
Abstract
The bed nucleus of the stria terminalis (BNST) is thought to generate anxiety-like states via its projections to autonomic and neuroendocrine regulatory structures of the brain. However, because most BNST cells are GABAergic, they are expected to inhibit target neurons. In contrast with this, infusion of calcitonin gene-related peptide (CGRP) into BNST was reported to potentiate anxiety while activating BNST targets. The present study aimed to shed light on this paradox. The CGRP innervation of BNST originates in the pontine parabrachial nucleus and targets its anterolateral sector (BNST-AL). Thus, we investigated the effects of CGRP on BNST-AL neurons using patch recordings in vitro in male rats. CGRP did not alter the passive properties of BNST-AL cells but increased the amplitude of IPSPs evoked by stimulation of the stria terminalis (ST). However, IPSP paired-pulse ratios were unchanged by CGRP, and there was no correlation between IPSP potentiation and variance, suggesting that CGRP acts postsynaptically. Consistent with this, CGRP hyperpolarized the GABA-A reversal of BNST-AL cells. These results indicate that CGRP increases ST-evoked GABA-A IPSPs and hyperpolarizes their reversal potential through a postsynaptic change in Cl− homeostasis. Overall, our findings suggest that CGRP potentiates anxiety-like behaviors and increases neural activity in BNST targets, by inhibiting BNST-AL cells, supporting the conclusion that BNST-AL exerts anxiolytic effects.
Introduction
The bed nucleus of the stria terminalis (BNST) is a poorly understood brain structure thought to play a critical role in fear and anxiety. Indeed, electrolytic (Gewirtz et al., 1998; Sullivan et al., 2004) or neurotoxic (LeDoux et al., 1988; Hammack et al., 2004; Duvarci et al., 2009) BNST lesions as well as reversible inactivation of BNST (Walker and Davis, 1997) impair the expression of classically conditioned fear responses to contexts. In keeping with this, BNST projects to hypothalamic (Prewitt and Herman, 1998; Dong et al., 2001; Dong and Swanson, 2006) and brainstem (Sofroniew, 1983; Holstege et al., 1985; Moga et al., 1989; Sun and Cassell, 1993) structures that generate the neuroendocrine, behavioral, and cardiovascular correlates of fear and anxiety.
Currently, it is unclear whether activation or inhibition of BNST is required to produce anxiety-like behaviors. On the one hand, the fact that lesion or inactivation of BNST produces anxiolytic effects suggests that increased BNST activity generates negative emotional states. On the other hand, BNST is mainly comprised of GABAergic neurons (Esclapez et al., 1993; Hur and Zaborszky, 2005; Poulin et al., 2009) and therefore presumably exerts inhibitory effects on its targets. In contrast with this, however, Sink et al. (2011) reported that intra-BNST injections of calcitonin gene-related peptide (CGRP) augment acoustic startle while increasing activity in targets of BNST. The present study was undertaken to address this apparent contradiction.
CGRP is a 37 amino acid peptide involved in autonomic functions and pain processing (for review, see van Rossum et al., 1997). The sole CGRP input to BNST originates in the pontine parabrachial nucleus (Shimada et al., 1985), which projects heavily to the anterolateral portion of BNST (BNST-AL) (Gustafson and Greengard, 1990; Alden et al., 1994; Dobolyi et al., 2005). Thus, to shed light on how BNST regulates anxiety, we studied the effect of CGRP on BNST-AL neurons recorded with the patch method in brain slices in vitro.
Materials and Methods
Slice preparation.
