
Abstract
Impaired signaling via CX3CR1, the fractalkine receptor, promotes recovery after traumatic spinal contusion injury in mice, a benefit achieved in part by reducing macrophage-mediated injury at the lesion epicenter. Here, we tested the hypothesis that CX3CR1-dependent changes in microglia and macrophage functions also will enhance neuroplasticity, at and several segments below the injury epicenter. New data show that in the presence of inflammatory stimuli, CX3CR1-deficient (CX3CR1−/−) microglia and macrophages adopt a reparative phenotype and increase expression of genes that encode neurotrophic and gliogenic proteins. At the lesion epicenter (mid-thoracic spinal cord), the microenvironment created by CX3CR1−/− microglia/macrophages enhances NG2 cell responses, axon sparing, and sprouting of serotonergic axons. In lumbar spinal cord, inflammatory signaling is reduced in CX3CR1−/− microglia. This is associated with reduced dendritic pathology and improved axonal and synaptic plasticity on ventral horn motor neurons. Together, these data indicate that CX3CR1, a microglia-specific chemokine receptor, is a novel therapeutic target for enhancing neuroplasticity and recovery after SCI. Interventions that specifically target CX3CR1 could reduce the adverse effects of inflammation and augment activity-dependent plasticity and restoration of function. Indeed, limiting CX3CR1-dependent signaling could improve rehabilitation and spinal learning.
SIGNIFICANCE STATEMENT Published data show that genetic deletion of CX3CR1, a microglia-specific chemokine receptor, promotes recovery after traumatic spinal cord injury in mice, a benefit achieved in part by reducing macrophage-mediated injury at the lesion epicenter. Data in the current manuscript indicate that CX3CR1 deletion changes microglia and macrophage function, creating a tissue microenvironment that enhances endogenous repair and indices of neuroplasticity, at and several segments below the injury epicenter. Interventions that specifically target CX3CR1 might be used in the future to reduce the adverse effects of intraspinal inflammation and augment activity-dependent plasticity (e.g., rehabilitation) and restoration of function.
- CX3CR1
- inflammation
- macrophages
- microglia
- plasticity
- spinal cord injury
Introduction
Spinal cord injury (SCI) elicits a protracted neuroinflammatory reaction throughout the lesion epicenter and in remote regions of the brain and spinal cord (Popovich et al., 1997; Hains and Waxman, 2006; Detloff et al., 2008; Hansen et al., 2013). These inflammatory cascades are dominated by microglia and monocyte-derived macrophages (MDMs) (Popovich and Longbrake, 2008; David and Kroner, 2011). Recent data indicate that as microglia and macrophages respond to cues in the lesion environment, stochastic activation of pattern recognition receptors (e.g., toll-like receptors) endows these cells with the ability to kill neurons and glia or enhance their growth potential (Gensel et al., 2009, 2015; Miron et al., 2013). Chemokine receptors are another class of receptors that may regulate divergent functions in microglia and MDMs after SCI.
Previously, we found that deletion of CX3CR1, a chemokine receptor that binds only to fractalkine (CX3CL1), improves functional recovery and reduces lesion pathology after SCI in mice (Donnelly et al., 2011). Improvements in locomotor function were due, in part, to reducing MDM-mediated neurotoxicity in CX3CR1-deficient (CX3CR1−/−) mice. However, deficiencies in CX3CR1 signaling also could endow microglia, both nearby and distal to the injury epicenter, with functions that directly promote neuroplasticity and functional recovery. Indeed, data show that to achieve optimal recovery after SCI, neuroinflammation needs to be reduced at the epicenter and also in distal spinal segments (Hansen et al., 2013).
After SCI, activated microglia dominate the lesion penumbra (Popovich and Hickey, 2001; Wang et al., 2015), but these cells also exist throughout the spinal cord and into the brain (Hains and Waxman, 2006; Detloff et al., 2008; Tan et al., 2008; Hansen et al., 2013). Whether activated microglia promote repair or exacerbate injury is likely to vary as a function of time postinjury, lesion severity, and location relative to the injury epicenter. Microglia are essential for lesion containment in acute injuries (Hines et al., 2009), but their protracted activation distal to the lesion, mediated in part via neuronal chemokines, can cause neuropathic pain and impair neurological function (Zhao et al., 2007; Tan et al., 2008; Hansen et al., 2013). Such aberrant effects of microglia can be explained by their ability to directly affect the formation and removal of synapses (synaptic plasticity) and the growth and maturation of axons or dendrites (Harkema et al., 2011; Paolicelli et al., 2011; Hoshiko et al., 2012; Pont-Lezica et al., 2014; Squarzoni et al., 2014; Baalman et al., 2015). Because CX3CR1 influences microglia effects on neural circuitry and because we previously found that recovery from SCI is improved in CX3CR1−/− mice (Donnelly et al., 2011), experiments in this report tested two distinct but interrelated hypotheses: (1) reduced inflammatory signaling in CX3CR1−/− microglia and MDMs enhances endogenous repair and plasticity at/nearby the lesion epicenter; and (2) reduced inflammatory signaling in CX3CR1−/− microglia remote from the injury site is associated with enhanced cellular repair and anatomical plasticity, including axonal sprouting and synaptogenesis.
New data in this report show that CX3CR1−/− microglia and MDMs augment endogenous repair, both at and distal to a SCI. At the lesion epicenter, a normally robust NG2 glial response is enhanced in CX3CR1−/− mice and is associated with enhanced sparing and sprouting of axons, including a subpopulation of serotonergic axons. In lumbar spinal cord, reduced inflammatory signaling in CX3CR1−/− microglia is associated with enhanced axon and synaptic plasticity and reduced dendritic pathology on spinal motor neurons.
Although damaged CNS axons do not regenerate efficiently, the CNS has the capacity to rewire connections within existing (surviving) neural circuitry, boosting spontaneous recovery and the capacity for neurorehabilitation (Bareyre et al., 2004; Courtine et al., 2008; Rosenzweig et al., 2010). Data in this report indicate that deleting CX3CR1, a chemokine receptor found exclusively on microglia and a subset of infiltrating MDMs (Cardona et al., 2006), can promote neuroprotection (sparing) and also augment plasticity in spared fibers after SCI.
Materials and Methods
Animals and surgery.
