studies investigating recovery from intranigral 6-OHDA lesions that were smaller than 70% reported that new axons grew through the median forebrain bundle to reinnervate the striatum within 16 weeks post-lesion (Finkelstein et al., 2000; Stanic et al., 2003). This is unlikely to have occurred under the conditions used here, as nigral lesions were larger than 70%, and reinnervation of the dorsal striatum was assessed at a much earlier time point (7 vs 16 weeks post-lesion). The dorsoventral density gradient of DAT-labeled axons we observed is suggestive of sprouting stemming from spared fibers in the ventral striatum, a phenomenon reported in an earlier study using retrograde tracer injections (Hansen et al., 1995). The sprouting fibers could originate from VTA neurons, from a few SNpc neurons that were spared by the 6-OHDA lesion, or both.
GlyT1 effects and the role of NMDA receptors
We found that the growth-promoting effect of the GlyT1 inhibitor ACPPB was dependent on the presence of NMDA receptors in dopaminergic neurons. There was a baseline reinnervation of the 6-OHDA-depleted striatum in cNR KO mice; however, that was not different from DATCre control mice, suggesting that the activity of axonal NMDA receptors may enhance axon branching more than axon elongation. The effects could be mediated by somatodendritic NMDA receptors or presynaptic, axonal NMDA receptors. Although there is at present no conclusive evidence for NMDA receptor expression on dopaminergic axons, several findings support their existence: dopamine release-regulating NMDA receptors appear to be present in synaptosomes derived from adult striatum (Krebs et al., 1989; Desce et al., 1992), an electron microscopy study found NMDA receptor immunolabel in dopaminergic axons and terminals in the adult ventral striatum (Gracy and Pickel, 1996), and we reported immunolabel of axons and axonal growth cones in postnatal cultures of mixed SN/VTA neurons (Schmitz et al., 2009). Axonal NMDA receptor expression in dopamine neurons may be developmentally regulated, as in other neuron types (Herkert et al., 1998; Lien et al., 2006; Corlew et al., 2007; Wang et al., 2011). It is also possible that presynaptic NMDA receptor expression is upregulated in sprouting dopamine axons following a 6-OHDA lesion, similar to another glutamatergic marker with developmentally decreasing expression in dopamine neurons, the vesicular glutamate transporter 2 (VGlut2; Dal Bo et al., 2008). Recently, it has been proposed that d-serine acts as the endogenous coagonist at synaptic NMDA receptors, whereas glycine acts at extrasynaptic NMDA receptors. In the presence of GlyT1 inhibitors, however, it was reported that enhanced glycine levels also caused activation of synaptic NMDA receptors that are more sensitive to d-serine, so that the observed GlyT1 inhibitor effects could be caused by both synaptic and extrasynaptic NMDA receptors (Gray and Nicoll, 2012; Papouin et al., 2012).
The finding that NMDA receptor activity in dopamine neurons promotes axonal sprouting poses the question of the source of the glutamatergic input. One possibility is that glutamatergic cortico- and thalamo-striatal projections affect dopaminergic axons that tend to make synapses onto medium spiny neurons in close proximity to these glutamatergic terminals, often onto the same spine (Bouyer et al., 1984; Moss and Bolam, 2008). A second possibility is that glutamate-releasing astrocytes play a role (Jourdain et al., 2007; Parpura and Zorec, 2010). A third is that dopamine axons release glutamate themselves as a growth-promoting feedback signal. Glutamate corelease from dopaminergic cells has been shown to occur in vitro (Sulzer et al., 1998) and in vivo (Chuhma et al., 2004; Hnasko et al., 2010; Stuber et al., 2010; Alsiö et al., 2011). As mentioned above, the expression of VGlut2 is relatively high in the dopaminergic VTA projection in early development, decreases in the adult brain, and is enhanced following 6-OHDA injury (Dal Bo et al., 2008; Bérubé-Carrière et al., 2009), so glutamate corelease from dopamine axons is a possible candidate.
