
Abstract
We identified a family in which a translocation between chromosomes X and 14 was associated with cognitive impairment and a complex genetic disorder termed “Genetic Epilepsy and Febrile Seizures Plus” (GEFS+). We demonstrate that the breakpoint on the X chromosome disrupted a gene that encodes an auxiliary protein of voltage-gated Na+ channels, fibroblast growth factor 13 (Fgf13). Female mice in which one Fgf13 allele was deleted exhibited hyperthermia-induced seizures and epilepsy. Anatomic studies revealed expression of Fgf13 mRNA in both excitatory and inhibitory neurons of hippocampus. Electrophysiological recordings revealed decreased inhibitory and increased excitatory synaptic inputs in hippocampal neurons of Fgf13 mutants. We speculate that reduced expression of Fgf13 impairs excitability of inhibitory interneurons, resulting in enhanced excitability within local circuits of hippocampus and the clinical phenotype of epilepsy. These findings reveal a novel cause of this syndrome and underscore the powerful role of FGF13 in control of neuronal excitability.
- animal model
- epilepsy
- excitatory inhibitory imbalance
- Fgf13
- GEFS+
- translocation
Introduction
Epilepsy is a common, serious neurological disorder affecting 1% of the population worldwide (Banerjee and Hauser, 2008). Familial aggregation and twin studies establish a causal role of genetic factors in many affected individuals (Kjeldsen et al., 2003; Helbig et al., 2008). Mutations of more than two dozen genes encoding mainly ion channels have been identified in rare multigenerational families (Hildebrand et al., 2013); collectively, these account for a small minority of patients with epilepsy.
“Genetic Epilepsy with Febrile Seizures Plus” (GEFS+) refers to a common genetic epilepsy syndrome with heterogeneous clinical phenotypes, including febrile seizures (FS), febrile seizures plus (FS+), mild generalized epilepsies, and severe epileptic encephalopathies (Scheffer and Berkovic, 1997, 2008). Focal epilepsies, such as temporal lobe epilepsy and frontal lobe epilepsy, occur but less commonly in GEFS+ families (Scheffer and Berkovic, 1997, 2008; Scheffer et al., 2005). The majority of GEFS+ families exhibit clinical genetic evidence of complex inheritance (Scheffer et al., 2005); notably, individuals within the same family carrying the identical mutation often exhibit striking variability in expressivity. Linkage analyses of rare, large autosomal dominant families with GEFS+ led to identification of mutations of β and α subunits of voltage-gated Na+ channels and the γ and δ subunits of GABAA receptors (Wallace et al., 1998, 2001; Escayg et al., 2000; Baulac et al., 2001; Dibbens et al., 2004). The mutant genes identified account for <20% of the GEFS+ cases (Scheffer and Berkovic, 2008). Identification of additional causative genes to account for the majority of GEFS+ families has been limited by the variability in expressivity of the heterogeneous clinical phenotypes and lack of large affected families. Genome-wide association studies have identified hundreds of genetic variants that are associated with complex diseases in humans (Manolio et al., 2009). These variants confer relatively small increments in risk and so far fail to account for the majority of the remaining risk factors (Manolio et al., 2009). Consistent with this, a recent exome sequencing of idiopathic generalized epilepsy patients followed by large-scale genotyping failed to identify a single rare variant associated with genetic risk (Heinzen et al., 2012). Among proposed alternative approaches to identify genes of complex genetic disorders, Manolio et al. (2009) suggested analyses of affected individuals with structural chromosomal variants as one strategy.
We identified a family with GEFS+ that was associated with a maternally transmitted balanced translocation between chromosomes X and 14. We demonstrate that the breakpoint on the X chromosome disrupted a gene that encodes an auxiliary protein of voltage-gated Na+ channels, fibroblast growth factor 13 (FGF13). We engineered Fgf13 null mutant mice and demonstrate that heterozygous female mice recapitulate key features of the clinical phenotype. Cellular electrophysiological studies of hippocampal neurons of Fgf13 mutant mice reveal an imbalance of excitatory and inhibitory synaptic input activity and suggest mechanisms by which this genotype causes these diverse phenotypes.
Materials and Methods
Subjects.
The proband and his family were interviewed for history of seizures and other neurological disorders and underwent a detailed neurological examination by one of the authors (D.V.L.). The institutional review board of Duke University approved the present study. Informed consent was obtained from all participants.
Cell lines.
Lymphoblastoid cell lines were established for individuals 301, 310, 311, 401, and 402 (see Fig. 1A) from peripheral blood lymphocytes using Epstein-Barr virus at Coriell Institute for Medical Research (Camden, NJ; http://www.coriell.org/). Lymphoblastoid cells were grown in RPMI 1640 containing 2 mm l-glutamine and 15% heat-inactivated bovine serum at 37°C under 5% carbon dioxide.
FISH using BAC and fosmid probes.
BACs and fosmids were identified through the Human Genome Browser database of the Genome Bioinformatics Group at the University of California at Santa Cruz (http://genome.ucsc.edu/) and the Ensembl genome data resources of the Sanger Institute Genome Database (http://ensembl.org/). BACs and fosmids were obtained from BACPAC resource (http://bacpac.chori.org). Bacterial cultures were prepared from a single colony picked from a plate in the presence of appropriate antibiotic, and DNA was purified using standard techniques. Approximately 1 μg of BAC or fosmid DNA was labeled with either Spectrum Green or Spectrum Orange by nick translation (Vysis). The probes were precipitated using COT1 and human placental DNA and resuspended in appropriate volumes of water and hybridization buffer and stored at −20°C. All BAC and fosmid coordinates are from the NCBI18/hg18 assembly.
Chromosome FISH .
Metaphase chromosome spreads were prepared from stimulated peripheral blood lymphocytes and lymphoblastoid cell lines according to standard cytogenetic techniques. Hybridization was performed as described in the instructions of the Nick Translation kit (Vysis). Briefly, before hybridization, slides containing the chromosome spreads were immersed in a 2 × SSC solution for 30 min at 37°C and then immediately dehydrated in an ethanol series of increasing concentration (70%, 85%, and 100%). Slides were denatured at 72°C in a 70% formamide solution, immediately dehydrated in a cold ethanol series, and dried at 50°C. Probes suspended in hybridization buffer were denatured at 75°C for 2 min. Denatured probes were applied to the dried slides, and coverslips were immediately applied and sealed with rubber cement. The slides were incubated overnight in a dark humid chamber at 37°C after which they were washed for 2 min in 0.4 × SSC/0.3% NP40 at 72°C and for 1 min in 2× SSC/0.1% NP40 at room temperature. Finally, slides were counterstained with DAPI and viewed with a Leica DMRXA microscope (Leica Microsystems) equipped with the Chroma 83000 filter set. Image acquisition was performed using a charge-coupled device camera with software from Applied Imaging.
PCR-based fine mapping and Fgf13 isoform detection.
Genomic DNA was isolated from 2 × 107 lymphoblastoid cells using Genomic DNA purification kit from Gentra Systems and was used as template for various PCR-based fine mapping. All PCR primer pairs used in this study were designed using the program Primer3 (http://primer3.ut.ee). In silico PCR (http://www.genome.ucsc.edu/cgi-bin/hgPcr?db=hg18) was performed for all possible PCR primer pairs designed for fine mapping of the translocation breakpoints. Only primer pairs generating a single amplicon in the human genome database were used in this study. The complete list of all primers used in this study is available upon request. Total RNA was isolated from 7 × 106 lymphoblastoid cells using RNeasy Plus Mini kit from QIAGEN and was used for Fgf13 isoform detection experiments. Total RNA was reversed transcribed using gene-specific primer, and the ensuing cDNA was amplified by PCR using isoform-specific primers.
Generation of Fgf13 knock-out animals.
A targeting vector was designed to include two regions of 1 kb and 10 kb homology around exon 2 of the Fgf13 gene on X chromosome (see Fig. 4A). The targeting vector contained an in-frame stop codon for the Fgf13 gene and a neomycin resistance cassette. Homologous recombination in C57BL/6 embryonic stem cells of the targeting vector at the Fgf13 gene locus resulted in a mutant allele that has a stop codon introduced into the 3′ end of the remaining portion of exon 2 and a neomycin-resistance gene cassette that replaces the deleted region of 620 bp (3′ half of exon 2 and intronic sequence). The C57BL/6 embryonic stem cells were subsequently microinjected into BALB/c blastocysts to generate germline chimeras. Male chimeras were bred with C57BL/6 females, and germ line transmission in female pups was established by PCR using wild-type and mutant-specific primers (wild-type: forward, TAACAAACACCTTCAGATCACTGG; reverse, AAACTATACAGCCGACAAGGCTAC; and mutant: forward, ACCATGATTACGCCAAGCAT; reverse, TTCCACAGCTGGTTCTTTCC). Fgf13 mutant female mice were bred for at least four generations with wild-type C57BL/6 males. Male offspring lacking the Fgf13 gene died at embryonic age 12.5, but heterozygous females appeared healthy and exhibited a normal lifespan. Age-matched and littermate control female wild-type and Fgf13 mutant mice were used in all of the experiments described.