Breeding pairs of CX3CR1GFP/GFP mice (hereafter referred to as CX3CR1−/− mice) were established from Dan Littman's original colony as described previously (Jung et al., 2000; Donnelly et al., 2011). CX3CR1−/− mice were generated by replacing the second exon of the cx3cr1 gene with the enhanced GFP reporter gene (Jung et al., 2000). Mice were backcrossed to a C57BL/6 background for >10 generations. Wild-type (WT) C57BL/6 mice were obtained from The Jackson Laboratory. A total of 104 male and female mice CX3CR1+/+, CX3CR1+/−, or CX3CR1−/− received a moderate (75 kdyn) mid-thoracic (T9) contusion SCI (Infinite Horizons device). With the exception of images shown in Figure 1F–I and the images and data in Figure 5L–N, all other comparisons were made between CX3CR1−/− and C57BL/6 WT mice. The aforementioned data were conducted as part of early experiments when CX3CR1+/− (heterozygote) mice were considered viable “control” groups as described by other laboratories using these same mice for evaluating basic microglia physiology (Davalos et al., 2005; Cardona et al., 2006). However, as more data emerged showing that loss of a single CX3CR1 allele impairs microglia function (Rogers et al., 2011), we moved to using only CX3CR1−/− and C57BL/6 WT mice for quantitative comparisons. Thus, the magnitude of myelin/axon sparing (see Fig. 1F,G) and number of synapses reported in CX3CR1+/− groups (see Fig. 5L–N) likely underestimate what would have been observed had we compared CX3CR1−/− mice with C57BL/6 mice. All mice were anesthetized with a ketamine (80 mg/kg, i.p.)/xylazine (10 mg/kg, i.p.) mixture. Postoperatively, animals were hydrated with 2 ml of saline (subcutaneous) and were given prophylactic antibiotic (0.1 ml gentamicin, s.c.) for 5 d. Bladders were voided manually twice per day for the duration of the study. Urine pH was monitored weekly, and mice were observed daily for signs of infection or abnormal wound healing at the site of surgery. All housing, surgical, and postoperative care procedures were performed in accordance with the Ohio State University Institutional Animal Care and Use Committee. After 4, 14, 28 or 56 d postinjury (dpi), spinal cords were prepared for immunohistochemistry (IHC), TEM, or Golgi Cox staining. To label proliferating cells, the thymidine analog BrdU was injected (50 mg/kg body weight in sterile saline; i.p.; Sigma-Aldrich) into a subset of mice immediately after injury then daily for 2 weeks.
Tissue processing.
For IHC, mice were killed at 0 (naive control; n = 3/group), 4, 14, 28 and 56 dpi (n = 5/genotype). All mice were given a lethal mixture of ketamine/xylazine (1.5× the surgical dose) and then perfused intracardially with PBS (0.1 m, pH 7.4) followed by 4% PFA in PBS. Perfused spinal cords were removed and postfixed for 2 h then rinsed and stored overnight in PBS. Samples were cryoprotected by immersion in 30% sucrose for 4 d. The 1 cm blocks of spinal cord tissue, centered over the injury site, were frozen on dry ice. Transverse serial (10 μm) and horizontal sections (18 μm) were cut on a Microm cryostat (HM 505E), collected on SuperFrost Plus slides (Thermo Fisher Scientific) then stored at −20°C.
For Golgi-Cox analyses, mice were perfused with PBS at 56 dpi mice (n = 5/group). Lumbar spinal cords were dissected, rinsed with double distilled water, and then immersed in a 1:1 mixture of FD solution A:B (FD Rapid GolgiStain) for 2 weeks at room temperature in the dark. Spinal cords were transferred to FD Solution C and kept in the dark at 4°C for 48 h. After replacing Solution C, spinal cords were frozen then embedded in disposable molds. Transverse serial sections (150 μm) were cut through each block on a Microm cryostat (HM 505 E). The sections were transferred to 24 well plates onto small drops of FD Solution C, rinsed in distilled water, then immediately stained as floating sections using Golgi-staining protocol (FD Rapid GolgiStain Kit catalog #PK401, FD NeuroTechnologies).
Additional mice were required for TEM analysis. CX3CR1+/− and CX3CR1−/− mice were perfused at 56 dpi (n = 3/group) with PBS, followed by 4% PFA/2% gluteraldehyde. The 5 mm segments of tissue below the lesion epicenter and lumbar enlargement were blocked in Epon-Spurr resin.
Antibodies and IHC.
Antibodies used for DAB immunoperoxidase or immunofluorescence staining are listed in Table 1. For immunoperoxidase labeling, spinal cord sections were rinsed in 0.1 m PBS and endogenous peroxidases quenched using a 4:1 solution of methanol and 30% hydrogen peroxide for 15 min in the dark. Sections were rinsed and blocked for nonspecific antigen binding using 4% BSA/0.1% Triton X-100/PBS (BP+) for 1 h. Next, sections were incubated in primary antibody overnight in humidified chambers at 4°C. The next day, sections were rinsed 3 times with PBS and incubated in biotinylated secondary (goat anti-rabbit 1:2000 or goat anti-chicken 1:1000 in BP+; Vector Laboratories) for 1 h at room temperature. Bound antibody was visualized using Elite-avidin-biotin enzyme complex (ABC; Vector Laboratories) for 1 h and DAB as a substrate (Vector Laboratories). The slides were dehydrated in 70%, 80%, 90%, and 100% ethanol and then coverslipped with Permount (Thermo Fisher Scientific). For BrdU IHC, sections were treated with 2N HCl at 37°C for 25 min to denature DNA before applying primary antibody. For DAB double-labeling, sections were overlaid with blocking solution (4% BSA+ BP+) for 1 h before applying the second primary antibody. Eriochrome cyanine was used to stain and distinguish intact myelin from gray matter and lesioned tissue as described previously (Kigerl et al., 2006). For immunofluorescent labeling, spinal cord sections were rinsed, blocked, and incubated in primary antibody as above. The next day, sections were rinsed 3 times with PBS and incubated in secondary antibody (1:1000 AlexaFluor-546, -33, -88; Invitrogen). After rinsing, slides were coverslipped with Immu-Mount (Thermo Scientific).
Antibodies used for immunohistochemistry
Quantifying IHC labeling: proportional area.
