
Abstract
Somatodendritically released peptides alter synaptic function through a variety of mechanisms, including autocrine actions that liberate retrograde transmitters. Cholecystokinin (CCK) is a neuropeptide expressed in neurons in the dorsomedial hypothalamic nucleus (DMH), a region implicated in satiety and stress. There are clear demonstrations that exogenous CCK modulates food intake and neuropeptide expression in the DMH, but there is no information on how endogenous CCK alters synaptic properties. Here, we provide the first report of somatodendritic release of CCK in the brain in male Sprague Dawley rats. CCK is released from DMH neurons in response to repeated postsynaptic depolarizations, and acts in an autocrine fashion on CCK2 receptors to enhance postsynaptic NMDA receptor function and liberate the retrograde transmitter, nitric oxide (NO). NO subsequently acts presynaptically to enhance GABA release through a soluble guanylate cyclase-mediated pathway. These data provide the first demonstration of synaptic actions of somatodendritically released CCK in the hypothalamus and reveal a new form of retrograde plasticity, depolarization-induced potentiation of inhibition.
SIGNIFICANCE STATEMENT Somatodendritic signaling using endocannabinoids or nitric oxide to alter the efficacy of afferent transmission is well established. Despite early convincing evidence for somatodendritic release of neurohypophysial peptides in the hypothalamus, there is only limited evidence for this mode of release for other peptides. Here, we provide the first evidence for somatodendritic release of the satiety peptide cholecystokinin (CCK) in the brain. We also reveal a new form of synaptic plasticity in which postsynaptic depolarization results in enhancement of inhibition through the somatodendritic release of CCK.
- cholecystokinin
- dorsomedial hypothalamus
- GABA
- nitric oxide
- NMDAR
- somatodendritic release
Introduction
Somatodendritic release of chemical messengers has emerged as a key mechanism for regulating synaptic activity. The most widely studied are fast-acting, labile transmitters such as endocannabinoids (eCBs) and nitric oxide (NO) that are liberated from the postsynaptic neuron and modulate neurotransmitter release from afferent terminals (Iremonger et al., 2013). There are also a number of reports, particularly in hypothalamic nuclei, that somatodendritically released neuropeptides also control synaptic function. In particular, there is clear evidence that arginine vasopressin, oxytocin, dynorphin, and enkephalin are released from the somatodendritic compartments of neurons in the hypothalamic paraventricular and supraoptic nuclei. Here, these peptides act as autocrine or paracrine signals to modulate neuronal excitability and alter behavior (Ludwig and Pittman, 2003; Landgraf and Neumann, 2004; Leng and Ludwig, 2008; Iremonger et al., 2011; Wamsteeker Cusulin et al., 2013). The autocrine actions often result in the liberation of a retrograde messenger that modulates presynaptic neurotransmitter release. For example, oxytocin released from the dendrites of neurons in the supraoptic nucleus triggers the synthesis of eCBs that quench glutamate (Hirasawa et al., 2004) or GABA (Oliet et al., 2007) release from afferent terminals. Whether similar autocrine actions are observed more broadly in the brain is not known.
The dorsomedial hypothalamic nucleus (DMH), a region implicated in satiety and stress, offers an opportunity to study putative interactions between autocrine and retrograde transmitters. We demonstrated previously that retrograde eCB and NO signals are important regulators of GABA signaling in the DMH (Crosby et al., 2011). In addition, DMH neurons express a multitude of peptides, particularly those that regulate food intake. Cholecystokinin (CCK), a satiety peptide and neuromodulator, is abundantly expressed in DMH neurons (Innis et al., 1979; Micevych et al., 1987; Otake, 2005; Wagner et al., 2013), which also express CCK receptors (CCKRs; Gaudreau et al., 1983; Durieux et al., 1988; Bi et al., 2004). Administration of CCK into the DMH reduces food intake (Blevins et al., 2000; Chen et al., 2008) and alters neuropeptide expression in these neurons (Bi et al., 2001), but how CCK alters neuronal function or synaptic drive to DMH neurons is not known.
In endothelial cells, CCK triggers NO production through CCK1Rs and CCK2Rs (Grossini et al., 2012). Since NO has robust actions at GABA synapses in the DMH (Crosby et al., 2011), one possibility is that CCK acts locally to enhance the production of NO. In addition, CCK enhances NMDA receptor (NMDAR) activity (Xiao et al., 2012), which is coupled to NO synthesis (Garthwaite et al., 1988; Bains and Ferguson, 1997; Nugent et al., 2007; Crosby et al., 2011), providing a possible cellular link between activation of CCKRs and NO production. Here, we show that repetitive postsynaptic depolarization liberates CCK from DMH neurons. CCK acts in an autocrine fashion to enhance NMDAR activity, resulting in NO production. In turn, NO acts as a retrograde signal to increase the probability of GABA release.
Materials and Methods
Animals.
All experiments were performed using male Sprague Dawley rats [postnatal day 21 (P21)–P30] obtained from Charles River Laboratories. Animals were maintained in specified pathogen-free conditions on a 12 h light/dark cycle at 22°C with food and water available ad libitum. Experimental procedures received approval from the University of Calgary Animal Care and Use Committee in accordance with guidelines established by the Canadian Council on Animal Care.
Brain slice preparation.