The genotype of each animal was assessed using tail genomic DNA as template and primers specific for detection of the wild-type and mutant alleles by PCR as described above. Genotyping was reconfirmed for every animal after the completion of the experiment. Mice were housed under a 12 h light/dark cycle with food and water provided ad libitum. Animals were handled according to the National Institutes of Health Guide for the Care and Use of the Laboratory Animals, and the experiments were conducted under an approved protocol by the Duke University Animal Care and Use Committee.
RNA harvesting and real-time PCR.
Hippocampi and cortex from 10 wild-type and 10 Fgf13 mutant littermate pairs between the P50 and P55 were rapidly dissected on ice, snap frozen in liquid nitrogen, and stored at −80°C. RNA was harvested using the Absolutely RNA kit (Stratagene); 500 ng of RNA was used for reverse transcription with random hexamer primers and Superscript II (Invitrogen). The cDNA generated was used for quantitative PCR (Power SYBR Green, ABI 7300 real-time PCR machine; Applied Biosystems) with the intron-spanning primers listed below. Each sample was measured in triplicate, and expression of the housekeeping gene Gapdh was used as a normalizing control for RNA quantity and sample processing. The primers used for the qPCR are as follows: Fgf13 forward, ACCAAAGACGAGGACAGCAC, Fgf13 reverse, GGTGTGAAATGTTCCGAGGT; Gapdh forward, CATGGCCTTCCGTGTTCCT and Gapdh reverse, TGATGTCATCATACTTGGCAGGTT.
Western blotting.
Cortices (20 mg) from 7 wild-type and 8 mutant female animals were placed in 500 μl of RIPA buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 5mM EDTA, 1% Triton X-100, 0.1% SDS, 1% Na-Deoxycholate, DNase1, 1mM PMSF, 1 μm pepstatin, 2.5 μg/ml aprotinin and leupeptin) in a 1.5 ml tube (on ice) and homogenized. After incubating homogenate on ice for 10 min, sample was centrifuged at 13,000 × g for 15 min. Supernatant was collected and stored at −80°C. Protein concentration was measured using the Bio-Rad DC protein assay kit; 20 μg of cortical extracts was subjected to 10% SDS-PAGE, transferred and probed with antibodies. Bands were visualized by reacting blots with ECL prime (GE Healthcare) and exposing to film. For quantitation, multiple exposures were taken to establish the linear range, and films were scanned for densitometry using ImageJ (http://rsbweb.nih.gov/ij/). All bands were normalized to actin in the same sample as a loading control. Primary antibodies used in this study include rabbit polyclonal anti-FGF13 (HPA002809, Sigma) at 1:500 dilution and mouse monoclonal anti-actin (Millipore Bioscience Research Reagents, #MAB1501R) at 1:2500 dilution. The size of the bands recognized by the FGF13 antibody (HPA002809, Sigma) approximates 22–28 kDa, a size predicted by the amino acid sequence of FGF13 isoforms. Detection of a single band of the proper size in Western blots of a recombinant FGF13 isoform in lysates of transfected HEK293T cells using the same FGF13 antibody supports the specificity of the antibody (www.proteinatlas.org/ENAG00000129682-FGF13/antibody). Secondary antibodies used in this study were peroxidase-labeled anti-rabbit IgG from Vector (#P-1000) at 1:10,000 and peroxidase-labeled anti-mouse IgG from Vector (#P-2000) at 1:10,000.
Fluorescent multiplex in situ hybridization (ISH).
Mouse brains were removed rapidly and quick-frozen in a beaker of isopentane equilibrated in a dry ice/methanol slurry and stored at −80°C until further processing. The frozen brains were embedded using Optimal Cutting Temperature compound in cryomolds. Coronal brain sections of 14 μm (bregma −1.36 mm to −2.56 mm) were sectioned and stored in slide boxes wrapped air tight with aluminum foil at −80°C until use. All cryomolds and brain sections were used within 3 months of processing. Brain sections (14 μm in thickness) were subjected to dual fluorescent ISH by using RNAscope (Advanced Cell Diagnostics) following the manufacturer's protocol (RNAscope Multiplex Fluorescent Assay for Fresh Frozen Tissues, User Manual catalog #320850, Advanced Cell Diagnostics) and as described by Wang et al. (2012). Probes were designed for the following genes; Fgf13 coding sequence (exons 2–5, 543 bases-NM_010200.2 and Ensembl transcript ID- ENSMUST00000033473), Fgf13-3′UTR (660 bases, NM_010200.2 and Ensembl transcript ID, ENSMUST00000033473), Gad-1 (982 bases spanning multiple exons NM_008077.2) and Gad-2 (858 bases spanning multiple exons NM_008078.1). Nuclei were stained with DAPI, and the sections were coverslipped using fluorescent mounting media (Prolong Gold antifade, Invitrogen). Images were collected on a Leica SP5 confocal microscope. In control experiments (data not shown), brain sections were incubated with probes of DapB labeled with different fluorophores, an mRNA of Escherichia coli that is not expressed in mammalian cells, or probes against ubiquitin C (Ubc) DNA directed RNA pol II (PolR2a) and peptidylproyl isomerase (Ppib), mRNAs that are expressed in mammalian cells. Single probe hybridization for each probe was performed and images collected in all channels to test for overlapping signals from one channel to the other. Multiprobe images were acquired by sequential scanning. Colocalization studies were further confirmed by ISH using Fgf13 3′UTR and Gad-2 RNAscope and riboprobes (data not shown). Representative images from multiple successful experiments (4 animals, multiple sections from each animal) are shown.
Hyperthermia-induced seizures.
Mouse core body temperature was recorded with a rodent rectal temperature probe placed at a depth of 17 mm and connected to a rodent temperature controller (TCAT-2DF, Physitemp). Behavior was monitored by a video camera (Victor Company of Japan). Command temperature ± 0.3°C was maintained using an infrared heat lamp positioned 12 inches above the floor of a 20.5 × 25 × 40 cm Plexiglas chamber. Baseline temperature was measured after the mice were allowed to acclimate to the chamber for 10 min. Mice were held at an initial set-point temperature of 37.5°C for 10 min. Body temperature was then elevated by 0.5°C increments. Mice were held at a given command temperature for 2 min before increasing to a new command temperature. Temperature was increased until a seizure or 42.5°C was reached (see Fig. 5A). At the end of hyperthermia induction, mice were placed on a cold surface and monitored for 10 min. Investigators were blinded to the genotype of the animals.
Surgery and kindling.
Procedures for surgery and kindling were performed as described previously (He et al., 2004) by an individual blinded to genotype of the animals. In the kindling model, repeated induction of brief, focal seizures by application of initially subconvulsive electrical stimuli to the amygdala leads to progressively longer and more widely propagated electrographic seizures that are accompanied by both partial and generalized tonic-clonic seizures (Goddard, 1967). Briefly, under pentobarbital (60 mg/kg) (Ovation) anesthesia, bipolar electrodes used for stimulation and recording were stereotactically implanted in the right amygdale, respectively. A ground electrode was placed over the dura, anterior to the coronal suture, and left of the sagittal suture. Following a postoperative recovery period of 2 weeks, the electrographic seizure threshold in the amygdala was determined and stimulations at the intensity of the electrographic seizure threshold were subsequently administered twice daily, 5 d per week as described previously (He et al., 2004). The behavioral manifestations of seizures were classified according to a modification of Racine's classification: 1, facial clonus; 2, head nodding; 3, unilateral forelimb clonus; 4, rearing with bilateral forelimb clonus; 5, rearing and falling (loss of postural control); 6, running or bouncing seizures; and 7, tonic hindlimb extension (Borges et al., 2003). Mice were stimulated until fully kindled as defined by the occurrence of 3 consecutive seizures of Class 4 or greater with limb clonus and/or tonus lasting at least 12 s. Accuracy of electrode placements was verified by histological analysis, and only animals with correct electrode placement in the amygdala were included in the statistical analysis for kindling experiment. Investigators were blinded to the genotype of the animals.
Spontaneous recurrent seizure monitoring.
Five Fgf-13 wild-type and five Fgf-13 mutant mice at age P40 were anesthetized with pentobarbital (60 mg/kg), and a bipolar electrode was stereotactically inserted into the left hippocampus using the following coordinates with bregma as reference: 2.0 mm posterior, 1.6 mm lateral, 1.5 mm below dura. The ground electrode was placed on top of the dura anterior to the hippocampal bipolar electrode. Hippocampal EEG telemetry (Grass Instrument) and time-locked video monitoring were performed using Harmonie software (Stellate Systems). After a postoperative recovery period of 1 week, animals underwent video EEG monitoring for 350–400 h over the span of 15–20 d. Spontaneous recurrent seizures were identified by review of video EEG files by two independent trained readers blinded to genotype of mice. Behavioral seizures were classified according to a modification of the Racine scale for mice (Borges et al., 2003).
In vitro electrophysiology.