To quantify proportional area (see Figs. 1, 4, 8) of neurofilament, 5-HT, p38MAPK, and IBA-1 images were digitized using a Zeiss Axioplan 2 Imaging microscope (Carl Zeiss) and then analyzed using an MCID Elite image analysis system (Imaging Research). Proportional area was determined by dividing the area occupied by immunoreactive cells or axons within a sample area or region of interest (ROI) by the total area of the ROI. For microglia (IBA-1; see Fig. 8), eight transverse sections spanning the L2-L3 and L4-L5 spinal segments were quantified. For p38MAPK (see Fig. 8), two transverse sections ∼6 mm from the epicenter were analyzed. Both were quantified at 56 dpi.
Number of neurofilament (NF)-positive axon profiles at lesion site.
Sections stained with anti-NF antibodies were digitized using a Zeiss Axioplan 2 Imaging microscope (Carl Zeiss). ImageJ was used to count axon profiles within an ROI spanning the area between spared myelinated axons and more centrally located regions of the spinal cord containing remnants of gray matter or connective tissue. Thousands of axon profiles were detected; however, only those achieving a continuous length of at least 4 μm length were counted (see Fig. 1J).
NG2 and NF.
Fluorescent images were captured on a FluoView FV1000 laser scanning confocal microscope (Olympus). For each animal, 15 images (20×) were captured from horizontal sections at the level of the central canal. Horizontal images were stitched together using Adobe Photoshop (version CS6) to create montages (see Fig. 3A,B). MetaMorph Image Analysis software (version 6.1) was used to quantify the proportional area of NG2 within an ROI (690 μm2) spanning the rostral-caudal interface centered on the lesion epicenter (see Fig. 3C). Horizontal sections double-labeled for NG2/NF were used to quantify the percentage NG2/NF colocalization (see Fig. 3I). Images of NG2 and NF were each given a threshold and the overlapping regions for NG2 and NF measured using MetaMorph Image Analysis software (version 6.1) within a square ROI (302 μm2).
NG2 cell proliferation.
NG2+/BrdU+ double-labeled cells were counted in the epicenter at 56 dpi in three sections spanning the dorsal-ventral axis (360, 630, and 900 μm from the dorsal spinal surface) of the spinal cord on 1-mm-long horizontal sections (see Fig. 3F). Only cells with a well-defined border surrounding an identified nucleus were counted. NG2+ macrophages or pericytes were excluded based on morphological criteria (McTigue et al., 2001).
5-HT+ axons.
Horizontal montages (∼6 mm long) of the spinal cord were generated with 20× images from three horizontal sections spanning the dorsoventral axis (360, 630, and 900 μm from the dorsal spinal surface) of the spinal cord gray matter. A box was drawn within an ROI at and rostral to the lesion epicenter at 56 dpi. ImageJ software (version 1.48d, National Institutes of Health) was used to quantify 5-HT+ axon density within these areas (see Fig. 4E). Time-dependent changes in 5-HT+ axon profiles were quantified at 4, 14, 28, and 56 dpi and were expressed relative to identical areas in naive sections (see Fig. 4H). 5-HT+ axon profiles within the ventral horn (lamina IX) were quantified at 56 dpi in each of two cross-sections from L3 and L4 lumbar spinal cord (see Fig. 4F,G). A z stack of each section was acquired at high-power (63× magnification/1.40 oil DIC); then 5-HT+ profiles were counted within an ROI containing at least two motor neurons (see Fig. 4I,J). A binary image was generated from each fluorescent z stack, and only 5-HT+ profiles >0.5 μm in length were counted (see Fig. 4K).
Synaptic puncta.
Synaptic puncta were analyzed using the Puncta Analyzer plug in developed for ImageJ 1.26 (kindly provided by Dr. Cagla Eroglu). Fluorescent z stack images were taken using an Olympus FluoView FV1000 and Leica TCS SP8 laser scanning confocal microscope. Measurements were performed on 2 or 3 images from L3 and L4, where most neurons innervating muscles responsible for locomotion are located. For synapsin (presynaptic receptor), images were captured at 60× (Z intervals 0.25 μm/10-μm-thick section) and analyses performed on ∼36–40 images/sample. Within ventral horn lamina IX, a square ROI was drawn (0.029 mm2) and all synaptic puncta within the ROI counted (see Fig. 5K).
Excitatory and inhibitory synaptic puncta.
Glutamic acid decarboxylase (GAD)1-GAD67 synaptic punta were measured on images taken at 40×/1.8 oil magnification on DIC using Z intervals of 0.30 μm in a 10-μm-thick section (see Fig. 6D). For VGAT and gephyrin measurements, fluorescent images were taken at Z intervals of 0.35 μm on 10-μm-thick sections (see Fig. 6N). VGlut1 is highly expressed in lamina VII and VGlut2 in lamina IX of the spinal cord. Combined labeling of these two presynaptic excitatory receptors allows for quantification of all excitatory synaptic contacts. We labeled postsynaptic receptors with Homer-1. Fluorescent z stack images were captured at 40×/1.8 oil on DIC at Z intervals of 0.30 μm over 10-μm-thick sections. Within lamina VII, a square (0.05 mm2) was drawn and the number of colocalized presynaptic and postsynaptic receptors VGLUT1 and Homer-1 were counted within this area (see Fig. 7I). The number of colocalized puncta for presynaptic and postsynaptic receptors VGlut2 and Homer-1 was measured on images taken at 40×/2.0 oil magnification and within lamina IX (see Fig. 7R).
TEM.
Ultrathin transverse sections (50–70 nm) were cut from blocks of spinal cord on an ultramicrotome (Ultracut MZ6; Leica). Sections were placed on formvar-coated copper grids and then contrasted with uranyl acetate and lead citrate. The samples were examined under a Tecnai Spirit G2 Bio-twin (Fei) transmission electron microscope operating at 80 kV. To measure synaptic covering, neurons with large cell bodies (>35 μm diameter) in the ventral motor pool and cut in the nuclear plane were identified as α motor neurons by the presence of C-type nerve terminals. The surface of the cells was then sequentially digitalized at a magnification of ×11.000. Images were mounted together using Corel Draw X5 Graphics software, and the total perimeter of neurons (μm) was measured. Synaptic terminals apposing motoneuron somata were identified and their length of apposition was measured using Image Toll software (version 3.0, University of Texas Health Center, Santo Antonio, TX) and then expressed as a percentage of synaptic coverage as described previously (Freria et al., 2012).