Animals were anesthetized with isofluorane, decapitated, and the brains quickly removed and placed in ice-cold slicing solution containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, and 75 sucrose saturated with 95% O2 and 5% CO2. Angled horizontal slices (250 μm) containing the DMH were obtained using a vibrating slicer (Leica), and slices were incubated in 32.5°C artificial CSF (ACSF) containing (in mm) 126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, and 10 glucose, saturated with 95% O2 and 5% CO2, for a minimum of 60 min. Slices remained in this incubation chamber until recording.
Electrophysiology.
Hypothalamic slices were placed in a recording chamber and superfused with 32.5°C ACSF at a flow rate of 1 ml/min. DMH neurons in the compact zone were identified and visualized with an Olympus upright microscope fitted with infrared differential interference contrast optics. Whole-cell electrophysiological recordings were made with borosilicate glass microelectrodes (tip resistance, 4.0–6.0 MΩ) filled with a recording solution containing the following (in mm): 108 potassium gluconate, 8 Na gluconate, 2 MgCl2, 8 KCl, 1 potassium EGTA, 4 potassium ATP, 0.3 sodium GTP, and 10 HEPES, corrected to pH 7.2 with KOH. In a subset of experiments, custom synthesized SNAP-25 blocking peptide or SNAP-25 scrambled peptide (Anaspec) was included in the intracellular solution (50 μm each). The SNAP-25 blocking peptide had a sequence of MEKADANKTRI (11 amino acid peptide corresponding to residues 182–192 of the SNAP-25 protein), and the SNAP-25 scrambled peptide was KANAKTDEIRM (same residue composition, but randomized). To disrupt G-protein signaling in another subset of experiments, 1 mm GDPβS was added to the above intracellular solution, and sodium GTP was removed. Recordings were accepted for analysis if changes in access resistance were <15%. Signals were amplified using the Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 1 kHz, digitized at 10 kHz using the Digidata 1322 (Molecular Devices), and stored for off-line analysis.
IPSCs were isolated by holding the postsynaptic neuron at −70 mV and by including DNQX (10 μm) in the perfusate to block AMPA and kainate receptor-mediated glutamatergic transmission. GABAergic fibers were stimulated extracellularly with a patch pipette filled with ACSF and positioned ∼50 to 100 μm from the recorded DMH neurons. IPSCs were evoked at a rate of 0.2 Hz, and paired-pulse responses were obtained by applying a pair of synaptic stimuli 50 ms apart. DMH neurons were repetitively depolarized in voltage-clamp mode from −70 to 0 mV for 100 ms at a frequency of 2 Hz, 240 times. Single synaptic stimuli were delivered following every fourth depolarization to assay synaptic strength. To prevent extracellular peptide degradation, the aminopeptidase inhibitor amastatin (10 μm) was included in the perfusate. Because depolarization triggers eCB synthesis and release and can lead to presynaptic suppression of neurotransmitter release, CB1 receptors (CB1Rs) were blocked with AM251 (5 μm) throughout these experiments.
In a subset of experiments, NMDAR currents were isolated by including DNQX (10 μm) to block AMPA-mediated currents and picrotoxin (100 μm) to block GABAA receptors. N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314; 5 mm) was included in the patch pipette solution to block voltage-gated Na+ channels. NMDAR currents were recorded at a holding potential of +40 mV using a pipette solution containing the following (in mm): 130 CsCl, 10 NaCl, 10 EGTA, 0.1 CaCl2, 4 K2ATP, and 0.3 Na3GTP, buffered with 10 HEPES. Cells were repetitively depolarized after stable baseline recording as described above.
In another series of experiments, DMH neurons were not repetitively depolarized, but instead were incubated in 0.1 μm CCK-8S (a sulfated form of the 8 amino acid variant of CCK) for 10 min following baseline recording, and changes in IPSCs or NMDAR currents were assessed.
Data analysis.
Evoked currents were analyzed using Clampfit 9 (Molecular Devices). The amplitude of the synaptic current was calculated from the baseline (current immediately before the evoked response) to the peak of each evoked response. Mean IPSC amplitudes (in 30 s epochs) were compared by expressing current amplitudes as a percentage of those obtained during the baseline recording period within each cell. The paired pulse ratio (PPR) was calculated by dividing the mean of the second peak IPSC amplitude by the mean of the first peak IPSC amplitude for every minute of recording. The coefficient of variation (CV) was determined by dividing the standard deviation of the IPSC amplitude by the mean amplitude. For clarity, the stimulus artifacts have been removed digitally from the traces depicted. IPSC and NMDAR current traces in all figures represent those averaged from the 5 min period immediately before (baseline) and 5 to 10 min following the end of repetitive depolarization.
Spontaneous IPSCs (sIPSCs; recorded in the absence of tetrodotoxin) that were observed in the intervals between consecutive applications of synaptic stimuli were analyzed using Mini Analysis software (Synaptosoft). The threshold for the detection of sIPSCs was five times the root mean square noise. sIPSC frequency was measured in events per minute, and amplitude was measured as the average amplitude in 1 min bins.
Statistical analysis.
Statistical analysis was performed on baseline measurements taken during the 5 min before repetitive depolarization and 5 to 10 min following the end of the depolarization. Results are expressed as means ± SEM. Significance was determined using a Student's paired t test in which data during the 5 to 10 min following depolarization were compared to baseline values, or a one-sample t test compared to a hypothetical value of 100%. Data were considered significant if the p value was <0.05.