Hippocampal slices were prepared from P21 to P30 days. Briefly, mouse brains were quickly removed and placed in ice-cold buffer containing the following (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, and 5 glucose. Following dissection of hippocampi, transverse slices (400 μm thick) were cut with a vibratome and kept in standard extracellular solution and continuously bubbled with 95% O2/5% CO2 at 36°C for at least 1 h before experiments. Slices were then transferred to a recording chamber for electrophysiological recordings. The standard extracellular solution contained the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 0.4 l-ascorbic acid, 3 myoinositol, and 2 Na-pyruvate, pH 7.3–7.4, 320 mOsm. The intracellular solution for current-clamp experiments contained the following (in mm): 158 K-gluconate, 10 mm KCl, 10 HEPES, 5 Na2-phosphocreatine, 5 MgATP, 0.5 NaGTP, and 0.2 EGTA, with pH adjusted to 7.2. Experiments were performed at room temperature. CA1 pyramidal neurons were whole-cell clamped at −60 mV (for sIPSC and mIPSC) or −70 mV (for sEPSC and mEPSC) using an Axon 700A amplifier (Axon Instrument). Data were digitized at 20 kHz, Clampfit 10.2, and Igor 5.0 was used for further data analysis.
After establishing a stable voltage-clamp configuration from CA1 pyramidal neurons, we switched to current-clamp mode. To evoke action potentials, depolarizing current steps of 50, 100, 120, 150, 170, 200, 220, 250, 270, 300, 320, 350, 370, 400, 420, 450, 470, 500, 550, and 600 pA were injected. Cell capacitances and input resistances were calculated by membrane test.
For sIPSCs and mIPSCs recordings, the intracellular solution contained the following (in mm): 140 CsCl, 10 HEPES, 5 Na2-phosphocreatine, 5 MgATP, 0.5 NaGTP, 5 EGTA, and 1.25 QX314, with pH adjusted to 7.2. sIPSCs were recorded in the presence of 20 μm CNQX and 50 μm d-AP5 in the ACSF. To isolate mIPSC, 1 μm TTX was added into the extracellular solution in addition to CNQX and d-AP5. For sEPSC and mEPSC recordings, the intracellular solution contained the following (in mm): 148 CsMeSO4, 10 CsCl, 10 HEPES, 5 Na2-phosphocreatine, 4 MgATP, 0.3 NaGTP, 5 EGTA, and 1.25 QX314, with pH adjusted to 7.2. sEPSCs were recorded in the presence of 10 μm bicuculline in the extracellular solution, whereas mEPSCs were recorded in the presence of both 10 μm bicuculline and 1 μm TTX in the extracellular solution. QX314, CNQX, d-AP5, TTX, and bicuculline were purchased from Tocris Bioscience.
Minis were semiautomatic selected using Mini Analysis Program 5.2.5 (Synaptosoft). All data are presented as mean ± SE. Investigators were blinded to the genotype of the animals.
PCR primers.
A partial list of the primers used in this study is listed below. A complete list is available upon request. Primers used in the mapping of the breakpoints: X13 forward, TGTATGGGAAGGAGGATGGA, X13 reverse, AAGCCTGATCTCACCCACTG; X14 forward, CAGTTTGCACACTCCTGGTC, X14 reverse, TTAGGGAATCCAGCCAGAGA; X15 forward, TTTCTTTCATTTTCTCAAAGCAGA, X15 reverse, GAAGGATCTGGAGGCTTTATGA; X13–14 forward, GTGGGCCATATGGTCTCTGT, X13–14 reverse, CCACAAATTTTAACCCTGTGC; X14–15 forward, CAGATGAGAATTAAGGCTCAGAGA, X14–15 reverse, GAGGCCAGAGAAGTCACCTG; 14–39 forward, ATGGGCCACTATCTTCCAGA; 14–40 forward, CGCATGAGATTTGGGAGAG. Primers used for amplification and sequencing of the 14-X and X-14 breakpoint junctions on the derivative 14 and X chromosomes: X-BP forward (X-chromosome-specific), ACACTCCTGGTCTGTAACACTCAA, X-BP reverse (chromosome-14-specific) GTGGGTCCACAATAACAGAAAAAT; 14-BP forward (chromosome-14-specific), CAGGACAGAAAGGAAATAAAATCTG, 14-BP reverse (X-chromosome-specific), TTAGGGAATCCAGCCAGAGA. Primers used for identification of Fgf13 isoforms from total RNA isolated from lymphoblastoid cell lines. Exon 3 reverse (RT primer): TGCCAAGTACAGCTTGGTTTGA; Fgf13-1S-forward, GAAATCCAATGCCTGCAAGT; Fgf13-1U-forward, TTGTTAAGGAAGTCGTATTCAGAGCCTC; Fgf13-1V-forward, GAGGAGAAAGATGCTTCTAAGGAGCCTC; Fgf13-1Y + 1V-forward, GTTCTGGATGACGCCCCCCCTGGCC; Fgf13-1Y and 1Y + 1V-forward, ATGTTACGACAAGATTCCATCCAATC; Fgf13-V reverse, CAGACCCACAGGGATGAGGTTAA; and exon 2 reverse, GTAAGTGCTGTCCTCATCTTTGGTGC.
Statistical analysis.
Survival analysis for hyperthermia-induced seizures was performed using the Kaplan–Meier method, with subsequent log-rank testing between curves. Area under the curve analysis was performed for kindling data using the ROC method. Remaining analyses were performed using t test or one-way ANOVA with post hoc Bonferroni's correction. Student's t tests were performed as specified in the text. F tests to assess difference in variances were used for t tests; for the sole significant F test in this manuscript (for sEPSC frequency, see Fig. 8), subsequent t test included Welch's correction for unequal variance. Analyses were performed using GraphPad Prism (version 5.04).
Results
Balanced reciprocal translocation in a GEFS+ family
The proband (numbered 401 in pedigree in Fig. 1A) is a 21-year-old Caucasian man who was well until age 2 when he experienced a simple febrile seizure. He remained well without medications until age 6 when he experienced a temporal lobe seizure (TLS) heralded by an aura of a bad smell. During the ensuing years, he repeatedly experienced TLS despite treatment with a diversity of antiseizure medications. He currently experiences clusters of seizures lasting a couple of hours approximately once each month despite antiseizure medications, including valproate, felbamate, and acetazolamide. His past medical history revealed cognitive impairment necessitating attendance at a special education school. Review of systems was unremarkable. His physical and neurological examinations were within normal limits. His typical seizure was captured by video EEG monitoring during a hospitalization at Duke University Medical Center in June 2004 and verified to be a TLS (Fig. 1C). He experienced 8 complex partial seizures within 1 d. Each seizure occurred during wakefulness, and seizure onset was evident by the patient's pressing a button reflecting his awareness of seizure onset. Within seconds thereafter, he was unresponsive to spoken word, exhibited a wide-eyed stare, and his right hand curled up in a dystonic posture. Additional studies included an EEG, which revealed independent epileptiform sharp waves and spikes over each temporal region but more prominently over the left temporal region (Fig. 1C). MRI revealed diffuse cortical atrophy, but no evidence of hippocampal sclerosis and no evidence of altered T1 or T2 signal over the hippocampi (Fig. 1D). The proband's sibling (402) is a 19-year-old man who was well until age 18 months when he experienced his first simple febrile seizure consisting of generalized jerking, which lasted 30 s. He experienced 3 febrile seizures in the next 6 months and was placed on phenobarbital. He developed afebrile seizures at age 4 despite the phenobarbital and was switched from phenobarbital to valproate; seizures persisted on valproate alone but remitted upon addition of acetazolamide. He has been seizure free on the combination of valproate and acetazolamide, except for recurrence of a generalized tonic-clonic seizure 3 years ago when a valproate taper was attempted. His past medical history and review of systems were unremarkable. His general physical and neurological examinations were within normal limits. Information from his parents revealed that his typical seizure consists of deviation of his eyes to the left followed by a generalized tonic-clonic seizure. His seizures have not been captured on video EEG monitoring. Whether initial deviation of eyes to left is associated with impairment of awareness is not clear. Additional laboratory studies included an EEG, which revealed runs of intermittent anterior temporal rhythmic δ activity and anterior temporal sharp waves independent in each temporal region, but clearly maximal on the left. Anterior intermittent rhythmic δ activity of this type has been described as characteristic of temporal lobe epilepsy (Geyer et al., 1999). Neither an MRI nor CT scan has been performed.