Neuronal dendrites and spines (from Golgi-Cox stain).
Forty motor neurons located in the lumbar (L2-L5) spinal cord ventral horn in CX3CR1+/+ and CX3CR1−/− mice were measured. From each neuron, the number of dendrites, perimeter of neuron cell body, longest dendrite, number of dendritic spines, and dendritic spine length were counted using Zeiss Axio Image M2 Microscope and Neurolucida neuron tracing software (MBF Bioscience; RRID: SCR_001775). Two dendrites per neuron, at least 20 μm away from the soma, were randomly chosen, and the length of dendritic spines was measured along a 50 μm length of the dendrite shaft. Neurons were considered “beaded” if they had at least three varicosities along the same dendrite (see Fig. 10). Thirty neurons from each group were counted. In beaded neurons, one dendrite per neuron, at least 10 μm away from the soma, was randomly chosen, and numbers of beaded structures along at least 100 μm length of dendrite were counted. Data are expressed as number of beaded neurons (normalized to dendrite length). In addition, the area of each varicosity on the same dendrite was examined, and the result was represented as area of varicosity (μm2). To measure the geometric characteristic of dendritic spines, 20 motor neurons located in the ventral horn of lumbar spinal cord (L2-L5) were analyzed in each group at 56 dpi. One dendrite at least 10 μm away from the soma was randomly chosen, and the length and width of individual dendritic spines along a 50 μm length of dendrite were measured. Spines were characterized as “mushroom” (mature) when they were >0.6 μm in width, “filopodial (Filo)” (immature) when they were >2 μm in length, or “thin” (immature) when they had a length to width ratio >3 μm.
Primary cortical neuron cultures.
Cortical neurons were isolated from neonatal mice at E12.5-E13. Using aseptic techniques, entire forebrains were removed and placed in cold Hibernate HE Ca2+ free (Invitrogen, A12476–01). The meninges were removed and brain pieces minced. Tissues were collected into sterile tubes and then incubated in Hibernate (Ca2+ free), papain (Worthington #3119- 20 U/ml) and DNase (σ D5025- 5 mg/ml) at 37°C for 30 min. Tissue was triturated through polished glass pipettes with progressively smaller-tip diameters. Debris was removed by allowing the pieces to settle between trituration steps. After trituration, cells were suspended in fresh media [Neurobasal A (Invitrogen) with 2% B27 supplement (Invitrogen), 1% Glutamax, and gentomicin 1:2000], and live cells were counted on a hemocytometer using trypan blue exclusion method. Cells were grown at 37°C/5% CO2 in poly-D-lysine (σ P7886 mk30K-70K; 0.25 mg/ml) and laminin (Sigma, L2020) 5 μg/ml-coated plastic culture plates seeded with glass coverslips. Cells were plated at 8 × 104 cells/cm2 and used 14 d after initial plating. For measuring varicosities along dendrites of cultured neurons, four pictures randomly chosen from 2 different coverslips were taken at 40× using a Zeiss Apotome microscope equipped with Axio camera MRM. Varicosities were counted on the longest process extending from each neuron. Varicosities were counted on eight neurons from each experimental group. Data are expressed as number of varicosities per dendrite length.
Primary microglia cultures.
Using aseptic technique, the forebrain and brainstem were removed from postnatal mouse pups P1-P2 (CX3CR1+/+ and CX3CR1−/−) and were placed in cold 10% glucose. Using fine forceps, meninges were removed and brains were incubated in trypsin EDTA 0.05% in a sterile tube for 20 min. Tissue was triturated through a 1 ml pipette; then cells were suspended in fresh media (DMEM-F12 Invitrogen 09045; 1% REPES Invitrogen Sigma H-0887; 10% FBS and gentomicin 1:2000 Invitrogen 15710–064). Live cells were counted on a hemocytometer using the trypan blue exclusion method and placed into 150 mm × 25 mm plates (Corning 430599). After 2.5 weeks, microglia were collected from the bottom of the astrocyte layer using 0.25% trypsin EDTA (Saura et al., 2003), then were reseeded into plate inserts (Millipore 0.4 μm pore) at 3.5 × 104 cells/cm2. For analysis of microglia-conditioned media (MCM), 5 × 105 cells were reseeded in fresh media (Neurobasal A; 2% B27 supplement, 1% Glutamax, and gentomicin 1:2000) onto 24-well plastic culture plates. MCM was collected after 24 h and diluted 1:1 with fresh media before adding MCM to neurons. After 24 h, coverslips of neurons cocultured with microglia or MCM were fixed using 2% PFA for 20 min, followed by a PBS wash. Blocking solution (100 μl of 0.1 m PBS/10% FBS/0.1% Triton X-100) was applied for 1 h at room temperature, followed by primary antibody β tubulin III (1:1000 in blocking solution) overnight at 4°C. Primary antibody was aspirated, and cells were washed in PBS. Secondary antibody anti-rabbit was applied 1:1000 for 1 h at room temperature.
Bone marrow-derived macrophages.
Previously, we showed that bone marrow-derived macrophages predict many of the phenotypic, molecular, and functional characteristics of the monocyte-derived macrophages that infiltrate the injured spinal cord (Longbrake et al., 2007; Kigerl et al., 2009). Accordingly, bone marrow-derived macrophages were generated from bilateral femurs and tibias of adult C57BL/6 and CX3CR1−/− mice (see Fig. 2) as described previously (Longbrake et al., 2007; Kigerl et al., 2009). Briefly, marrow cores were flushed then differentiated into macrophages as described previously (Longbrake et al., 2007; Kigerl et al., 2009). Macrophages (5 × 104 cells/cm2) were seeded onto sterile glass coverslips and then were incubated at 37°C/5% CO2 for 12 h, after which macrophages were stimulated with media only (control) or inflammatory stimuli: LPS (100 ng/ml; Sigma-Aldrich) plus IFNγ (20 ng/ml; eBioscience) for 24 h. Twenty-four hours later, cells were either fixed with 4% PFA or media was removed from the culture and Trizol (Invitrogen) was added to isolate RNA. Fixed cells were stained by overlaying cells with blocking solution for 1 h; then AlexaFluor Phalloidin 546 (1:200; catalog #A22283 Invitrogen) was applied for 2 h at room temperature. After rinsing, all coverslips were placed onto slides with Immu-Mount. Fluorescence images were taken at 20× using a Zeiss Apotome microscope equipped with a Zeiss Axio camera MRM. Cell area and perimeter were assessed using MCID Elite image analysis system. Form factor (FF) was calculated using the following formula: FF = 4π area/(perimeter)2 according to (Wilms et al., 1997). The FF of a perfect circle is equal to 1; morphologies that deviate from a perfect circular have a FF <1. Based on FF, we identified macrophages as bipolar, unipolar, elongated, flat or round.