Procedures were approved by the Institutional Animal Care and Use Committee of Rutgers University, in compliance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services). Male Lewis rats (4–7 weeks old) were anesthetized with avertin (300 mg/kg, i.p.), followed by isoflurane. After abolition of reflexes, they were perfused with an ice-cold solution containing (in mm) as follows: 126 choline chloride, 2.5 KCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 10 glucose. The brains were sliced with a vibrating microtome (300 μm) while submerged in the same solution. The slices were then kept in an oxygenated chamber containing artificial CSF (aCSF; as above except for the substitution of 126 mm NaCl for choline chloride, pH 7.2, 300 mOsm). The temperature of the chamber was kept at 34°C for 20 min and then returned to room temperature. One hour later, slices were then transferred to a recording chamber perfused with oxygenated aCSF at 32°C (6 ml/min).
Electrophysiological recordings.
Whole-cell recordings of BNST-AL neurons were obtained under visual guidance using 5–8 mΩ pipettes pulled from borosilicate glass capillaries. The intracellular solution contained (in mm): 130 K-gluconate, 10 HEPES, 10 KCl, 2 MgCl2, 2 ATP-Mg, and 0.2 GTP-tris(hydroxymethyl)aminomethane, pH 7.2, 280 mOsm. The liquid junction potential was 10 mV with this solution. However, membrane potential (Vm) values mentioned below were not corrected for the junction potential. We used an Axoclamp-2B amplifier (Molecular Devices) and digitized the data at 10 kHz with a Digidata-1200 interface controlled by pClamp-8.1 (Molecular Devices).
To characterize the electroresponsive properties of the cells, we applied series of current pulses (±10 pA increments; 500 ms; 0.2 Hz) from −55 and −70 mV; this revealed that all of the neurons recorded in this study correspond to the previously described Type 1 or Type 2 neurons (Hammack et al., 2007; Rodríguez-Sierra et al., 2013), with no difference in CGRP responsiveness between them. A pair of tungsten-stimulating electrodes (intertip spacing, 200 μm) was placed in the stria terminalis (ST; see Fig. 1A) and used to deliver brief current pulses (0.1 ms; 0.03 Hz). Cells were kept at −55 mV unless stated otherwise. When testing the effects of CGRP on ST-evoked EPSP amplitudes, stimulation intensity (0.1–0.8 mA) was adjusted to obtain the highest subthreshold response amplitudes. When testing CGRP effects on IPSPs, the stimulation intensity was adjusted to elicit IPSPs of ∼5 mV amplitude so that the IPSP peak would not approach the GABA-A reversal potential. Input resistance (Rin) was calculated as the average voltage response to −10 pA current injections during the stimulation protocol. A 10 min baseline recording was obtained before CGRP application. CGRP was then applied for 10 min. Before versus after peptide comparisons were made using responses obtained 5–10 min before versus 15–20 min after onset of CGRP application (separate averages of 10 ST-evoked responses). Antagonists were added to the perfusate solution 15 min before CGRP application and were present throughout the recordings.
Drugs.
CGRP (rat), SB-268262 (N-methyl-N-(2-methylphenyl)-3-nitro-4-(2-thiazolylsulfinyl)-benzamide), picrotoxin, CNQX disodium salt, and (±)-3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP) were obtained from Sigma. CGRP8–37 was obtained from Tocris Bioscience.
Data analysis.
Data were analyzed offline using Clampfit version 9.2. All data are reported as mean ± SEM. SEM calculations were modified for repeated designs as described by Cousineau (2005). For statistical analyses, we conducted one-way ANOVA with Tukey's Honestly significant difference paired post hoc tests as well as paired t tests.