Results
Postsynaptic repetitive depolarization triggers potentiation of GABA synapses that requires somatodendritic peptide release
To determine whether somatodendritic CCK modulates neuronal activity in the DMH, we obtained whole-cell recordings from DMH neurons in slices from male Sprague Dawley rats. We first tested whether we could evoke changes in synaptic activity using a postsynaptic depolarization protocol that was shown previously to elicit somatodendritic peptide release in the hypothalamus (Iremonger and Bains, 2009). Neurons were depolarized in voltage-clamp mode from −70 to 0 mV for 100 ms at a frequency of 2 Hz, 240 times (Fig. 1A,B). This repetitive depolarization elicited a robust potentiation of GABA synapses as assessed by examining IPSC amplitude (160.7 ± 19.3% of baseline; n = 12; 12 animals; p = 0.009; Fig. 1C,D), now referred to as depolarization-induced potentiation of inhibition (DPI). To determine whether DPI has a presynaptic or postsynaptic locus, we examined several variables including PPR, CV, and the frequency and amplitude of sIPSCs. Following repetitive depolarization, we observed a decrease in PPR (91.5 ± 3.6% of baseline; n = 12; 12 animals; p = 0.037; Fig. 1E) and CV (87.2 ± 6.0% of baseline; n = 12; 12 animals; p = 0.046; Fig. 1E), and an increase in sIPSC frequency (123.8 ± 9.4% of baseline; n = 12; 12 animals; p = 0.014; Fig. 1F), but no change in sIPSC amplitude (104.4 ± 4.6% of baseline; n = 12; 12 animals; p = 0.357; Fig. 1F). Together, these findings suggest a depolarization-induced increase in GABA release from the presynaptic terminal.

Repetitive depolarization triggers potentiation of GABA synapses in the DMH. A, Schematic representation of experimental setup (top) and repetitive depolarization protocol (bottom). B, Schematic representation of DMH illustrating the approximate position of recording and stimulating electrodes in the compact zone (CZ) of the DMH. f, Fornix. C, Sample traces of IPSCs before and after repetitive depolarization (top) and a single representative cell (bottom) illustrating potentiation following repetitive depolarization. Traces in all figures are taken from the same cell and averaged for 5 min immediately before (Baseline) and 5–10 min following the end of depolarization (Post depol.). The stimulation artifacts in this and all subsequent figures have been digitally removed for clarity. Calibration: 25 pA, 10 ms. The dotted line represents the average of baseline recording. The shaded area represents the time and duration of depolarization. D, Summary data showing potentiation of GABA synapses following repetitive depolarization (n = 12). The dotted line indicates the baseline, and the solid colored line indicates the duration at which we conducted statistical analysis compared to baseline. Values are mean ± SEM. E, Summary PPR (left) and CV (right) data showing decreases following repetitive depolarization (the black bars in these and subsequent figures indicate the means). F, Summary sIPSC data showing an increase in frequency and no change in amplitude following repetitive depolarization. Inset, Representative sIPSC traces taken before and after repetitive depolarization. Calibration: 25 pA, 0.5 s. *p < 0.05.
We next asked whether enhancement of presynaptic GABA release is due to somatodendritic peptide release from DMH neurons. There is considerable evidence that somatodendritic peptides are stored in vesicles and that their release requires SNARE-dependent exocytosis (Iremonger and Bains, 2009; Tobin et al., 2012). SNAP-25 is a member of the SNARE family and assembles with other proteins to form the SNARE complex that is required for vesicular exocytosis (Jahn et al., 2003). We therefore examined whether postsynaptic SNARE-dependent exocytosis is required for DPI by including a SNAP-25 blocking peptide (50 μm) in the patch pipette to inhibit postsynaptic vesicular fusion. We then assessed the effect of postsynaptic depolarization on IPSCs in the presence of the SNAP-25 blocking peptide and found that repetitive depolarization had no effect on IPSC amplitude (96.5 ± 11.5% of baseline; n = 7; five animals; p = 0.775; Fig. 2A), PPR (102.0 ± 8.2% of baseline; n = 7; five animals; p = 0.809), or CV (100.4 ± 3.2% of baseline; n = 7; five animals; p = 0.856). In contrast, in the presence of a SNAP-25 scrambled peptide (50 μm) in the patch pipette, DPI persisted with an increase in IPSC amplitude (125.8 ± 4.5% of baseline; n = 8; five animals; p = 0.0004; Fig. 2B), that was associated with a decrease in PPR (91.5 ± 3.8% of baseline; n = 8; five animals; p = 0.038), but no significant change in CV (99.3 ± 4.2% of baseline; n = 8; five animals; p = 0.830). These findings implicate postsynaptic vesicular fusion and release in the potentiation of GABA synapses.