A maternally transmitted apparently balanced translocation between the X chromosome and chromosome 14 is associated with febrile seizures, cognitive impairment necessitating special education, and temporal lobe epilepsy. A, A four generation pedigree exhibiting balanced translocation. Red symbols represent the presence of febrile seizures. Green symbols represent the presence of cognitive impairment necessitating special education classes. Black symbols represent the presence of temporal lobe epilepsy. B, GTW-banded partial karyotypes from the mother (301) and the proband (401) reveal the apparently balanced reciprocal translocation between chromosomes 14 and X [t(X; 14) (q27; q21)]. Chromosomes are arranged left to right showing the normal X chromosome (mother only), derivative X, normal chromosome 14, and derivative 14. C, EEG pattern of seizure of proband detected during video EEG monitoring. Behavioral and EEG features during segments (S1–S5) before, during, and following the seizure. S1, Segment 1: Patient is awake with eyes open and listening to nurse 5 s before seizure onset. EEG is characterized by symmetric low-voltage mixed frequencies. High-amplitude slow waves in electrodes FP1 and FP2 reflect eye movements, and low-voltage fast activity in electrodes T3 and T4 reflects muscle activity. S2, Segment 2: Patient was talking to nurse and suddenly turned and pushed button (arrow) notifying onset of seizure at which time EEG is dominated by eye movement, muscle, and movement artifacts. S3, Segment 3: Fifteen seconds following seizure onset, patient is shifting restlessly, flailing right arm purposely, and is unresponsive to nurse's questions. EEG exhibits rhythmic θ activity of higher amplitude over the left hemisphere. S4, Segment 4: Eighty-two seconds following seizure onset, patient blinks and relaxes. Rhythmic θ has terminated, and EEG now exhibits eye movement artifact, muscle artifact, and low-voltage activity with slowing more prominent in left compared with right parasagittal region. Patient was oriented to person, place, and time ∼4 min after seizure onset and did not recall any events transpiring during the seizure. S5, Segment 5: Interictal EEG recorded ∼7 h following the seizure denoted in S1-S4. Arrow indicates the occurrence of a high-amplitude sharp transient in the left temporal region. D, Fluid attenuated inversion recovery image of the proband reveals mild diffuse atrophy (arrowhead) but no evidence of hippocampal sclerosis (arrow). Scale bar, 50 mm. L, Left; R, right.
The proband's mother (301; Fig. 1A) is a 45-year-old white woman who is in good health. Apart from febrile seizures as an infant, there is no history of epilepsy or cognitive impairment. Her neurological examination is normal. Her family history includes a brother (302) who has no history of seizures or epilepsy or cognitive impairment; he has three children, the oldest of whom (403) had febrile seizures as an infant. In addition, the proband's mother's maternal aunt (203, now deceased) was cognitively impaired but did not have seizures or epilepsy; she married and had six children, two of whom (303 and 306) are also cognitively impaired but have no history of seizures or epilepsy. There is no family history of seizures, epilepsy, or cognitive impairment in his father (310) or father's extended family (Fig. 1A). The family history together with the phenotypes of the proband (401) and his brother (402) are consistent with the diagnosis of GEFS+.
Review of medical records revealed that the proband (401) and his brother (402) inherited the balanced translocation, t(X; 14) from the mother (301). Normal karyotype was found in the father (310) and the maternal uncle (302). To confirm information contained in the medical records, Giemsa Trypsin Wright (GTW)-banding was performed on metaphase chromosome spreads. Cytogenetic analysis revealed the presence of an apparently balanced reciprocal translocation between the X chromosome and chromosome 14, t(X; 14) (q27; q21) (Fig. 1B) in the proband, his brother, and his mother.
FISH identifies genomic DNA spanning the breakpoints
To localize the X chromosome breakpoint junction, metaphase chromosome spreads prepared from lymphoblastoid cell lines established from the peripheral blood lymphocytes of individuals 301, 310, 311, 401, and 402 (Fig. 1A) were analyzed by FISH using 11-X chromosome-specific BAC clones as probes (X-A through X-K, RP11-79C15, RP11-486A7, RP11-97N5, RP1-20J23, RP11-112H23, RP1-260J9, RP11-59720,RP1-106C24, RP11-158N4, RP6-220J16, and RP6-88D7, respectively; Fig. 2C). Multiple Spectrum Orange- and Spectrum Green-labeled combination dual-probe hybridizations revealed a BAC clone (X-H) RP1-106C24 that spanned the breakpoint (Fig. 2A). The location of the breakpoint within RP1-106C24 was further narrowed with dual-color FISH using five human fosmid clones (F1-F5, G248P86009A3, G248P83222B11, G248P88496C3, G248P80529F11, and G248P87924F2, respectively), permitting identification of a 43.6 kb fosmid F4 (G248P80529F11) that hybridized to the normal X and both of the derivative chromosomes (data not shown), thereby reducing the region of the breakpoint to ∼17 kb (Fig. 2C). A similar strategy was used to localize the chromosome 14 breakpoint by dual-probe hybridizations using chromosome 14-specific BAC clones (14A-14K- RP11-81F13, RP11-79M10, RP11-159P3, RP11-341B22, RP11-245O17, RP11-139N24, RP11-787D15 and RP11-696F1, RP11-453F20, RP11-321F3, and RP11-368A1, respectively; Fig. 2B). Three distinct BACs spanning the breakpoint (BACs 14C, 14D, and 14E; Fig. 2E) were identified that narrowed the breakpoint region to ∼20 kb (Fig. 2E).

Mapping the translocation breakpoints on X-chromosome and chromosome 14. A, Dual-color FISH analysis using X-chromosome-specific BACs. BAC-XH (RP1-106C24 labeled with Spectrum Green-dUTP) and BAC-XF (RP1-260J9 labeled with Spectrum Orange-dUTP) probes were hybridized to metaphase spreads of the proband's mother exhibiting the translocation. Green and orange signals are present on the normal X chromosome and the derivative X chromosome [der(X)]. In addition, the green signal is also present on the derivative chromosome 14 [der (14)], demonstrating that the BAC-XH (RP1-106C24) spans the breakpoint of translocation (inset). B, Dual-color FISH analysis using chromosome 14-specific BACs. BAC-14C (RP11-245O17, labeled with Spectrum Green-dUTP) and BAC-14J (RP11-321F3 labeled with Spectrum Orange-dUTP) probes were hybridized to metaphase spreads of the proband's mother exhibiting the translocation. The green signal is present on the normal 14, the derivative 14 [der (14)], and the derivative X chromosome [der(X)]. The orange signal is present on the normal chromosome 14 and derivative X, demonstrating that BAC-14C (RP11-245O17) spans the breakpoint of translocation (inset). C, The relative positions of the BACs (BACs X-A to X-K, vertical lines) are shown on Xq25-q27.1 (top line); the region harboring the breakpoint. BAC-H (RP1-106C24 shown as red line) was found to span the breakpoint (Fig. 1B). To pinpoint the location of the breakpoint within BAC-H, dual-color FISH experiments were performed using fosmids 1–5, narrowing the region of the breakpoint to ∼17 kb and demonstrating that fosmid F4 (red line) spanned the breakpoint. PCR primers (X1-X20 and X1–2 to X19–20) were designed as shown in the figure and used to amplify the control genomic DNA (father; Fig. 1A) and the translocation harboring genomic DNA (proband's genomic DNA Fig. 1A). D, The presence of the amplicons using primers X13–14 and X14–15 in the translocation harboring genomic DNA of the proband (P) together with the absence of amplicon using primers X14 (depicted by arrow) narrowed the region of the breakpoint to ∼400 bp. Using the control genomic DNA of the father (F), PCR amplicons with all the primers were generated. E, The relative positions of BACs 14A-14F used as probes for dual-color FISH experiments are presented as horizontal lines. These experiments revealed three distinct BACs spanning the breakpoint, BACs 14C, 14D, and 14E. Information on the coordinates of these three BACs narrowed the breakpoint to a region approximating 20 kb as depicted by the vertical lines in the expanded segment of chromosome 14 in the lower part of figure. PCR primers using chromosome 14 sequences (14-F1 to 14-F40) in the forward direction and PCR primers either X-14R or X15-R in the reverse direction were designed to identify an amplicon harboring the translocation breakpoint using the proband's genomic DNA as the template. F, The chromosome 14 breakpoint is located between the primers 14-F40 and X-14R. Using primers designed as depicted in A, primer pair combination 14-39F-X-14R (lane 1), 14-40F- X-14R (lane 2), 14-39F- X-15R (lane 3), and 14-40F -X15R (lane 4), amplicons were generated using genomic DNA of proband (401; Fig. 1A) as the template. PCR using genomic DNA of the father (310; Fig. 1A) as the template did not generate the amplicons seen with the proband's DNA.
PCR and DNA sequencing reveal disruption of gene encoding Fgf-13 on the negative strand of X chromosome
A nested PCR strategy centered on the ∼17 kb in the 3′ end of Fosmid F4 (Fig. 2C) was used to pinpoint the breakpoint on the X chromosome. Using the father's DNA as template, amplicons of the predicted size were detected for all of the primer pairs. In contrast, using the proband's DNA as template, amplicons of the predicted size were obtained for all but one primer pair-X14 (Fig. 2D). A similar strategy narrowed the breakpoint on chromosome 14 (Fig. 2E,F). PCR using forward primers from the 20 kb narrowed breakpoint region on chromosome 14 paired with reverse primers X-14R and X-15R (Fig. 2E) from X chromosome revealed amplicons harboring the breakpoint on chromosome 14 using the proband's DNA as template, but not the unaffected father (Fig. 2F). DNA sequencing of the amplicons generated from all affected individuals revealed coordinates of the breakpoints on chromosomes 14 and X (Fig. 3A,B). The breakpoint on the X chromosome lies 39.3 kb 3′ of exon Y and 78.9 kb 5′ of exon U within the Fgf13 gene (Fig. 3C). In contrast, the breakpoint on chromosome 14 resides 2.2 Mb 3′ to Lfnn5 and 0.8 Mb 5′ to Klhl28 (Fig. 3C).