Results
Effect of CGRP on the electroresponsive properties and synaptic responses of BNST-AL cells
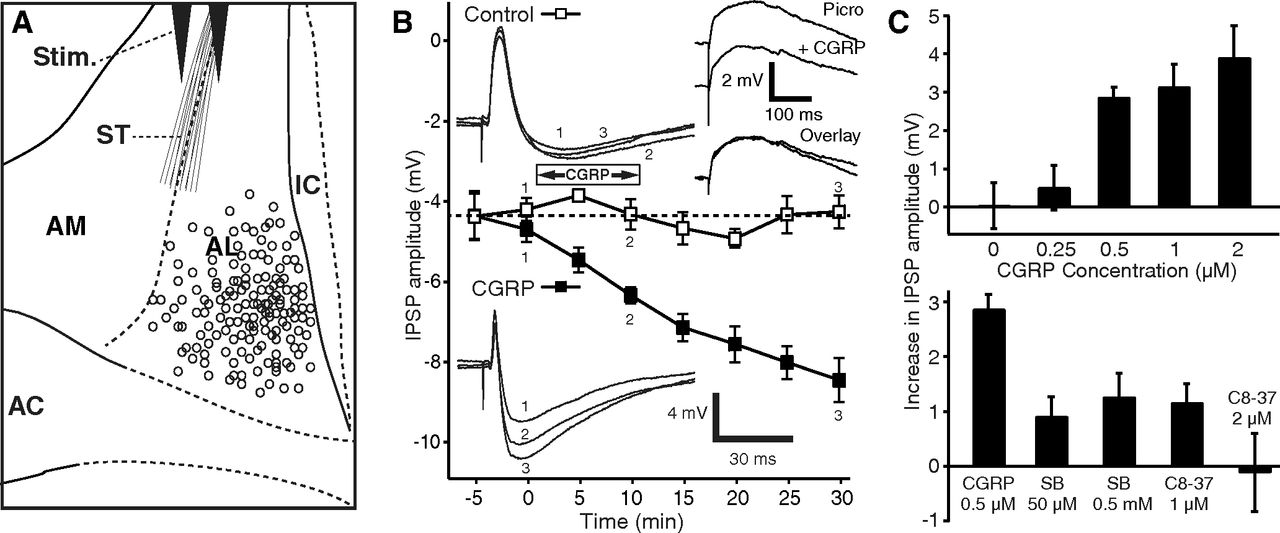
We obtained patch recordings of 184 BNST-AL cells (Fig. 1A) that had stable resting potentials and generated overshooting action potentials upon depolarization. We first tested whether CGRP (0.5–2 μm) alters the passive properties, firing pattern, or spike characteristics of BNST-AL cells but found no effect (Table 1). Because it was reported that CGRP increases EPSP amplitudes in central amygdala neurons at their resting potential (Han et al., 2005, 2010), we next examined the impact of CGRP on ST-evoked EPSPs in the presence of picrotoxin (100 μm). Picrotoxin completely abolished ST-evoked IPSPs in 90% of tested neurons (36 of 40), allowing us to examine the influence of CGRP (1 μm) on isolated EPSPs. However, EPSP amplitudes were unaffected by CGRP (difference of −0.7 ± 0.6 mV, n = 12, p = 0.25; Fig. 1B, top right inset).

CGRP potentiates ST-evoked IPSPs. A, Scheme showing stimulating (Stim.) and recording (circles) sites. B, Time course of CGRP effect on IPSPs (solid squares represent CGRP 0.5 μm; empty squares represent control cells). CGRP steadily increases ST-evoked IPSP amplitudes, and this effect outlasts the period of CGRP application. Insets on left, Examples of ST-evoked responses in a control cell (top) and one exposed to CGRP (bottom). Inset on top right, Examples of EPSPs isolated with picrotoxin (100 μm) before versus after a 10 min application of CGRP (1 μm). C, Top, Dose dependency of CGRP effect on IPSPs. After CGRP application (15 min), IPSP amplitudes increased to 260 ± 49% for 2 μm (n = 9, p = 0.002), 174 ± 22% for 1 μm (n = 4, p = 0.01), and 170 ± 15% for 0.5 μm (n = 5, p = 0.0005) of preapplication values. At a concentration of 250 nm CGRP had no significant effect (n = 8, p = 0.41). Eight cells were tested with no CGRP application. C, Bottom, Two CGRP receptor Type 1 antagonists reduced the effect of CGRP, compared with when CGRP (500 nm, n = 5) was applied alone (SB268262, 50 μm, n = 12, p < 0.001; 500 μm, n = 7, p < 0.01; CGRP8–37, 1 μm, n = 4, p < 0.001; 2 μm, n = 5, p = 0.01). AC, Anterior commissure; IC, internal capsule.