For PCR analysis and subsequent analysis by real-time PCR, RNA was isolated according to the manufacturer's protocol and then treated with DNase I (1 μg/μl) to eliminate genomic DNA contamination (Invitrogen); 1–2 μg of DNase-treated RNA was primed with random hexamers (1 μm, Applied Biosystems) and reverse transcribed using Super-Script II reverse transcriptase (Applied Biosystems). Primer pairs were designed using ABI Prism Primer Express 2.0. Primer sequence specificity was confirmed by performing BLAST analysis for highly similar sequences against known sequence databases. Forward and reverse primer sequences for each gene are listed in Table 2. RNA profiles were analyzed using primers specific to the gene of interest and SYBR Green master mix (Applied Biosystems). All PCR was performed using an Applied Biosystems 7300 system. Melting point analyses were performed for each reaction to confirm single amplified products. Gene expression was extrapolated from standard curves generated concurrently for each gene using a control cDNA dilution series. Expression was normalized to 18S for each sample. Data were calculated using the ΔΔCt method (Schmittgen and Livak, 2008) and expressed as fold change from control macrophages cells (gene/18S ratio for control samples/unstimulated macrophages). Data in Figure 2 are representative of one of three independents experiments.
Primer sequences for real time RT-PCR
Data analysis.
All investigators involved in data acquisition or analysis were blind to group designation or experimental manipulations. Blinding was accomplished by assigning animals a unique identification (ID) number before injury. These unique numbers were maintained on all samples throughout the period of assessment. An individual that was not involved with data analysis or acquisition made all ID assignments. Data were analyzed using GraphPad Prism 5 and are presented as mean ± SEM. Neurofilament, RT-PCR data, NG2 cell counts, NG2 proportional area, number of 5-HT+ axons, synaptic covering, inhibitory and excitatory synapses, IBA-1 proportional area, axonal varicosities, and dendritic spines were analyzed by Student's (unpaired) two-tailed t test. All other data, including macrophage FF (see Fig. 2E,F), 5-HT density (see Fig. 4E,H), number of puncta synapses (see Fig. 5K), IBA-1 PA (see Fig. 8E), and dendritic spines morphology (see Fig. 9L–N), were analyzed by two-way ANOVA with Bonferroni's post hoc test.
Results
After experimental spinal contusion injury, CX3CR1 expression is dynamically regulated in CNS myeloid cells (microglia and MDMs) and abolishing CX3CR1 signaling in these cells improves functional recovery with significant neuroprotection evident at the injury site for at least 4–6 weeks after injury (Donnelly et al., 2011). Extending previously published data from our laboratory, new data in Figure 1 show significant protection of myelinated axons within the injured spinal cord of CX3CR1-deficient (CX3CR1−/−) mice at 8 weeks after injury (Fig. 1A,H,I). Also notable in these lesions were numerous thin axonal sprouts (∼4–10 μm) that span the area between spared axons in the ventrolateral white matter and more centrally located regions of “spared” gray matter (Fig. 1F,H,J). Axonal “sprouts” were more prevalent in CX3CR1−/− mice (Fig. 1J) and could indicate the presence of a lesion environment that is less toxic and/or more supportive of cell growth and migration.

Impaired CX3CR1 signaling confers neuroprotection (myelin and axons) and enhances endogenous repair after SCI. A–D, Eriochrome cyanine (EC, blue) and neurofilament (NF200, brown) labeling in CX3CR1+/+ and CX3CR1−/− mice at 56 dpi. Light microscopic images from representative mice (A, C) and schematic maps of those images illustrate the mean differences in spared myelin (EC) and axon (NF+ immunoreactivity) area. E, Greater NF labeling was observed in CX3CR1−/− mice at 8 weeks after injury. F, H, Representative images of NF labeling in CX3CR1−/− (F) and CX3CR1+/− mice (H) ∼0.8 mm caudal to the lesion site. G, I, Electron microscopic images of sections cut from tissue blocks adjacent to those used in F, H confirm genotype-dependent differences in axon sparing (G, CX3CR1+/−; I, CX3CR1−/−). F, H, Boxed regions represent the regions of spared white matter from which images in G, I were captured. J, Numbers of NF+ axon profiles (“streamers”) were measured (arrows in H; ∼4–10 μm in length) within regions between spared myelinated axons and central gray matter in the lesion core. NF+ axon profiles were increased significantly in CX3CR1−/− spinal cord sections. Graphs are representative of single independent experiments (n = 5 +/+ and n = 6 −/− mice/group). *p < 0.05 (Student's unpaired two-tailed t test). **p < 0.01 (Student's unpaired two-tailed t test). Scale bars: H, 5 μm; I, 50 μm. Data are mean ± SEM.
Inflammatory stimuli elicit a “tissue-repair” phenotype in CX3CR1-deficient macrophages
Intracellular signaling networks control cell protrusion, adhesion, and tension, which in turn affects nuclear organization, chromatin condensation, and histone modification; all can impact the genetic programs that influence macrophage phenotype and function (Bakal et al., 2007; McWhorter et al., 2013). To determine whether CX3CR1 deficiency induces morphological and molecular changes in CNS myeloid cells, which might predict functions that enhance cell (axon) growth and/or limit neurotoxicity, macrophages were prepared from WT and CX3CR1−/− mice. Cells were stimulated with IFNγ + LPS to induce an inflammatory “M1” phenotype, one that causes neurotoxicity and axonal die-back in models of SCI (Popovich et al., 1999; Horn et al., 2008; Busch et al., 2009). Unstimulated (M0) macrophages served as controls. Resting or stimulation-induced changes in cell morphology and gene expression were compared between genotypes.