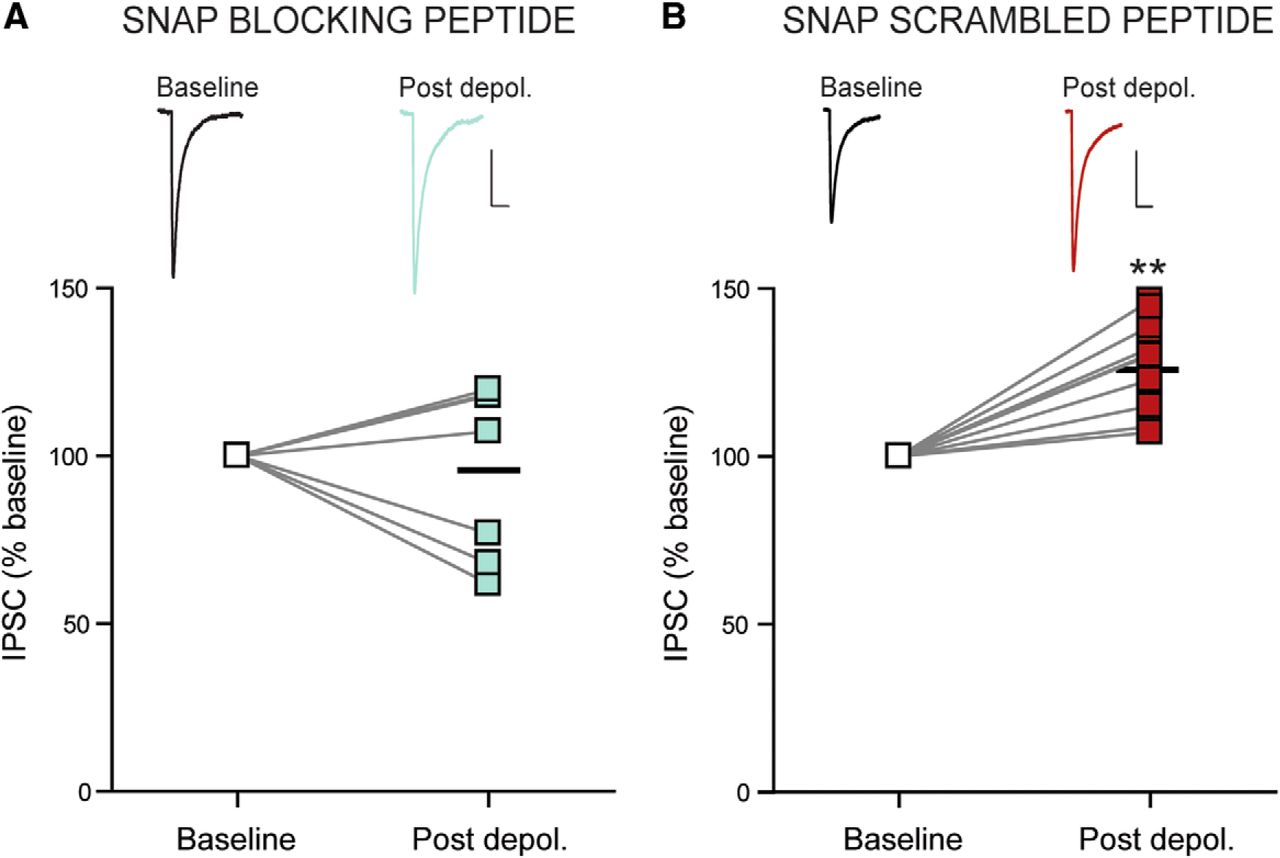
Postsynaptic vesicular release is required for DPI. A, Sample traces of IPSCs before and after repetitive depolarization (top) and summary data from each cell (bottom; n = 7) showing no potentiation in the presence of the SNAP-25 blocking peptide (50 μm; included in the patch pipette). B, Sample traces of IPSCs before and after repetitive depolarization (top) and summary data from each cell (bottom; n = 8) showing potentiation in the presence of the SNAP-25 scrambled peptide (50 μm; included in the patch pipette). Calibrations: 25 pA, 10 ms. **p < 0.01 versus baseline.
CCK-induced autocrine activation of CCK2Rs mediates DPI
We next determined which transmitter is responsible for DPI. There is unquestionable evidence that DMH neurons express the peptide CCK (Innis et al., 1979; Micevych et al., 1987; Otake, 2005; Wagner et al., 2013), and CCK increases GABA release in other brain regions (Kombian et al., 2005; Deng and Lei, 2006; Chung and Moore, 2007; Földy et al., 2007; Karson et al., 2008). Thus, to determine whether repetitive depolarization releases CCK to augment IPSCs, we depolarized DMH neurons in the presence of CCKR antagonists. We first examined whether CCK2R activation is required for DPI. In the presence of the CCK2R antagonist 2-[2-(5-bromo-1H-indol-3-yl)ethyl]-3-(1-methylethoxy)phenyl]-4-(3H)-quinazoline (LY-225910; 1 μm), GABA synapses failed to potentiate, as assessed by examining IPSC amplitude (94.9 ± 9.2% of baseline; n = 7; three animals; p = 0.604; Fig. 3A), PPR (97.8 ± 5.9% of baseline; n = 7; three animals; p = 0.475), and CV (104.7 ± 7.7%; n = 7; three animals; p = 0.460). Similarly, we observed no change in the frequency (79.0 ± 23.3% of baseline; n = 7; three animals; p = 0.27) or amplitude (106.9 ± 24.1% of baseline; n = 7; three animals; p = 0.80) of sIPSCs with continuous blockade of CCK2Rs. CCK1Rs are also expressed in the DMH; therefore, we examined the effect of postsynaptic depolarization on GABA release in the presence of the CCK1R antagonist lorglumide (1 μm). Under these conditions, we observed a variable increase in IPSC amplitude (141.3 ± 29.2% of baseline; n = 6; three animals; p = 0.206; Fig. 3B), with no significant change in PPR (88.2 ± 9.5% of baseline; n = 6; three animals; p = 0.173), or CV (118.8 ± 18.9% of baseline; n = 6; three animals; p = 0.480). Importantly, there was no difference in IPSC amplitude following postsynaptic depolarization in control slices compared to lorglumide (p = 0.571, Student's t test). Thus, we conclude that repetitive depolarization releases CCK that acts primarily on CCK2Rs to augment presynaptic GABA release.