Effect of CGRP (1 μm) on the electroresponsive properties of BNST-AL neuronsa
In contrast, in the absence of picrotoxin, CGRP significantly increased the amplitude of ST-evoked IPSPs (Fig. 1B, solid squares and bottom inset) in a dose-dependent fashion (n = 26; ANOVA, F(4,29) = 5.48, p = 0.02; Fig. 1C, top). The potentiation of ST-evoked IPSPs outlasted the CGRP application period by >20 min (Fig. 1B). Because the increase in IPSP could have resulted from the gradual equilibration of the intracellular Cl− concentration with that of the pipette solution, we examined whether the IPSP amplitudes increased spontaneously over time (n = 8), with no peptide application. However, no significant time-dependent changes were observed (Fig. 1B, empty squares, inset; n = 8, p = 0.35). Furthermore, two different CGRP Type 1 receptor antagonists, SB268262 (n = 19) and CGRP8–37 (n = 9), reduced or completely abolished the IPSP potentiation produced by CGRP (Fig. 1C, bottom).
Mechanisms underlying the potentiation of ST-evoked IPSPs by CGRP
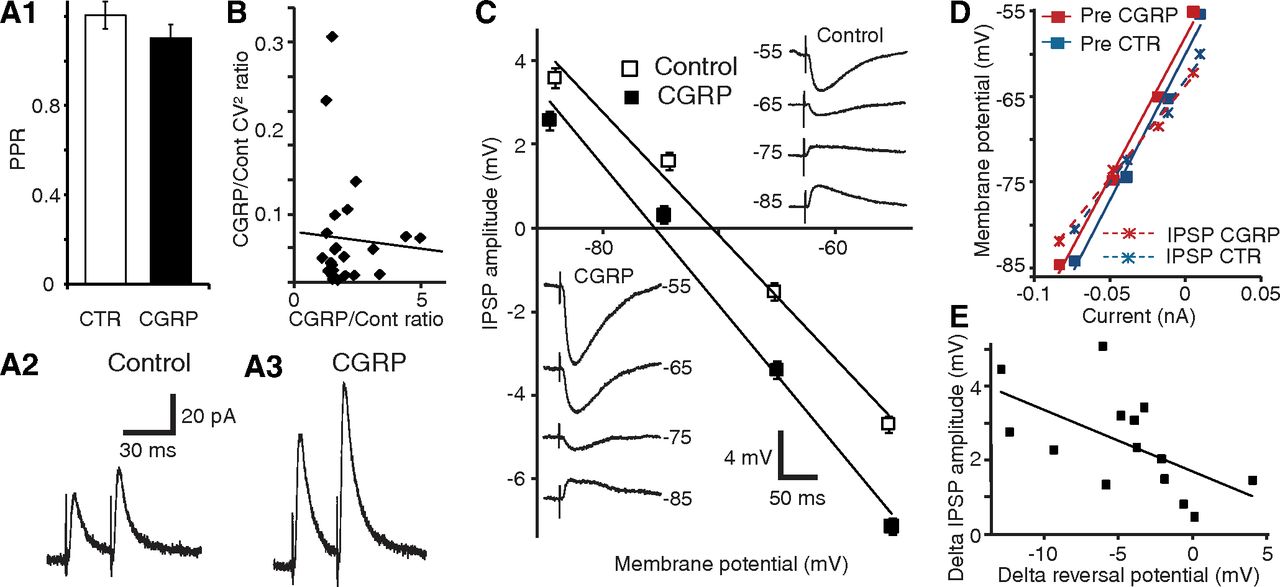
To investigate whether the increase in ST-evoked IPSPs produced by CGRP is dependent on presynaptic or postsynaptic mechanisms, we conducted three analyses. First, we compared the paired-pulse ratio (PPR) of ST-evoked IPSCs before versus after CGRP application (Fig. 2A). In such analyses, two stimuli of equal intensity are applied in brief succession (50 ms), leading to an enhancement or reduction of the response elicited by the second stimulus. Changes in PPR are commonly thought to reflect alterations in transmitter release probability (Creager et al., 1980; Manabe et al., 1993). PPR tests were performed in the presence of the glutamate receptor antagonists CNQX (10 μm) and CPP (10 μm). In control conditions, a small average paired-pulse facilitation was observed, but it did not reach significance (p = 0.32). Addition of CGRP (1 μm) did not alter the PPR of ST-evoked IPSPs (Fig. 2A; PPR difference of 0.1 ± 0.1; n = 6; p = 0.43), suggesting that CGRP acts postsynaptically to potentiate the IPSPs.