GlyT1 inhibitors and Parkinson's disease
At the time of onset of motor signs in Parkinson's disease ∼30% of the dopaminergic SNpc neurons are already lost (Cheng et al., 2010). This may reflect a high level of redundancy in the dopaminergic innervation of the striatum (Matsuda et al., 2009), but could also indicate that a certain degree of compensatory sprouting occurs (Lee et al., 2008). In contrast, dopaminergic fiber loss may actually precede cell death and there may be already ∼50% fiber loss in the putamen at the onset of motor signs (Cheng et al., 2010). The most studied approach to induce compensatory sprouting in dopamine neurons is delivery of glial derived neurotrophic factor (GDNF), which has been observed to induce sprouting in a Parkinson's disease patient (Love et al., 2005). GDNF is thought to have both a survival- and a growth-promoting action (Rosenblad et al., 2000; Kirik et al., 2004; Rangasamy et al., 2010). While GDNF treatment prevents degeneration and/or provides functional recovery in toxin-based (MPTP and 6-OHDA) models in rodents and monkeys, it was recently reported to have little effect in a mouse model of Parkinson's disease featuring overexpression of α-synuclein (Decressac et al., 2011).
If functional sprouting could be induced in dopamine axons that are spared in Parkinson's disease, GlyT1 inhibitors would be potential candidates for therapy; similarly, they could promote the establishment of functional connections in transplantation approaches. GlyT1 is expressed in brain areas with high densities of glycinergic synapses in brainstem and cerebellum as well as in the forebrain in glial and neuronal cells (Zafra et al., 1995; Cubelos et al., 2005). Analysis of glia-specific GlyT1 knock-out mice indicated that GlyT1 is important for glycinergic transmission in early development but not in the adult brain (Eulenburg et al., 2005, 2010). Thus, GlyT1 inhibitors in adult mice mostly act to modulate glycine-binding to NMDA receptors in the forebrain and do not interfere with glycinergic transmission. Several positron emission tomography imaging studies using various radiotracers for GlyT1 in rodents, rhesus monkeys, and humans report an intermediate signal in caudate/putamen: weaker than in pons, thalamus, and cerebellum, but stronger than in multiple cortical areas (Herdon et al., 2010; Hamill et al., 2011; Borroni et al., 2013; Martin-Facklam et al., 2013). GlyT1 inhibitors are being developed by the pharmaceutical industry, mostly intended for treatment of cortical NMDA receptor hypofunction in schizophrenia (Javitt, 2008; Pinard et al., 2010). A role for NMDA receptors in Parkinson's disease has been mostly discussed with respect to levopoda-induced dyskinesias that are ameliorated by weak NMDA receptor antagonists. These effects may, however, rather occur through actions on other receptors than NMDA receptors (Calabresi et al., 2010). In a recent pilot study, Parkinson's patients treated with d-serine, which binds to the NMDA receptor glycine site, showed improvement as assessed by the Unified Parkinson's Disease Rating Scale and the Positive and Negative Symptoms Scale after 6 weeks of treatment (Gelfin et al., 2012), suggesting that GlyT1 inhibitors may be beneficial as adjuvant treatment in Parkinson's disease (Heresco-Levy et al., 2013).
Footnotes
- Received June 26, 2012.
- Revision received September 10, 2013.
- Accepted September 13, 2013.
This work was supported by the JPB Foundation, the Michael J Fox Foundation, the National Parkinson's Foundation, the Parkinson's Disease Foundation (PDF), and the Udall Center of Excellence at Columbia University. Z.K. received a summer student fellowship from PDF. We are grateful to Tinmarla Oo, Tatyana Kareva, and Robert Burke for training and advice in stereotactic 6-OHDA injections; to Shane Grealish and Eilis Dowd for advice on behavioral tests; to Eugene Mosharov for help with HPLC; and to Paul Witkovsky for valuable comments on this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Yvonne Schmitz or David Sulzer, Department of Neurology, 650 W 168th Street BB308, New York, NY 10032. ys290{at}columbia.edu or ds43{at}columbia.edu
- Copyright © 2013 the authors 0270-6474/13/3316778-12$15.00/0