The area and perimeter of macrophages were used to calculate FF (Wilms et al., 1997). FF is a morphometric measure of circularity; a perfect circle has FF = 1, whereas a line has FF = 0. Phalloidin staining was used as it reveals actin-rich filipodial extensions from the soma of both WT and CX3CR1−/− macrophages (Fig. 2A–D). Under control (M0) conditions, 85% of WT and CX3CR1−/− macrophages exhibited bipolar or unipolar morphologies at similar frequencies (Fig. 2E). When activated with inflammatory (M1) stimuli, most WT macrophages adopted an elongated or flat morphology with few filipodia (Fig. 2F). Conversely, inflammatory stimuli failed to elicit this “activated” morphology in CX3CR1−/− macrophages; most remained smaller with a unipolar morphology and FF similar to that of unstimulated macrophages.

Inflammatory stimuli elicit a tissue-repair phenotype in CX3CR1−/− macrophages. A–D, Morphology (phalloidin stain for actin) of control (unstimulated) (A, B) macrophages or macrophages activated with inflammatory stimuli (IFNγ + LPS) (C, D). Inflammatory macrophages are easily distinguished by their large, flattened cell bodies with few filipodia. E, F, Genotype-specific changes in responsiveness of macrophages to inflammatory signals. Although CX3CR1 deletion has no effect on resting (unstimulated/M0) macrophage morphologies (E), only CX3CR1 WT (+/+) macrophages consistently show dramatic FF changes (indicative of activation) in response to inflammatory signaling (F). G–L, qRT-PCR analyses of inflammatory macrophages generated from +/+ or −/− mice. Graphs are mean data from one of three independent replicate experiments. Data were calculated using the ΔΔCt method (gene/18S ratio for control unstimulated macrophages). Inflammatory macrophages derived from −/− mice show higher expression of genes encoding neurotrophic and gliogenic proteins. Data are mean ± SEM obtained from n = 3 replicate wells/group. *p < 0.05 (Student's unpaired two-tailed t test). **p < 0.01 (Student's unpaired two-tailed t test).
Such distinct morphological differences are likely associated with unique gene expression profiles. We evaluated resting and inflammatory-induced changes in TGFβ1, IGF1, FGF2, BDNF, NT3, and MMP9. These genes are released by macrophages and have potent effects on axon growth (Zhou and Shine, 2003), NG2 cell proliferation and differentiation (McTigue et al., 1998; Wu et al., 2010; Liu and Shubayev, 2011; Wennström et al., 2014), and tissue remodeling (Lu et al., 2011). At rest (no stimulation), baseline gene expression was similar in CX3CR1−/− and WT macrophages (data not shown). However, in response to inflammatory stimuli, marked changes in gene expression were noted for a subset of genes. Notably, TGFβ1, IGF1, and FGF2 mRNA expression increased significantly only in CX3CR1−/− macrophages (Fig. 2G–I). Other genes (BDNF, NT3, and MMP9) were largely unaffected by the absence of CX3CR1 (Fig. 2J–L). Based on previously published data (Donnelly et al., 2011) and data in Figure 2, we can conclude that inflammatory stimuli fail to elicit a neurotoxic phenotype in CX3CR1−/− macrophages. Instead, deletion of CX3CR1 enhances the neurotrophic and gliogenic potential of intraspinal macrophages.
CX3CR1-deficient macrophages create an environment that supports NG2 glia and the growth or stability of axons
After SCI, NG2 glia increase at the interface between the macrophage-rich lesion core and adjacent spared tissue (Lytle et al., 2006, 2009; Tripathi and McTigue, 2007; Wu et al., 2010). Based on data in Figures 1 and 2, we predicted that the altered inflammatory milieu created by CX3CR1−/− macrophages would affect the endogenous NG2 glia response after SCI.
After SCI, the density of NG2 glia was significantly greater across the rostrocaudal extent of the chronic (56 dpi) contusion lesion in CX3CR1−/− mice compared with WT mice (Fig. 3A–C). To determine whether this develops as a result of enhanced NG2 cell proliferation at a time coinciding with the onset and peak of intraspinal macrophage activation, mice were pulsed with BrdU daily for 2 weeks starting immediately after SCI. NG2 cell proliferation increased significantly in CX3CR1−/− mice relative to WT mice; total number of NG2+/BrdU+ cells increased ∼20% at the lesion border in CX3CR1−/− mice (Fig. 3D–F).

CX3CR1−/− macrophages create an environment that supports NG2 glia and the growth or stability of axons. A, B, NG2 staining of horizontal spinal cord sections from (A) CX3CR1+/+ or (B) CX3CR1−/− mice at 56 dpi. C, Graph represents area of NG2 labeling in a 302 μm2 box centered on the epicenter. Schematic indicates measurement area. D, E, Double-labeling for BrdU (red) and NG2+ cells (green) near the lesion border. F, Total number of NG2+/BrdU+ cells in a 1 mm section centered over the epicenter. Cells were quantified through the dorsoventral axis of the spinal cord in multiple planes of section as illustrated in the schematic. G–K, Double-labeling of neurofilament (blue) and NG2 (red) at the rostral interface of the lesion epicenter. I, Percentage area of NG2 and NF colocalization. Schematic of spinal cord indicates measurement area. J, K, High-power magnifications of boxes in G, H. Note NG2+ cells wrapping axons (K). Scale bars: B, 150 μm; D, E, 20 μm; G, H, 30 μm; J, K, 10 μm. Graphs are representative of a single experiment. Data are mean ± SEM; n = 5 +/+ and n = 4 −/− mice/group. *p < 0.05 (Student's unpaired two-tailed t test). **p < 0.01 (Student's unpaired two-tailed t test).
Although NG2 proteoglycans can inhibit growth and synaptic transmission in some axons (Tan et al., 2006; Petrosyan et al., 2013), NG2+ glia also can stabilize or limit die-back of injured axons and can serve as substrates for growing axons, especially serotonergic (5-HT) axons (McTigue et al., 2006; Busch et al., 2010). Double-labeling for NG2 and axons (NF) revealed a marked increase in colocalization between NG2 glia and axons at the lesion border (Fig. 3G–K). Consistent with previous reports from our group (McTigue et al., 2006), NG2 cells could be seen wrapping or closely apposed to axons (Fig. 3J,K).