CGRP potentiates ST-evoked IPSPs through a postsynaptic mechanism. A1, PPR before and 15 min after CGRP application (n = 6). A2, A3, Average traces from the same cell before (2) and after (3) CGRP application. Despite strong ST-evoked IPSC potentiation, there is no difference in the PPR. B, CV2 analysis. Ratio of CV2 during CGRP to control (y-axis) versus ratio of CGRP to control IPSP amplitudes (x-axis, n = 18). C, CGRP causes a negative shift of the GABA-A reversal potential. CNQX (10 μm) and CPP (10 μm) present throughout. Linear fits of IPSP amplitude (y-axis) as a function of Vm (x-axis) before (empty squares) and after (solid squares) CGRP (mean ± SEM, 14 cells). Insets, Average IPSP amplitude of a representative cell before (top right) and after (bottom left) CGRP. D, Linear fits of Vm (y-axis) versus current (x-axis) before the ST stimulus (solid squares) and at the IPSP peak (×), before (blue) and after (red) CGRP. E, CGRP-induced changes in IPSP amplitudes (y-axis) and GABA-A reversal potentials (x-axis) are correlated.
Second, we studied IPSP variability using the data obtained in the dose–response experiments of Figure 1C. This variability is known to reflect the probabilistic process underlying transmitter release and can be estimated by computing the coefficient of variation (CV, SD/mean). By plotting the ratio of experimental to control CV2 against the ratio of experimental to control response amplitudes, the dependence of presynaptic versus postsynaptic function can be determined (Bekkers and Stevens, 1990; Manabe et al., 1993). In these prior studies, a positive correlation between the two was shown to reflect a presynaptic mechanism, whereas a horizontal regression is indicative of a purely postsynaptic action. We found no significant relationship between CV2 and IPSP amplitudes (Fig. 2B; r = −0.06, p = 0.77, n = 14), again pointing to a postsynaptic locus of CGRP action.
Third, we tested whether CGRP alters the reversal potential (EGABA-A) of ST-evoked IPSPs in the presence of the glutamate receptor antagonists CNQX (10 μm) and CPP (10 μm). CGRP (1 μm) caused a significant negative shift of EGABA-A (Fig. 2C; difference 4.31 ± 1.25 mV; control, −72.66 ± 1.9 mV; CGRP, −76.97 ± 1.99 mV; p = 0.004, n = 14). Importantly, this effect was not associated with a change in the Rin drop caused by the IPSPs (Fig. 2D; −11 ± 11 mΩ; p = 0.33), suggesting that the CGRP-induced augmentation in IPSP amplitude is largely dependent on an increased Cl− driving force. Indeed, there was a significant correlation between the CGRP-induced changes in IPSP amplitudes and EGABA-A (Fig. 2E; n = 14, r = −0.59, p = 0.03). In contrast, in a different sample of control cells without CGRP application, no time-dependent shift in GABA-A reversal potential was observed (−0.1 ± 0.9 mV, n = 14, p = 0.89).