Translocation disrupts a gene on the X chromosome that encodes a member of the fibroblast growth factor family, Fgf13. A, PCR amplification X-14 and 14-X translocation junctions in all affected individuals. X-14 junction: Using primers X-BP F (X-chromosome-specific) and X-BP R (chromosome 14-specific), a ∼350 bp junction amplicon was generated from affected individuals harboring the translocation (lanes 1, 3, and 4, individuals 301, 401, and 402; Fig. 1A) but not from the unaffected individuals (lanes 2 and 5, individuals 310 and 302; Fig. 1A) using genomic DNA as template. 14-X junction: Using primers, 14-BP F (chromosome 14-specific) and 14-BP R (X-chromosome-specific), a ∼700 bp junction amplicon was generated from affected individuals harboring the translocation (lanes 1, 3, and 4, individuals 301, 401, and 402; Fig. 1A) but not from the unaffected individuals (lanes 2 and 5, individuals 310 and 302; Fig. 1A) using genomic DNA as template. B, Identification of the precise positions of the breakpoints on the derivative X and 14 chromosomes: Sequencing and BLAST analysis of the 350 and 700 bp junction amplicons generated from the genomic DNA of 301 revealed the breakpoint on X chromosome at position 137727985 and at position 43651276 on chromosome 14. The junction sequences of 401 and 402 were identical to the sequence from 301. All coordinates are from the human genome browser at http://www.genome.ucsc.edu/ (NCBI36/hg18 assembly). The breakpoint on X-chromosome disrupts the negative strand-encoded gene Fgf13. C, Physical map of the X-chromosome and chromosome 14 in the region of the translocation breakpoints. Locations of the breakpoint on chromosomes X (top). Location of the FGF13-isoforms and the breakpoint on the negative strand of X chromosome are shown. The positions of the exon 1's (i.e., 1V, 1Y, 1U, 1S) and exons 2–5 were constructed using the published mRNA and protein sequence and the human genome browser at http://www.genome.ucsc.edu/. The location of the breakpoint between exons 1U and 1Y was determined by DNA sequencing and located at position 137727985 on the X chromosome. Location of the breakpoint on the chromosome 14 (bottom). The annotated genes closest to the breakpoint at position 43651376 bp are KLHL5, a BTB (POZ) domain-containing 5 protein, and LRNF5 (leucine rich repeat and fibronectin Type III). The annotated genes were identified from the UCSC browser using multiple gene prediction programs, including RefSeq, Uniport, GenBank, Comparative Genomics, Consensus CDS, and others. In contrast to these annotated genes, some hypothetical genes closer to the breakpoint were predicted by the above listed programs, but whether these putative genes are expressed remains to be established. D, Detection of Fgf13 isoforms by RT-PCR using total RNA isolated from the lymphoblastoid cell lines established from the unaffected individual 310 and affected individual 401. Amplicons reflecting isoforms 1S and 1U were generated by RT-PCR from the lymphoblastoid cell line total RNA of both individuals, whereas amplicons reflecting isoforms 1V, 1VY, and 1VY + 1Y were generated only from the unaffected individual 310. The absence of 1V, 1VY, and 1VY + 1 Y isoforms in the affected individual confirms the position of the breakpoint on X chromosome. The amplicons were sequenced and their identity confirmed by BLAST analysis.
These results predict that the translocation will eliminate expression of FGF13 isoforms 1V, 1Y, and 1V + 1Y but not 1S and 1U in affected males (Fig. 3C). To test this prediction, RNA was isolated from lymphoblastoid cell lines of the proband (401; Fig. 1A) and unaffected father (310; Fig. 1A), reverse-transcribed, and amplified with one primer specific to sequence in exon 2 and a second primer specific to isoforms 1S, 1U, 1V, 1V + 1Y, or 1VY. Amplicons reflecting the presence of all isoforms were obtained from RNA isolated from the father (Fig. 3D). By contrast, amplicons of isoforms 1S and 1U, but not 1Y, 1VY, and 1Y + 1VY, were obtained from RNA isolated from the proband (Fig. 3D). Similar results were obtained for the affected brother (402; Fig. 1A). Together, these results confirm the absence of Fgf13 mRNA for isoforms 1Y, 1VY, and 1Y + 1VY in total RNA of lymphoblastoid cell line of individuals with the translocation as predicted by the location of the breakpoint on the X chromosome.
Gender-specific consequences of targeted ablation of Fgf13 gene
Identification of the DNA sequences disrupted by the translocation led us to hypothesize that interruption of the Fgf13 gene underlies the phenotypic abnormalities in the affected family members. To begin to test this hypothesis, we engineered a mouse lacking the Fgf13 gene by removing the 3′ half of exon 2 that is shared by all isoforms and the adjoining intronic sequence (620 bases) (Fig. 4A) by homologous recombination. Male chimeras generated were bred with C57BL/6 wild-type females, and germ line transmission in female pups was established by Southern blotting (Fig. 4B) and PCR using wild-type and mutant-specific primers (Fig. 4C). Female mice harboring the mutation were bred with C57BL6 (Charles River) males. Male offspring lacking the Fgf13 gene die at embryonic age 12.5, revealing a requirement of FGF13 for embryonic development. Heterozygous females appear healthy and exhibit a normal lifespan. Subsequently, all experiments were performed using age- and litter-matched wild-type and mutant female mice. qPCR analysis of hippocampal and cortical mRNA from 50-d-old, age- and litter-matched wild-type and mutant mice revealed a reduction of Fgf13 mRNA content in the mutants approximating 50% (Fig. 4D). Similar results were obtained by Western blot analysis of the cortical protein lysates (Fig. 4E). These results support the conclusion that ablation of one Fgf13 allele reduces expression of Fgf13 mRNA and protein approximating 50% in the heterozygous female mice. In addition, the lethality of male embryos carrying the mutant X chromosome demonstrates a requirement of FGF13 during development.
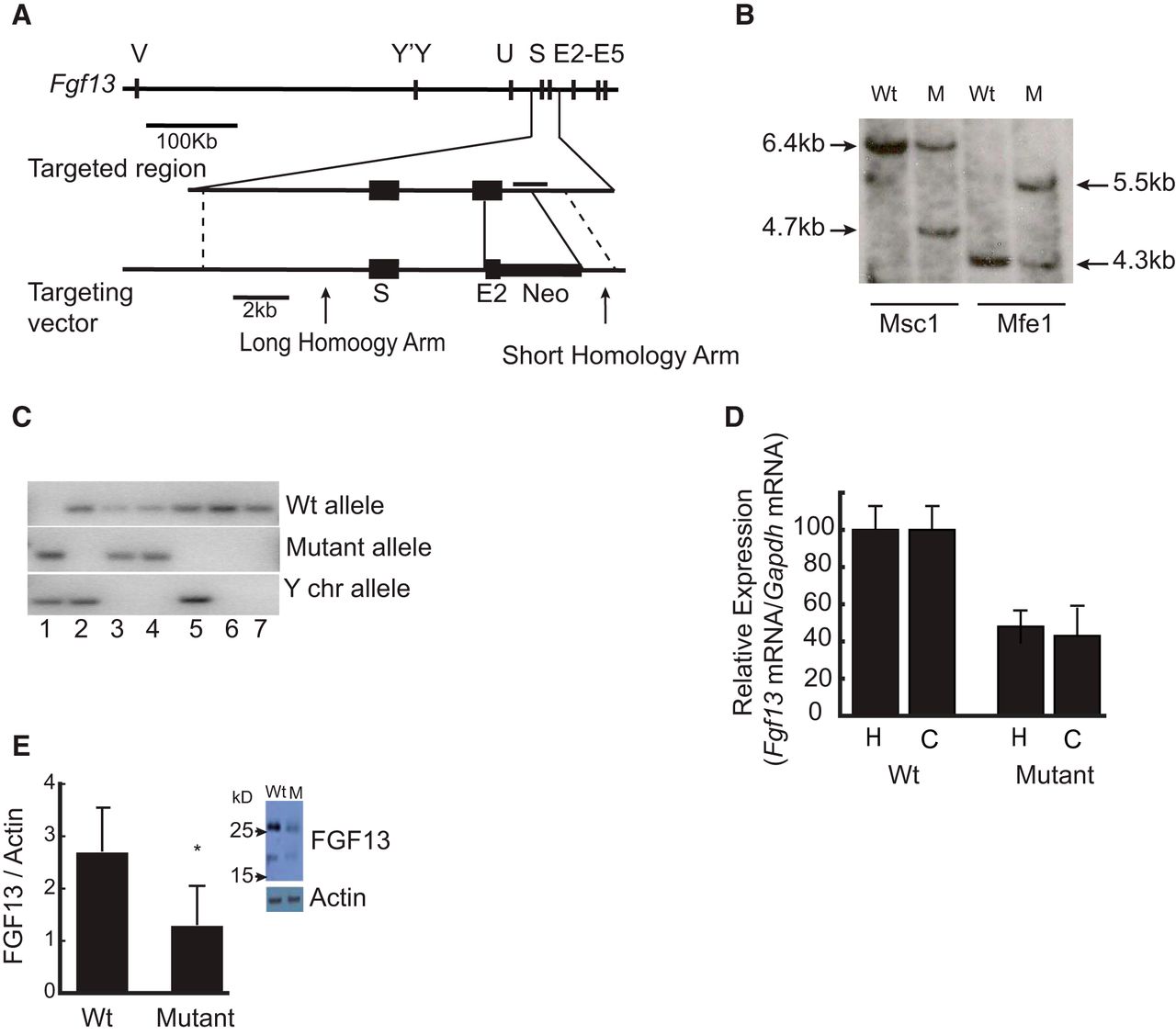
Targeted ablation of Fgf13 gene leads to embryonic lethality in mutant male mice and reduction in the expression of the mRNA and protein in the mutant female mice. A, The strategy for the generation of the Fgf13 knock-out mice. The gene structure of mouse Fgf13 gene along with the targeting locus and targeting vector is shown schematically. Fgf13 mutant mice were generated by homologous recombination. Targeting vector was designed to remove the 3′ region of the exon 2 and a 600 bp intronic sequence common to all isoforms and replace it with a neo-cassette and an in-frame stop codon after the last codon of the remaining exon 2. B, Southern blot confirmation of the modification of the fgf13 locus in the mutant female mice using MSc1 and Mfe1 restriction enzyme digests of the genomic DNA. Horizontal line in the targeting locus schematic indicates the position of the 1.35 kb probe used for the southern hybridization. C, PCR validation of the wild-type, heterozygous, and homozygous genotypes of the E12.5 embryos and the determination of the sex of the embryos. Lane 1 represents an Fgf13 mutant male from nonviable embryo (homozygous); lanes 2 and 5, wild-type males; lanes 3 and 4, Fgf13 mutant females (heterozygous); lanes 6 and 7, wild-type females. D, Quantitation of pan Fgf13 mRNA by qPCR of hippocampal and cortical total RNA from mutant female mice (n = 10) and wild-type, litter- and age-matched female mice (n = 10). Data are mean ± SD. p < 0.001 (unpaired t test). Hippocampal RNA, *p = 1.6e-8 (t = 9.64, df = 18. Cortical RNA, **p = 4.0e-9 (t = 10.53, df = 18). E, Relative levels FGF13 protein in the cortical lysates of the mutant female mice (n = 8) compared with the wild-type female control mice (n = 7). Data are mean ± SD. p < 0.005. *p = 0.0048 (unpaired t test, t = 4.06, df = 7). Inset, Representative Western blot of sample from wild-type and mutant mouse.