Enhanced 5-HT axon growth at and distal to the lesion epicenter in CX3CR1−/− mice
When injured, serotonergic (5-HT+) axons have an unusual propensity to grow new branches from the proximal regions of their injured axons (Jacobs and Fornal, 1997; Schmidt and Jordan, 2000; Hawthorne et al., 2011); however, recent data indicate that injured 5-HT+ axons exhibit true regenerative growth with little contribution from local sprouting (Jin et al., 2016). Here, 5-HT+ axons were labeled then quantified in lumbar spinal cord as a function of time postinjury. Overall, the density of 5-HT axons was then analyzed via two-way ANOVA with genotype and location (rosral/epicenter) as independent variables. There were significant main effects of both genotype (F(1,16) = 13.85, p = 0.0019) and location relative to the site of injury (F(1,16) = 9.24, p = 0.0078; Figure 4A–E). When evaluated over time relative to naive genotype-matched spinal cords, 5-HT+ axons were reduced at all times in both groups (Fig. 4H). However, by 56 dpi, the density of 5-HT+ axons increased >30% in CX3CR1−/− mice, especially in the ventral horn (Fig. 4F–H). A closer look at these axons revealed obvious differences in both the length and morphology of the 5-HT+ fibers with more and longer 5-HT+ profiles present in CX3CR1−/− spinal cords (Fig. 4I–K). Based on the repopulation kinetics of 5-HT labeling and the known plasticity and regenerative potential of 5-HT+ axons (Saruhashi et al., 1996; Müllner et al., 2008; Takeoka et al., 2009; Hawthorne et al., 2011; Jin et al., 2016), these are likely “new” regenerated axons.

The density and distribution of serotonergic (5-HT) axons increase at and below the injury epicenter in SCI CX3CR1−/− mice. A, B, Representative horizontal sections from CX3CR1+/+ (A, C) and CX3CR1−/− (B, D) mice. Rostrocaudal orientation is left-to-right. C, D, High-magnification images of boxes in A, B. C, D, Red asterisks indicate lesion epicenter. Note the higher density of 5-HT+ axons at the rostral interface in CX3CR1−/− mice. E, Schematic illustrates the horizontal planes through the dorsoventral axis of the spinal cord where 5-HT immunoreactivity was quantified. Graph represents average 5-HT density across three horizontal sections both rostral and at the lesion epicenter. F, G, Lumbar spinal hemi-cords showing 5-HT labeling in transverse sections in CX3CR1+/+ (F) and CX3CR1−/− mice (G). H, Quantification of 5-HT density in the ventral horn as a function of time postinjury. I, J, High-power images of boxes in F, G. K, The number of 5-HT+ profiles in lumbar ventral horn. E, H, Graphs were generated from single experiments and were analyzed using two-way ANOVA with Bonferroni post tests. *p < 0.05. **p < 0.01. G, n = 5 +/+ and 5−/− mice/group. H, n = 3 mice at day 0 and n = 4/group at 4, 14, 28, and 56 dpi. K, Representative graph from two independent experiments using n = 5 +/+ and −/− mice/group each time. ***p < 0.001 (Student's unpaired two-tailed t test). Scale bars: B, 250 μm; D, 200 μm; G, 100 μm; J, 40 μm. Data are mean ± SEM.
Enhanced synaptic plasticity in lumbar spinal cord of CX3CR1−/− mice
We next tested whether the increase in axonal plasticity noted at and distal to the injury epicenter in CX3CR1−/− mice was associated with changes in synaptogenesis. First, synapsin immunohistochemistry was used to quantify changes in presynaptic coverage on lumbar ventral horn motor neurons from 4 to 56 dpi. The number of synapsin-positive puncta were analyzed using two-way ANOVA with genotype and time postinjury as independent variables. There were significant main effects for both genotype (F(1,30) = 5.81, p = 0.0223) and time postinjury (F(4,30) = 18.79, p < 0.0001). At 4 dpi in both CX3CR1+/+ and CX3CR1−/− mice, synaptic density decreased ∼70% compared with uninjured levels (Fig. 5A–K). Over the next 10 d, new synapses formed in both genotypes; however, only in CX3CR1−/− mice did new synapses form throughout the evaluation period. Indeed, by 56 dpi, synapsin labeling returned toward baseline (preinjury) levels only in CX3CR1−/− mice (Fig. 5K). A significant increase in presynaptic coverage of motor neurons after SCI in CX3CR1−/− mice was confirmed via electron microscopy (Fig. 5L–N). The overall (genotype × time postinjury) interaction was not significant.