If CGRP acts postsynaptically to enhance IPSPs by increasing the Cl− driving force, one would expect manipulations that interfere with the cells' Cl− homeostasis to disrupt CGRP's effects. A major regulator of the intracellular Cl− concentration in neurons (for review, see Kaila, 1994) is the potassium-chloride cotransporter (KCC), which mediates Cl− extrusion (Misgeld et al., 1986; Thompson et al., 1988). Consistent with this, VU-0240551 (40 μm), a selective KCC2 blocker, depolarized EGABA-A by 4.1 ± 1.1 mV (n = 9, p = 0.007) and prevented the effects of CGRP on IPSP amplitudes (0.5 ± 0.5 mV; n = 7, p = 0.31) and EGABA-A (−0.3 ± 0.7 mV; p = 0.72; Fig. 3).

{kind=link}
{kind=link}
{kind=link}
KCC2 antagonist VU-0240551 (VU) prevents the effects of CGRP on IPSP amplitudes and reversal potentials. IPSP amplitude (y-axis) as a function of membrane potential (x-axis) in cells recorded in the absence (empty diamonds) versus the presence (filled diamonds) of the KCC2 antagonist VU-0240551 (40 μm). Top right inset, Amplitude of IPSPs before (empty bars) versus after CGRP (filled bars) in the absence (left) versus presence (right) of VU-0240551. Bottom left inset, GABA-A reversal potential before (empty bars) versus after CGRP (filled bars) in the absence (left) versus presence (right) of VU-0240551.
An alternative interpretation for CGRP effects, namely, that it results from an enhancement of GABA-B IPSPs, appears unlikely for the following reasons. First, as mentioned just above, inhibiting KCC2 blocked the CGRP effect. Second, our IPSP measurements were performed at the peak of the GABA-A IPSPs, ∼30 ms from response onset, well before the development of GABA-B responses in other cell types. Third, most BNST-AL cells lacked overt GABA-B responses, with picrotoxin abolishing ST-evoked IPSPs in 36 of 40 cells. Last, in the rare BNST-AL cells with picrotoxin-resistant IPSP components, addition of CGRP failed to alter the residual IPSP amplitudes (reduction of 0.4 ± 0.04 mV, n = 2).
Discussion
This study aimed to characterize the influence of CGRP on BNST neurons. Pontine parabrachial neurons constitute the sole source of CGRP to BNST, and they project to its anterolateral sector (Alden et al., 1994) where there are no glutamatergic, only GABAergic/peptidergic, cells (Poulin et al., 2009). In light of these data, the finding that intra-BNST infusions of CGRP enhance startle and neuronal activation in BNST-AL targets (Sink et al., 2011) suggested that CGRP inhibits BNST-AL neurons. However, this inference is in apparent contradiction with the generally accepted view that BNST activity exerts an anxiogenic influence (Davis et al., 2010). Here, we observed that CGRP inhibits BNST-AL neurons. Below, we consider the mechanisms and significance of CGRP's inhibitory influence on BNST-AL for the regulation of fear and anxiety.
CGRP potentiates GABA-A inhibition through a postsynaptic regulation of Cl− homeostasis
To the best of our knowledge, there are no prior reports of CGRP's influence on BNST neurons. However, in other parts of the nervous system, a variety of cell type-specific effects were reported. For example, CGRP inhibits high-threshold voltage-gated Ca2+ currents in neurons of nucleus tractus solitarius (Hosokawa et al., 2010) but enhances them in DRG cells (Ryu et al., 1988). It causes a membrane hyperpolarization in some cell types (Kajekar and Myers, 2008) and the opposite (Gokin et al., 1996) or no change in others (Meng et al., 2009). In CA1 pyramidal cells, CGRP inhibits the slow Ca2+-dependent K+ current (Haug and Storm, 2000). Consistent with this, in central amygdala neurons, CGRP reduces spike frequency adaptation. In the same cell type, CGRP also causes a postsynaptically mediated increase in glutamatergic EPSCs at rest (Han et al., 2005, 2010).