CCK-induced activation of CCK2Rs mediates DPI. A, Sample traces of IPSCs before and after repetitive depolarization in the presence of the CCK2R antagonist LY-225910 (1 μm; top) and summary data showing that LY-225910 completely blocks potentiation of GABA synapses following repetitive depolarization (bottom; n = 7). Calibration: 25 pA, 10 ms. B, Summary data showing the percentage change in IPSC amplitude following repetitive depolarization in the following conditions: control (n = 12), LY-225910 (n = 7), lorglumide (CCK1R antagonist; 1 μm; n = 7), and GDPβS (1 mm; in the patch pipette; n = 6). **p < 0.01 versus baseline.
Having determined that CCK is released somatodendritically to activate CCK2Rs, we next asked whether these receptors are localized on the presynaptic GABA terminal, or, as has been shown previously for some hypothalamic peptides, on the postsynaptic cell of origin with the peptide acting in an autocrine manner. To test the latter possibility, we examined the impact of postsynaptic G-protein signaling on the ability of GABA afferents to undergo potentiation. When GDPβS (1 mm) was included in the patch pipette to disrupt postsynaptic G-protein signaling, potentiation of GABA synapses was completely abolished (86.4 ± 13.2% of baseline; n = 6; four animals; p = 0.350; Fig. 3B), as were the changes in PPR (98.8 ± 9.0% of baseline; n = 6; four animals; p = 0.586) and CV (99.2 ± 7.7% of baseline; n = 6; four animals; p = 0.640). Thus, it appears that when CCK is released by postsynaptic depolarization, it acts in an autocrine fashion on the cell from which it is released.
Exogenous application of CCK enhances GABA release through a CCK2R- and NO-mediated pathway
We have demonstrated that somatodendritic CCK increases GABA release through a CCK2R-mediated mechanism. Exogenous application of CCK should therefore elicit a similar increase in GABA transmission. Consistent with this idea, a 10 min application of 0.1 μm CCK-8S potentiated IPSCs (142.9 ± 11.7% of baseline; n = 12; eight animals; p = 0.003; Fig. 4A,B) and triggered a decrease in PPR (93.4 ± 4.6% of baseline; n = 12; eight animals; p = 0.048; Fig. 4C) and CV (87.9 ± 5.6% of baseline; n = 12; eight animals; p = 0.048; Fig. 4C), an increase in sIPSC frequency (198.0 ± 30.0% of baseline; n = 12; eight animals; p = 0.008; Fig. 4D), but no significant change in sIPSC amplitude (116 ± 8.0% of baseline; n = 12; eight animals; p = 0.071; Fig. 4D). This potentiation was completely blocked when CCK-8S was applied in the continuous presence of the CCK2R antagonist LY-225910 (103.2 ± 5.1% of baseline; n = 5; four animals; p = 0.548; Fig. 4E); there was also no change in PPR (98.5 ± 6.5% of baseline; n = 5; four animals; p = 0.548) or CV (109.9 ± 12.3% of baseline; n = 5; four animals; p = 0.414). If CCK is acting on the postsynaptic neuron, it must be triggering the release of a substance that is liberated postsynaptically and diffuses to the presynaptic terminal to alter GABA release. We demonstrated previously that NO, when liberated from DMH neurons, enhances GABA release from afferent terminals (Crosby et al., 2011). Thus, to determine whether NO mediates the CCK-induced potentiation of GABA release, we examined the ability of CCK to increase IPSC amplitude in the continuous presence of the broad-spectrum NO synthase (NOS) inhibitor N-ω-nitro-l-arginine methyl ester (l-NAME; 200 μm). Under these conditions, CCK-8S failed to potentiate GABA release (89.8 ± 4.9% of baseline; n = 7; seven animals; p = 0.084; Fig. 4F) or alter PPR (99.9 ± 0.9% of baseline; n = 7; seven animals; p = 0.891) or CV (114.2 ± 23.3% of baseline; n = 7; seven animals; p = 0.481), suggesting NO is required for CCK-induced enhancement of GABA transmission.

Exogenous application of CCK-8S enhances GABA release through a CCK2R- and NO-mediated pathway. A, Sample traces of IPSCs before and during application of CCK-8S (0.1 μm; top) and a single representative cell (bottom) illustrating potentiation in response to CCK-8S application (duration of application indicated by colored bar). Calibration: 25 pA, 10 ms. B, Summary data from each cell showing potentiation of IPSCs with CCK-8S application (n = 12). C, Summary PPR (left) and CV (right) data showing decreases with CCK application. D, Summary sIPSC data showing an increase in frequency and no change in amplitude with CCK application. Inset, Representative sIPSC traces taken before and during CCK-8S application. Calibration: 25 pA, 0.5 s. E, Summary data from each cell showing no change in IPSCs with CCK-8S application in the continuous presence of LY-225910 (CCK2R antagonist; n = 7). F, Summary data from each cell showing no change in IPSCs with CCK-8S application in the continuous presence of l-NAME (NOS inhibitor; n = 7). *p < 0.05; **p < 0.01 versus baseline.