Fgf13 mutant mice exhibit age-dependent sensitivity to hyperthermia-induced seizures
One feature common to the proband (401), his brother (402), and their mother (301) was a history of seizures with elevated temperature as infants. To determine whether loss of one copy of the Fgf13 gene might underlie this phenotype, we tested susceptibility to hyperthermia-induced seizures at three ages, namely, P15, P30, and P60 (Fig. 5A). Elevations of body temperature to 42°C–42.5°C induced behavioral seizures of Class 3 or greater in 9 of 11 (82%) Fgf13 mutant mice at age P15 but not in wild-type littermate control mice (Fig. 5B). Behavioral seizures consisted of clonic forelimb jerking of abrupt onset and termination, a typical episode lasting >40 s. This sensitivity was age-dependent because hyperthermia failed to evoke seizures in mutant mice at ages P30 or P60 (Fig. 5B,C). The age-dependent susceptibility to hyperthermia-induced seizures in Fgf13 mutant mice recapitulates the age-dependent febrile seizures in the proband's mother, the proband, and his brother, individuals with disruption of Fgf13 due to balanced translocation.

Age-dependent susceptibility to hyperthermia-induced seizures in Fgf13+/− mice. A, Schematic representation of the hyperthermia-induced seizure protocol. Black arrows indicate the time at which the mouse enters the hyperthermia chamber and the time at which the heat source is turned off. Shaded arrowhead on top of the line indicates the time at which the 10 min acclimatization to the chamber is over and the heat source was turned on. Arrowheads indicate the time at which a new temperature in increments of 0.5°C was set starting from 37.5°C. Triangles represent the point at which the temperature set-point was achieved. Bars on top of the line represent the duration of 2 min the animals were held at a particular temperature. B, Percentage of Fgf13+/− mice and wild-type mice at P15, P30, and P60 remaining seizure free plotted against core body temperature. Filled diamonds represent P15 mutant animals. Filled square line represents wild-type animals. P30 and P60 animals did not exhibit any seizures at any given temperature and were identical to the wild-type animals. Log-rank test revealed a significant difference between the P15 wild-type (n = 8) and mutant (n = 11) animal seizure-free rates (p = 0.0006; χ2 = 11.73, df = 1). All experiments were performed in a blinded manner to the genotype of the animals. C, Percentage of P15 Fgf13+/− exhibiting seizures. Percentage of wild-type animals and Fgf13+/− mutant animals exhibiting seizures at different ages (P15, P30, and P60) is shown. Wild-type: P15, n = 8; P30, n = 6; P60, n = 4; mutant: P15, n = 11; P30, n = 5; P60, n = 5.
Fgf13 mutant mice exhibit enhanced susceptibility to epilepsy
A second phenotype exhibited by the proband was a history of recurrent temporal lobe seizures beginning in childhood and persisting in adulthood. To determine whether a mutation of Fgf13 might underlie this phenotype, we used the kindling model to compare the susceptibility to epilepsy in Fgf13 mutant and wild-type control mice. No significant differences were detected in the current required to induce the initial electrographic seizure (164 ± 31 and 156 ± 33 μA for Fgf13+/+ [n = 8 animals] and Fgf13+/− [n = 13], respectively, mean ± SEM, Student's t test, p = 0.87 [t = 0.16, df = 19]). However, enhanced seizure responses were evident Fgf13 mutants compared with wild-type control mice (Fig. 6A). Whereas the initial stimulation evoked a brief electrographic seizure (<10 s) without overt behavioral seizure in wild-type mice, this stimulation evoked longer electrographic seizures (>25 s) accompanied by behavioral seizures of Class 2 or greater in Fgf13 mutant mice (Fig. 6A,B). Subsequent stimulations evoked seizures of greater duration and behavioral intensity in the Fgf13 mutant compared with wild-type mice (Fig. 6A,B). Fewer stimulations were required to induce completion of kindling (as defined by occurrence of three consecutive seizures of Class 4 or greater) in Fgf13 mutants compared with wild-type control mice (5.5 ± 0.7 and 15.0 ± 1.9 for Fgf13 mutant and wild-type, respectively, mean ± SEM, p = 2.5e-5, df = 19).

Fgf-13+/− mice exhibit enhanced rate of kindling development and spontaneous recurrent seizures. A, Kindling development is presented as behavioral seizure class (y-axis). Stimulation number (x-axis) refers to the number of stimulations that evoked an electrographic seizure with duration of at least 5 s. Bottom line, Wild-type animals (n = 8). Top line, Fgf13+/− mutant (n = 13) animals. Data are mean ± SEM. Area under the curve analysis for seizure class demonstrated a significant difference (p = 0.002) between wild-type and mutant animals (area under ROC curve = 0.804; SEM = 0.074; df = 17). B, Electrographic seizure duration (seconds) as a function of stimulation number in Fgf13+/− (n = 13) and wild-type mice (n = 8). Bottom line, Wild-type animals. Top line, Fgf13+/− mutant animals. Data are mean ± SEM. Area under the curve analysis for seizure duration demonstrated a significant difference (p = 5.6e-22) between wild-type and mutant animals (area under ROC curve = 0.985; SEM = 0.015; df = 17). C, Spontaneous recurrent seizures in Fgf13 mutant mice. Representative EEG recordings from hippocampus of two wild-type (n = 5) and two mutant animals (n = 5) are shown in C1-C2 and C3-C4, respectively. C1 and C2 are representative EEG recordings from hippocampus of wild-type littermate control female mice. C3 and C4 are representative EEG recordings from hippocampus of littermate mutant mice. All five mutant mice exhibited seizures providing statistically significant evidence (χ2, p = 0.0015) for spontaneous recurrent seizures in the Fgf13 heterozygous mice Mutant animal represented in C3 exhibited 30 spontaneous recurrent seizures (SR) and the animal represented in C4 exhibited 6 spontaneous recurrent seizures. Detection of seizures was performed by individual blinded manner to the genotype of the animals.
The enhanced sensitivity to stimulus-evoked seizures in the kindling model raised the possibility that the Fgf13 mutant mice may be exhibiting spontaneous seizures in the absence of electrical stimulation. Episodic behaviors typical of Class 4 or 5 seizures were observed in several Fgf13 mutant mice but not in wild-type mice, strengthening the possibility of spontaneous seizures in mutant animals. To directly test this possibility, we implanted electrodes into the left hippocampus of five wild-type and five Fgf13 mutant age-matched animals and conducted video EEG monitoring. Each of the five Fgf13 mutant mice exhibited spontaneous recurrent seizures (30, 6, 18, 1, and 26, respectively), whereas no seizures were detected in either of the wild-type control mice. Examples of two wild-type and two mutant animals are presented in Fig. 6C. The behaviors of these spontaneous seizures were similar to Classes 3–6 observed during the kindling experiments. The average duration of electrographic seizures accompanying these behavioral seizures was 28, 31, 26, 11, and 32 s for the five mutant animals, respectively. Seizures were predominantly of behavioral Class 5 and 6 in mutant animals. In sum, Fgf13 mutant mice exhibited spontaneous recurrent seizures similar to those observed in the proband and his brother.