Given the functional kinship and anatomical similarities between BNST and the central amygdala, one might expect CGRP to exert similar effects at the two sites. Yet, this is not what we observed. In BNST-AL cells, firing rate/pattern, passive properties, spike characteristics, and EPSP amplitudes were unaffected by CGRP. Instead, CGRP produced a robust potentiation of ST-evoked IPSPs, and this effect was reduced or blocked by CGRP Type 1 receptor antagonists.
Several observations indicate that the IPSP potentiation produced by CGRP is dependent on a postsynaptic mechanism. First, CGRP did not alter the PPR, and there was no correlation between the ratios of experimental to control IPSP variance versus amplitude. Second, the Rin drop associated with the IPSP was not altered by CGRP. Third, CGRP shifted EGABA-A negatively, and the amplitude of this shift correlated with the magnitude of IPSP potentiation. Last, CGRP's effect on IPSP amplitudes and reversal potentials was blocked by prior application of a KCC inhibitor. Although the mechanisms of CGRP action on chloride homeostasis are currently unclear, given the results obtained in other cells types (for review, see Kahle et al., 2010), an upregulation of potassium chloride transporters by phosphoregulation is likely involved.
Significance for the role of BNST in fear and anxiety
Although its name implies otherwise, BNST is in fact comprised of a collection of nuclei that form contrasting connections with fear effector neurons (Bota et al., 2012). As reviewed above, early lesion and inactivation studies targeting BNST as a whole supported the view that BNST promotes the genesis of long-lasting anxiety-like states (Walker and Davis, 1997). However, more recent reports using techniques that allow selective manipulations of different BNST regions (Kim et al., 2013) or of different cell types within these regions (Jennings et al., 2013) revealed that BNST is functionally heterogeneous. Consistent with this, neurons in BNST-AL and the anteromedial part of BNST (BNST-AM) show opposite behaviors in relation to classically conditioned fear (Haufler et al., 2013): BNST-AL neurons fire at lower rates during high than low fear states, whereas BNST-AM cells do the opposite. The reduced activity of BNST-AL cells during fear is consistent with our observations that CGRP inhibits BNST-AL neurons and the fact that intra-BNST CGRP infusions potentiate acoustic startle while increasing neuronal activity in BNST-AL targets (Sink et al., 2011). Together, these findings suggest that BNST-AM and BNST-AL exert opposite influences on fear output networks, with BNST-AM neurons promoting and BNST-AL cells inhibiting fear and anxiety. Moreover, because BNST-AL contributes inhibitory projections to BNST-AM (Turesson et al., 2013), it is likely that antagonistic interactions take place between the two regions.
In contrast to the above, a different interpretation was recently offered for CGRP's anxiogenic effects. Indeed, Sink et al. (2013) reported that virally mediated knockdown of CRF expression as well as systemic or intra-BNST infusions of CRF1 antagonists interfered with the startle potentiation produced by intra-BNST CGRP infusions. Based on these results, the authors suggested that CGRP acts by increasing the activity of CRF-positive BNST neurons. Given that these cells constitute a small subset of BNST-AL cells and that the CGRP effects described here were seen in the vast majority of tested cells, it remains unclear what the relative importance of the CRF and GABA effects is in mediating CRGP's anxiogenic influence.
Footnotes
- Received August 13, 2013.
- Revision received October 14, 2013.
- Accepted November 5, 2013.
This work was supported by National Institutes of Mental Health Grant R01 MH-098738 to D.P.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Denis Pare, Center for Molecular and Behavioral Neuroscience, Rutgers, State University of New Jersey, 197 University Avenue, Newark, NJ 07102. pare{at}andromeda.rutgers.edu
- Copyright © 2014 the authors 0270-6474/14/340060-06$15.00/0