Retrograde NO signaling and NMDAR activation are necessary for DPI
To determine whether NO also mediates the depolarization-induced enhancement of GABA release, we performed two experiments. First, we examined whether the potentiation of GABA synapses was impacted by blocking the production of NO. In the presence of l-NAME, repetitive depolarization failed to enhance IPSC amplitude (83.4 ± 12.8% of baseline; n = 7; four animals; p = 0.251; Fig. 5A) or alter PPR (102.0 ± 3.3% of baseline; n = 7; four animals; p = 0.574) or CV (86.4 ± 9.1% of baseline; n = 7; four animals; p = 0.232). This suggests that DMH neurons synthesize NO in response to intense depolarization. Accumulating evidence suggests that NO enhances GABA release through a presynaptic intracellular signaling cascade that originates with the “NO receptor” soluble guanylate cyclase (sGC; Nugent et al., 2007; Crosby et al., 2011). Furthermore, sGC and other presynaptic NO machinery required for modulation of neurotransmitter release are localized in the DMH (El-Husseini et al., 1999). Therefore, we next tested whether activation of sGC was required for the potentiation of GABA synapses in response to postsynaptic depolarization. Consistent with a role for retrograde NO signaling, blockade of sGC with the sGC inhibitor 1H-(1,2,4)oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; 10 μm) completely blocked the increase in IPSC amplitude (74.3 ± 11.9% of baseline; n = 5; two animals; p = 0.082; Fig. 5B) and the change in PPR (108.8 ± 15.2% of baseline; n = 5; two animals; p = 0.838) and CV (101.3 ± 14.3% of baseline; n = 5; two animals; p = 0.743). These data suggest that retrograde NO signaling mediates DPI in the DMH. NO synthesis is often coupled to NMDAR activation (Bains and Ferguson, 1997; Nugent et al., 2007) due to a physical link between NO synthase and NMDARs via the scaffolding protein postsynaptic density-95 (Brenman et al., 1996). We therefore asked whether NMDAR activation plays a role in postsynaptic depolarization-induced NO signaling. In the presence of the NMDAR antagonist APV (50 μm), GABA synapses failed to potentiate in response to repetitive depolarization (103.3 ± 12.3% of baseline; n = 7; three animals; p = 0.796; Fig. 5C), with no associated changes in PPR (97.2 ± 11.9% of baseline; n = 7; three animals; p = 0.600) or CV (117.6 ± 18.2% of baseline; n = 7; three animals; p = 0.522). These data suggests that, in addition to CCK2R activation, NMDAR activation is also necessary for DPI.

NMDAR activation and retrograde NO signaling are necessary for DPI. A, Sample traces of IPSCs before and after repetitive depolarization in the presence of the NO synthase inhibitor l-NAME (200 μm; top) and summary data from each cell showing l-NAME completely blocks potentiation of GABA synapses following repetitive depolarization (bottom; n = 6). B, Sample traces of IPSCs before and after repetitive depolarization in the presence of the sGC inhibitor ODQ (10 μm; top) and summary data from each cell showing ODQ completely blocks potentiation of GABA synapses following repetitive depolarization (bottom; n = 6). C, Sample traces of IPSCs before and after repetitive depolarization in the presence of the NMDAR antagonist APV (50 μm; top) and summary data from each cell showing APV completely blocks potentiation of GABA synapses following repetitive depolarization (bottom; n = 7). Calibrations: 25 pA, 10 ms.
Postsynaptic repetitive depolarization enhances NMDAR-mediated currents through a CCK2R-dependent mechanism
We next analyzed the effect of repetitive depolarization on NMDAR currents. NMDAR currents were recorded at +40 mV in the presence of the voltage-gated Na+ channel blocker QX-314 and with AMPA, kainate, and GABAA receptors blocked. We confirmed that the currents elicited at +40 mV were NMDAR-mediated currents by applying APV. Application of APV (50 μm) completely abolished these currents (Fig. 6A), confirming that they are mediated by NMDARs. To determine whether NMDAR currents are altered in response to postsynaptic depolarization, we assessed the amplitude of these currents before and after repetitive postsynaptic depolarization. Baseline NMDAR currents were recorded at +40 mV, and membrane potential was then quickly returned to −70 mV to deliver the repetitive depolarization protocol. In response to this depolarization, there was a robust potentiation of NMDAR currents (131.1 ± 5.6% of baseline; n = 7; four animals; p = 0.001; Fig. 6B–D). These data indicate a depolarization-induced increase in the function of NMDARs. To determine whether CCK2Rs are required for this potentiation in NMDAR currents, we repeated this experiment with LY-225910 and failed to observe any depolarization-induced potentiation of NMDAR currents (99.8 ± 4.62%; n = 7; four animals; p = 0.962; Fig. 6E). Because we observed a variable increase in GABA release in response to CCK1R blockade, we further probed the role of this receptor subtype by examining NMDAR currents in the presence of the CCK1R antagonist lorglumide. With CCK1Rs blocked, we observed an increase in NMDAR currents (119.1 ± 7.0% of baseline; n = 5; three animals; p = 0.052; Fig. 6E), suggesting these receptors likely do not play a major role in enhancing NMDAR activity in DMH neurons. Also, there was no significant difference in NMDA currents between control and lorglumide conditions (p = 0.202, Student's t test). Finally, we tested whether exogenous application of CCK-8S enhances NMDAR currents. Consistent with this idea, we observed a significant increase in NMDAR currents in response to CCK-8S (120.3 ± 3.5% of baseline; n = 5; four animals; p = 0.004; Fig. 6F). Together our findings suggest that CCK acts as an autocrine signal to activate CCK2Rs on DMH neurons following repeated depolarization, resulting in enhanced NMDAR activity and subsequent retrograde NO signaling to stimulate presynaptic GABA release (Fig. 7).