Fgf13 mRNA is expressed in excitatory and inhibitory neurons in mouse hippocampus
As a first step to determine how elimination of one Fgf13 allele confers susceptibility to hyperthermia-induced seizures and recurrent temporal lobe seizures, we sought to identify the brain regions and cells within which the Fgf13 gene is expressed. Notably, no overt abnormalities were evident in Nissl-stained coronal sections of adult Fgf13 mutant mice compared with wild-type controls (data not shown). ISH conducted on a coronal section of adult wild-type mouse brain (Fig. 7) with probes complementary to sequence of exons 2–5 of Fgf13 revealed the highest levels of expression in hippocampus and pyriform cortex with lower levels in thalamic nuclei and neocortex (Fig. 7B). We further sought to establish the cellular locales of Fgf13 expression within the hippocampus because of its high levels of expression together with the central role of hippocampus in both hyperthermia-induced seizures (VanLandingham et al., 1998; McClelland et al., 2011) and recurrent temporal lobe seizures (Engel et al., 1997). Fgf13 mRNA expression was enriched in excitatory CA3 and CA1 pyramidal cells as well as dentate granule cells (Fig. 7B). Review of a higher-power image confirmed the presence of Fgf13 mRNA in these neurons and revealed scattered cells in strata radiatum, oriens, and lacunosum moleculare consistent with expression within interneurons of the CA1 region (Fig. 7C, arrowheads). Multiplex ISH identified the expression of Fgf13 mRNA within inhibitory neurons as evidenced by its colocalization with Gad1 mRNA, a marker for interneurons (Fig. 7D–F). In sum, these experiments reveal expression of Fgf13 mRNA within both excitatory and inhibitory neurons of adult hippocampus.

Fgf13 is expressed in excitatory and inhibitory neurons in mouse hippocampus. A–C, ISH of an adult mouse coronal section using branched nucleotide probes for Fgf13. DAPI staining of the coronal section on which the Fgf13 ISH was performed is shown in A. Fgf13 ISH with branched nucleotide probes shows high expression of the mRNA in multiple regions of the brain, particularly in hippocampus, and pyriform cortex with moderate expression in neocortex and thalamus (B). An enlarged image revealed scattered cells in hippocampus strata oriens (blue arrowheads), radiatum (yellow arrowheads), and lacunosum moleculare (green arrowheads) consistent with expression within interneurons of the CA1 region (C). Scale bars: A, B, 1000 μm. D–F, Dual-color ISH using Gad-1(green) and Fgf13 (red) probes demonstrating the expression of Fgf13 in both excitatory neurons and inhibitory neurons as seen in the merged image by the colocalization of the signals in the CA1 region of the mouse hippocampus. Yellow and red arrows indicate the colocalization of Fgf13 and Gad-1 in interneurons in stratum oriens and stratum radiatum, respectively. Scale bar, 100 μm.
Altered excitatory and inhibitory synaptic inputs to CA1 pyramidal cells of Fgf13 mutant mice
To begin to understand the cellular mechanisms by which elimination of one Fgf13 allele promotes enhanced susceptibility to recurrent temporal lobe seizures, we examined the synaptic and intrinsic properties of CA1 pyramidal cells in whole-cell recordings of hippocampal slices isolated from wild-type and Fgf13 mutant mice. A reduction in the frequency of IPSCs was found in slices of Fgf13 mutant mice compared with controls (mutant 6.1 ± 0.8 Hz [n = 12] vs wild-type 8.0 ± 0.5 Hz [n = 14], p < 0.05; Fig. 8A,B); no significant differences in the amplitude of these currents were found (mutant 35.18 ± 3.88 pA [n = 12] vs wild-type 43.85 ± 4.45 pA [n = 14], p > 0.05; Fig. 8B). Analyses of mIPSCs revealed a reduction in amplitude (mutant 26.1 ± 2.2 pA [n = 18] vs wild-type 35.6 ± 2.1 pA [n = 19], p < 0.01; Fig. 8B,C) but not frequency (mutant 5.5 ± 0.7 Hz [n = 18] vs wild-type 5.2 ± 0.6 Hz [n = 19] p > 0.05; Fig. 7C) in Fgf13 mutant mice compared with wild-type controls. Further analyses of excitatory synaptic events revealed an increased frequency of sEPSCs in Fgf13 mutant mice compared with wild-type controls (mutant 1.1 ± 0.3 Hz [n = 14] vs wild-type 0.6 ± 0.1 Hz [n = 11], p < 0.05; Fig. 8D,E). No differences in either amplitude or frequency of mEPSCs were found (Fig. 8F).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Synaptic properties of CA1 pyramidal neurons in wild-type and Fgf13 mutants. A, Representative whole-cell recordings of sIPSCs and mIPSCs from hippocampal CA1 pyramidal neurons in wild-type and Fgf13+/− mutant animals. B, C, Cumulative fraction curves of interevent intervals (left), average frequency (middle), and amplitude (right) of sIPSCs (B) and mIPSCs (C) in wild-type and Fgf13+/− mutant cells. Data are mean ± SEM. *p < 0.05. **p < 0.01. For sIPSC recordings, n = 14 cells from n = 14 slices for wild-type, and n = 12 cells from n = 12 slices for mutant. sIPSC frequency difference: p = 0.02 (t = 2.15, df = 24); sIPSC amplitude difference: p = 0.07 (t = 1.50, df = 24). For mIPSC recordings, n = 19 cells from n = 19 slices for wild-type, and n = 18 cells from n = 18 slices for mutant. mIPSC frequency difference: p = 0.35 (t = 0.40, df = 35); mIPSC amplitude difference: p = 0.002 (t = 3.18, df = 35). D, Representative sEPSCs and mEPSCs from wild-type and Fgf13+/− mutant hippocampal CA1 pyramidal neurons. E, F, Cumulative fraction curves of interevent intervals (left), average frequency (middle), and amplitude (right) of sEPSCs (E) and mEPSCs (F) in wild-type and Fgf13+/− mutant cells. p < 0.05 (unpaired t test). All experiments were performed in a blinded manner to the genotype of the animals. Data are mean ± SEM. *p < 0.05. For sEPSC recordings, n = 11 cells from n = 11 slices for wild-type, and n = 14 cells from n = 14 slices for mutant. sEPSC frequency difference: p = 0.04 (t = 1.87, df = 16.6, t test with Welch's correction due to significant F test for variance comparison [F = 8.82, p = 0.002]); sEPSC amplitude difference: p = 0.48 (t = 0.06, df = 23). For mEPSC recordings, n = 13 cells from n = 13 slices for wild-type and n = 15 cells from n = 15 slices for mutant. mEPSC frequency difference: p = 0.38 (t = 0.30, df = 26); mEPSC amplitude difference: p = 0.33 (t = 0.46, df = 26).
Examination of the intrinsic properties of CA1 pyramidal cells revealed no differences between wild-type and Fgf13 mutant mice. No significant differences in resting membrane potential (wild-type −70.8 ± 0.8 mV [n = 11] vs mutant −69.1 ± 1.2 mV, [n = 11], mean ± SEM, unpaired t test, p = 0.51 [t = 0.67, df = 20], n = number of cells from equal number of slices from each genotype), cell capacitance (wild-type 81.2 ± 6.3 pF vs mutant 69 ± 5.7 pF, unpaired t test, p = 0.14 [t = 1.52, df = 20]), or input resistance (wild-type 153.9 ± 18.2 MΩ vs mutant 188.1 ± 19.6 MΩ, unpaired t test, p = 0.20 [t = 1.34, df = 20]) were found. Injection of increasing amounts of current-induced similar firing of action potentials in CA1 pyramidal cells in slices from wild-type and Fgf13 mutant mice (data not shown). Collectively, these findings demonstrate that CA1 pyramidal cells of Fgf13 mutant mice receive reduced inhibitory synaptic input and increased excitatory input, abnormalities that may result in increased excitatory output of this local circuit and result in enhanced susceptibility of the hippocampal network to seizures.
Discussion
We analyzed a unique maternally transmitted balanced translocation between chromosomes X and 14 that was associated with the syndrome of GEFS+ in a nuclear family. Five principal findings emerged. First, FISH analyses combined with nested PCR and DNA sequencing revealed that the breakpoint on the X chromosome disrupted a gene that encodes a member of the FGF family, Fgf13. The breakpoint within Fgf13 disrupted expression of three of its five isoforms. Second, targeted ablation of the Fgf13 gene in mice led to lethality in male embryos and reduced expression of Fgf13 mRNA and FGF13 protein in heterozygous female mice. Third, the mutant female mice exhibited age-dependent sensitivity to hyperthermia-induced seizures and recurrent temporal lobe seizures. Fourth, Fgf13 mRNA was richly expressed in both excitatory and inhibitory neurons of hippocampus of adult mice. Fifth, whole-cell recordings of CA1 pyramidal neurons in hippocampal slices isolated from Fgf13 mutant mice revealed decreased inhibitory and increased excitatory synaptic inputs compared with wild-type control mice. Collectively, these findings demonstrate that eliminating one allele of Fgf13 is sufficient to induce an unusual phenotype of neuronal hyperexcitability in mice with features similar to the nuclear family, thereby supporting the conclusion that the mutation of Fgf13 resulting from the translocation caused the syndrome of GEFS+.
FGF13 was discovered as part of an effort to identify novel genes of interest expressed in human retina (Smallwood et al., 1996). Partial sequencing of 5000 cDNAs in a library prepared from human retina revealed a subset of clones containing sequence homology to known FGFs. Database searches combined with degenerate PCR led to identification of a distinct and highly conserved branch of the FGF family, a subfamily that includes FGFs 11, 12, 13, and 14 (Ornitz and Itoh, 2001; Goldfarb, 2005). Multiple isoforms of FGF 11, 12, 13, and 14 are generated through the use of alternate start sites of transcription and splicing of one or more of a series of 5′ exons (Munoz-Sanjuan et al., 2000). Isoforms 1A and 1B (1S and 1U for FGF13) are generated by each of the FGFs, but FGF13 generates three additional isoforms, 1V, 1Y, and 1VY (Munoz-Sanjuan et al., 2000). The functional significance of these various isoforms is incompletely understood. Notably, the mutation of FGF13 introduced into the mouse germline in the present study eliminated expression of all five isoforms, whereas the mutation resulting from the translocation eliminated isoforms V, Y, and VY yet preserved isoforms S and U (Fig. 3D). It seems likely that the presence of isoforms S and U in the proband and his brother prevented the embryonic lethality evident in male mice with the null mutation. The presence of isoforms S and U in the derived chromosome may also account for the lesser severity of the seizure disorder in the proband's mother compared with the heterozygous female mice. That said, isoforms V, Y, and VY undoubtedly serve critical functions as evident in the phenotypic abnormalities in each of the three individuals with the translocation. FGF13 was recently shown to polymerize tubulins and stabilize microtubules (Wu et al., 2012). Genetic deletion of Fgf13 in subsets of neurons resulted in neuronal migration abnormalities in hippocampus in an age-dependent manner, the effects evident at P7 but not later in development or adulthood (Wu et al., 2012); our morphological studies were restricted to animals P30 and older and confirmed the absence of any abnormalities. Notably, the above paper (Wu et al., 2012) did not report the occurrence of seizures in the Fgf 13 mutant mice, a phenotype that may have escaped detection due to the intermittent occurrence of these brief events. Fgf13 has been advanced as a candidate gene for X-linked mental retardation mapped to Xq26 (Malmgren et al., 1993; Gecz et al., 1999); the impaired cognitive function evident in the proband advance Fgf13 as one gene in this interval capable of producing this phenotype.
The phenotypic manifestations of this nuclear family are consistent with the syndrome of GEFS+ (Scheffer et al., 2005; Scheffer and Berkovic, 2008). Notably, the occurrence of partial seizures as exhibited by the proband (Fig. 1C) has been recognized as one feature of GEFS+ (Scheffer and Berkovic, 2008). Causal mutations of a diversity of genes have been implicated in the syndrome of GEFS+, the most common consisting of the SCN1A subunit of voltage-gated Na+ channels (Escayg et al., 2000; Yu et al., 2006; Catterall, 2014). Mutations of an auxiliary subunit of SCN1A, namely, SCN1B, and of the γ2 and δ subunits of the GABAA receptor can also cause this syndrome (Wallace et al., 1998, 2001; Escayg et al., 2000; Baulac et al., 2001; Dibbens et al., 2004). One function ascribed to the FGF11–14 family is that of auxiliary proteins of voltage-gated Na+ channels (Wittmack et al., 2004; Lou et al., 2005; Rush et al., 2006; Laezza et al., 2007, 2009; Goetz et al., 2009; Wang et al., 2011). FGFs can regulate trafficking of pore forming (α) subunits of Na+ channels to the axon initial segment and modulate fast inactivation (Goldfarb et al., 2007; Goetz et al., 2009; Wang et al., 2011; Xiao et al., 2013). FGF13 binds directly to a sequence in the intracellular C-terminal region of multiple α subunits, including SCN1A (Goetz et al., 2009), and can be pulled down efficiently with a carboxy-terminal fragment of SCN1A (Wang et al., 2011). Structural analyses of crystal packing contacts of the C terminus of an α subunit with a 149-residue fragment of FGF13 that carries the structural features of the homology core domain of all FGF 11–14 isoforms, revealed a conserved surface on the core domain that mediates channel binding in vitro and in vivo (Goetz et al., 2009). Notably, mutations of the channel binding surface of FGF13 impaired its ability to colocalize with Na+ channels at the axon initial segment of hippocampal neurons (Goetz et al., 2009) and also impaired FGF-mediated modulation of fast inactivation of Na+ channels (Xiao et al., 2013). Reduced expression of FGF14 resulted in decreased content of α subunits of multiple Nav at the axon initial segment of Purkinje cells, the reduced content of these subunits likely contributing to the reduced excitability of these neurons (Xiao et al., 2013).
How might the absence of one Fgf13 allele modify cellular and local circuit function to produce increased neuronal excitability evident in the heterozygous female mice? Our ISH findings reveal Fgf13 expression in both excitatory neurons, particularly CA3 and CA1 pyramidal cells, and inhibitory interneurons within hippocampus. Our cellular physiological analyses reveal reduced frequency of sIPSCs and increased frequency of sEPSCs within CA1 pyramidal cells in recordings from hippocampal slices acutely isolated from FGF13 heterozygous female mice. Interestingly, recordings from CA1 pyramidal cells in hippocampal slices acutely isolated from SCN1A heterozygous mice revealed identical findings, namely, reduced frequency of sIPSCs and increased frequency of sEPSCs (Han et al., 2012). A parsimonious explanation is that the abnormalities detected in the CA1 pyramidal cells represent a consequence of reduced excitability of inhibitory interneurons, the impaired synaptic inhibition resulting in enhanced firing of CA1 pyramidal neurons. Notably, selective elimination of SCN1A from interneurons, but not principal cells, is sufficient to induce the seizure phenotype (Martin et al., 2010; Cheah et al., 2012; Dutton et al., 2013; Ogiwara et al., 2013), supporting this proposal. Reduced excitability of inhibitory interneurons could be one consequence of impaired trafficking of the α subunit of SCN1A to the axon initial segment (Kole et al., 2008; Wimmer et al., 2010; Kole and Stuart, 2012), the predicted consequence of reduced expression of FGF13 (Laezza et al., 2007; Goetz et al., 2009; Wang et al., 2011). Alternatively, reduced expression of FGF13 could impair fast inactivation of Na+ channels of interneurons, thereby limiting their sustained repetitive firing (Goldfarb et al., 2007; Dover et al., 2010). Our suggestion of impaired function of interneurons together with the fact that mutations of γ and δ subunits of GABAA receptors can cause GEFS+ (Baulac et al., 2001; Dibbens et al., 2004) raises the possibility that impaired synaptic inhibition is a mechanism common to diverse genetic causes of this syndrome. That said, we recognize that alternative mechanisms may contribute to the hyperexcitability observed here, including interactions of FGF13 with voltage-gated calcium channels (Hennessey et al., 2013; Yan et al., 2013), as well as loss of FGF13-mediated long-term inactivation of Na+ channels (Dover et al., 2010; Goldfarb, 2012).
The discovery that a mutation of Fgf13 can cause GEFS+ sheds light on the genetics of this syndrome. Causal mutations have been identified in <20% of families, raising the possibility that mutations of Fgf13 may be identified in some of the remaining 80% of families and potentially in sporadic cases as well. A striking feature of the majority of GEFS+ families is the clinical genetic evidence of complex inheritance. Within a family, the phenotypic manifestations are notable for striking variability in expressivity, expressed perhaps as a simple febrile seizure in one member yet a devastating epileptic encephalopathy in another (Scheffer and Berkovic, 1997, 2008). Inheritance of other genes is thought to be one factor contributing to the varied expressivity. It will be interesting to determine whether variants of Fgf13 contribute to the striking variation in expressivity within GEFS+ families.
Footnotes
- Received August 19, 2014.
- Revision received April 27, 2015.
- Accepted May 1, 2015.
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS056217 to J.O.M., T.L. was supported by Howard Hughes Medical Institute Medical Research Fellows Program. We thank the members of the family reported here for participation in the study; Wei Qian for maintaining and genotyping the animals; Dr. Aita Megumi (University of North Carolina In Situ Hybridization core facility) for help with ISH; Dr. Vladimir Gukkasyan (University of North Carolina Neuroscience Department) for help with image analysis; Drs. Geoffery Pitt, Richard Mooney, Wayne Frankel, and Gautam Narasimhan for input on the manuscript; Ethan Ludmir for the help with statistical analysis; Dr. Saurabh Sinha for help with presentation of patient information; and all members of the J.O.M. laboratory during the course of this work.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. James O. McNamara, Department of Neurobiology, 401 Bryan Research Building, Research Drive Box 3676, Duke University Medical Center, Durham, NC 27710. jmc{at}neuro.duke.edu
- Copyright © 2015 the authors 0270-6474/15/358866-16$15.00/0