Repetitive depolarization triggers potentiation of NMDA currents. A, Sample traces of NMDAR-mediated currents before (black) and during incubation with the NMDAR antagonist APV (50 μm; blue). B, Sample traces of NMDAR-mediated currents before (left) and after repetitive depolarization (right). C, Single representative cell illustrating potentiation in NMDAR-mediated currents following repetitive depolarization. D, Summary data from each cell showing potentiation in NMDAR-mediated currents following repetitive depolarization. E, Summary data from each cell showing percentage change in NMDA current amplitude following repetitive depolarization in the following conditions: control (n = 7), LY-225910 (CCK2R antagonist; n = 7), and lorglumide (CCK1R antagonist; n = 5). F, Summary data from each cell showing that exogenous application of CCK enhances NMDA current amplitude (n = 5). **p < 0.01 versus baseline. Calibrations: 25 pA, 10 ms.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of somatodendritic CCK release and subsequent DPI. Postsynaptic depolarization (1) triggers somatodendritic CCK release (2). CCK activates postsynaptic CCK2Rs (3), which increase NMDAR activity (4). NMDAR activation triggers NO production through coupling to neuronal NOS (5), and NO acts as a retrograde signal (6) to enhance GABA release from the presynaptic terminal (7).
Discussion
Here, we demonstrated that depolarization of DMH neurons triggers the somatodendritic release of CCK, which acts as an autocrine signal to enhance NMDAR function and liberate NO. Subsequent retrograde NO signaling stimulates the release of GABA from presynaptic terminals in the DMH. These observations provide two critical pieces of information: first, they provide, to the best of our knowledge, the first report of somatodendritic release of the neuropeptide CCK in the brain. Second, they describe a novel form of synaptic regulation in which postsynaptic depolarization enhances synaptic inhibition. The latter observation is particularly intriguing because it provides an example of a feedback system in which a neuron quenches its own excitability by increasing the release of GABA from its afferents. This stands in contrast to the widespread inhibition of neurotransmitter release reported in multiple brain regions following postsynaptic depolarization and the subsequent liberation of eCBs (Wilson and Nicoll, 2002).
Our data suggest that DMH neurons provide a local source of CCK through somatodendritic release following postsynaptic depolarization. The CCK peptide is expressed in neurons in the DMH (Innis et al., 1979; Micevych et al., 1987; Otake, 2005; Wagner et al., 2013), and although DMH neurons receive input from both intrahypothalamic and extrahypothalamic regions that may express CCK, there is no direct evidence of CCK terminals impinging upon DMH neurons. It remains unknown whether CCK is somatodendritically released from neurons in other CNS regions; however, there is direct evidence for CCK expression in dendrites (Paloff et al., 1996; Wyeth et al., 2012). There is also evidence that CCK can be endogenously released from axons of hippocampal interneurons following the same repetitive depolarization protocol used in our study (Deng et al., 2010).
We report that somatodendritic CCK enhances GABA release onto DMH neurons, a finding corroborated with exogenous application of CCK-8S. CCK has been shown to increase GABA release in other brain regions (Kombian et al., 2005; Deng and Lei, 2006; Chung and Moore, 2007; Földy et al., 2007; Karson et al., 2008), with many reports suggesting this effect is mediated by presynaptic CCKRs. Although our data also suggest a presynaptic locus, our observations indicate a model in which somatodendritic CCK acts as an autocrine signal to activate postsynaptic CCK2Rs and liberate the retrograde messenger, NO. Indeed, our finding that DPI is blocked with inclusion of GDPβS in the patch pipette suggests that CCK2Rs are localized on the postsynaptic neuron. Blockade of CCK2Rs in our study completely abolished the increase in GABA release observed following repetitive depolarization and exogenous application of CCK-8S, indicating that CCK2R activation is required for CCK-induced modulation of GABA release; this is supported by evidence of expression of CCK2Rs in the DMH (Gaudreau et al., 1983; Durieux et al., 1988). Although CCK1Rs are also localized on DMH neurons (Moran et al., 1986; Miyasaka and Funakoshi, 2003), our observations suggest that these receptors do not reliably participate in enhancing NMDAR currents or increasing GABA release. We do note, however, that there was some degree of variability when CCK1Rs were blocked. This variability could be due to the heterogenous population of neurons in the DMH (Crosby et al., 2011), with CCK1Rs potentially contributing to NO liberation at a fraction of the synapses. Alternatively, it may indicate that, in some conditions, CCK1Rs and CCK2Rs act in a synergistic manner (Mohammad et al., 2012).
The physiological consequences of CCK's actions in the DMH have yet to be resolved. CCK is a well-established satiety signal, and microinjection of CCK directly into the DMH triggers a long-lasting reduction in food intake (Blevins et al., 2000; Chen et al., 2008). There is also evidence that CCK downregulates the orexigenic neuropeptide Y in DMH neurons (Bi et al., 2001); however, our data suggest that additional, more rapid CCK actions exist in the DMH. It is possible that CCK inhibits NPY-expressing neurons in the compact zone of the DMH, thus preventing the release of NPY into downstream targets including the paraventricular nucleus of the hypothalamus. In addition to playing a role in satiety, the DMH is one of the key players in regulating the stress response (Crosby and Bains, 2012; Stamper et al., 2015), and CCK2R activity has been associated with activation of the hypothalamic–pituitary–adrenal axis (Abelson and Young, 2003; Malendowicz et al., 2003). Thus, it is possible that our proposed signaling pathway plays a role in the response to stress.
We demonstrate that activation of CCK2Rs triggers an increase in NMDAR currents in DMH neurons. Although the mechanism underlying this phenomenon is not yet known, it could be due to an increase in the surface expression of NMDARs or the composition or phosphorylation of NMDAR subunits. There is widespread evidence that NMDARs are regulated by numerous G-protein coupled receptors (for review, see Rojas and Dingledine, 2013), including type I (Benquet et al., 2002; Cheng et al., 2013) and type II (Skeberdis et al., 2001) metabotropic glutamate receptors, orexin A receptors (Borgland et al., 2006), and muscarinic receptors (Lu et al., 1999). Furthermore, previous work demonstrated that activation of CCK2Rs in hippocampal neurons enhances NMDAR function via activation of the phospholipase C and protein kinase C (PKC) pathway (Xiao et al., 2012). Indeed, numerous studies have demonstrated a link between PKC and potentiation of NMDAR currents, with potential mechanisms including phosphorylation of the NR2 subunit resulting in enhanced channel activity (Liao et al., 2001) and phosphorylation of SNAP-25 leading to NMDAR delivery to the cell surface (Lau et al., 2010).
We and others have demonstrated that NO enhances GABA release from presynaptic terminals in the hypothalamus (Horn et al., 1994; Bains and Ferguson, 1997; Stern and Ludwig, 2001; Di et al., 2009; Crosby et al., 2011) and throughout the CNS (Nugent et al., 2007; Fenselau et al., 2011; Lange et al., 2012). The source of this NO is likely neuronal, since blockade of NMDARs, which are coupled to neuronal NO synthase (Brenman et al., 1996), prevents NO-induced GABA release. Interestingly, NMDARs can also potentiate GABA transmission through postsynaptic mechanisms. Activation of NMDARs in hypothalamic magnocellular neurons can potentiate GABAAR-mediated currents independent of NO signaling (Potapenko et al., 2013). In the hippocampus, NMDAR activation enhances GABA signaling via SNARE-dependent exocytosis of postsynaptic GABA receptors (Marsden et al., 2007). In contrast, our data indicate that the link between NMDAR activity and potentiation of GABA transmission is NO, which acts presynaptically to enhance GABA release.
Importantly, our findings demonstrate that postsynaptic depolarization leads to presynaptic enhancement of inhibition. This stands in contrast to the well-established eCB-mediated depolarization-induced suppression of inhibition (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). eCB signaling through CB1Rs was continuously blocked in the present study to eliminate any dampening effect eCBs may have on GABA release. In the future, it will be interesting to examine whether eCBs alter the magnitude or expression of DPI observed in this study. Indeed, we demonstrated previously that eCB signaling precludes NO activity at GABA synapses in the DMH following bursts of afferent activity (Crosby et al., 2011). Our findings are also in contrast to reports of somatodendritic neuropeptide release that result in suppression of inhibition. For example, somatodendritic release of oxytocin from hypothalamic neurons suppresses GABA transmission in an eCB-dependent manner (Oliet et al., 2007). Our findings are in agreement with one report that postsynaptic depolarization can potentiate synaptic inhibition in the cerebellum (Duguid and Smart, 2004). In this previous study, the authors reported that retrograde glutamate signaling at presynaptic NMDARs on interneuron terminals enhances GABA release onto Purkinje cells.
Overall, our findings reveal a new form of retrograde synaptic plasticity in which somatodendritic CCK release potentiates presynaptic GABA drive through a CCK2R and NMDAR-mediated pathway involving retrograde NO signaling. Due to the widespread expression of neuronal CCK and the ubiquitous nature of NO, these findings could have major implications for neuronal communication and retrograde plasticity throughout the CNS.
Footnotes
- Received July 29, 2014.
- Revision received August 9, 2015.
- Accepted August 20, 2015.
This work was supported by operating grants from the Canadian Institutes for Health Research and personnel awards to J.S.B. and Q.J.P. Scholarship support to K.M.C. was provided by the Natural Sciences and Engineering Research Council, Alberta Innovates Health Solutions, and a Hotchkiss Brain Institute Obesity Initiative Studentship. We thank Mio Tsutsui and Cheryl Sank for their technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Quentin J. Pittman, Hotchkiss Brain Institute, University of Calgary, 3330 Hospital Drive, Northwest Calgary, Alberta T2N 4N1, Canada. pittman{at}ucalgary.ca
- Copyright © 2015 the authors 0270-6474/15/3513160-11$15.00/0