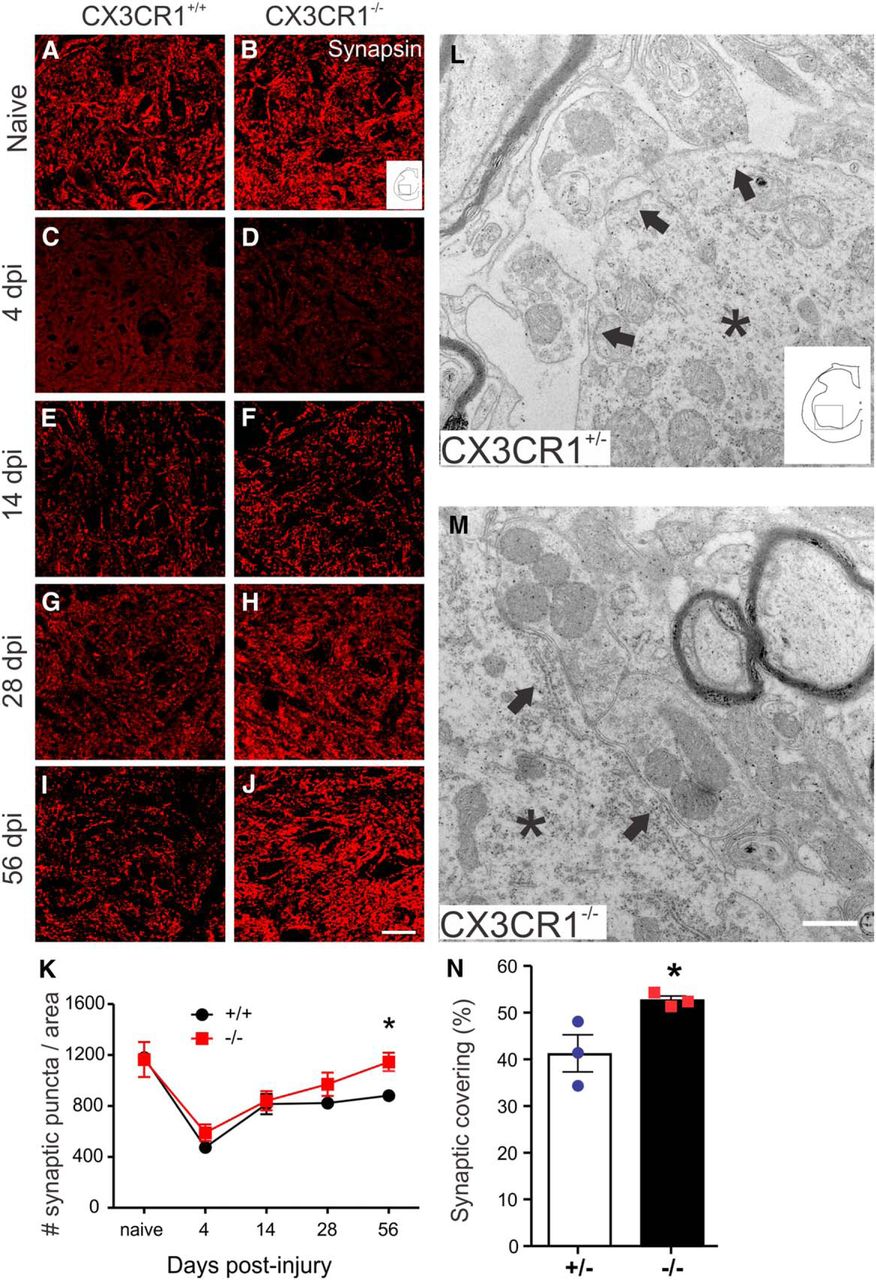
Synaptic plasticity is enhanced in spinal cord lumbar ventral horn after SCI in CX3CR1−/− mice. Representative images illustrate time-dependent changes in synapsin labeling in lumbar spinal cord of CX3CR1+/+ (A, C, E, G, I) and CX3CR1−/− (B, D, F, H, J) mice. K, Quantification of synapsin-positive puncta as a function of time after SCI within ventral horn. L, M, Ultrastructural images of synapses apposed to lumbar motor neurons in CX3CR1+/− (L) and CX3CR1−/− (M) mice. Asterisks indicate motor neuron cytoplasm. Arrows indicate presynaptic receptors contacting motor neuron surfaces. N, Percentage of motor neuron circumference covered by synapses. K, N, Graphs generated from independent experiments, in (K) naive and 4 dpi, n = 3 +/+ and −/− mice/group; 14 dpi, n = 5/group; 28 and 56 dpi, n = 5/group and n = 4/group. N, n = 3 mice/group. K, Data analyzed using two-way ANOVA with Bonferroni post tests. N, Data were analyzed using Student's unpaired two-tailed t test. *p < 0.05. Scale bars: J, 20 μm; M, 0.5 μm. Data are mean ± SEM.
Previously, we found that forelimb/hindlimb coordination was significantly improved after SCI in CX3CR1−/−-deficient mice (Donnelly et al., 2011). Serotonergic axons play a critical role in modulating interlimb coordination, notably by strengthening inhibitory synaptic transmission to motor neurons (Ciranna, 2006; Bos et al., 2013; Gackière and Vinay, 2014). Given the increase in plasticity of 5-HT+ fibers around lumbar motor neurons in CX3CR1−/− mice (Fig. 4) and the dramatic increase in synaptic coverage of these neurons (Fig. 5), we next examined the relative density of inhibitory and excitatory presynaptic and postsynaptic densities on lumbar (laminae IX) spinal cord motor neurons.
First, immunolabeling for l-GAD, an enzyme involved in the synthesis of the inhibitory neurotransmitter GABA, was used to quantify GAD67+ puncta. Significantly more GAD67+ puncta were detected in CX3CR1−/− mice, with overall numbers approaching levels found in naive, uninjured mice (Fig. 6A–D). We extended these analyses by double-labeling for VGAT (vesicular GABA transporter, presynaptic marker) and gephyrin, a protein responsible for organizing both glycine and GABA receptor subunits, both of which mediate postsynaptic inhibition in the spinal cord (Tretter et al., 2012). Using this antibody combination, a significant increase of presynaptic/postsynaptic inhibitory puncta was detected on motoneurons in the lumbar spinal cord of CX3CR1−/− mice (Fig. 6E–N).

Plasticity of inhibitory synapses is enhanced in spinal cord lumbar ventral horn after SCI in CX3CR1−/− mice. GAD67 labeling in transverse sections in the lumbar (L3/L4) spinal cord of (A) naive, (B) CX3CR1+/+, and (C) CX3CR1−/− mice at 56 dpi. D, Quantification of GAD67+ synaptic puncta in the ventral horn of CX3CR1+/+ and CX3CR1−/− mice. E, H, K, VGAT (presynaptic receptor, red) and (F, I, L) gephyrin (postsynaptic receptor, green) double-labeling in naive (E–G), SCI CX3CR1+/+ (H–J), and CX3CR1−/− (K–M) mice. G, J, M, Merged images represent VGAT and gephyrin colocalization. N, Quantification of VGAT-gephyrin colocalized synaptic puncta in the ventral horn of WT (+/+) and CX3CR1-deficient (−/−) mice. Schematic of spinal cord illustrating lamina IX of lumbar spinal cord where synaptic puncta were quantified. Graphs generated from a single experiment; n = 5 +/+ and n = 4 −/− mice/group. *p < 0.05 (Student's unpaired two-tailed t test). Scale bars: C, M, 20 μm. Data are mean ± SEM.
Motor synergies are also controlled by excitatory input to laminae IX neurons from motor cortex and segmental sensory pathways (Cowley and Schmidt, 1997; Alvarez et al., 2004; Landry et al., 2004; Persson et al., 2006; Rossignol et al., 2009; Levine et al., 2014). Therefore, combinations of the presynaptic markers VGlut1 and VGLut2 and postsynaptic marker Homer-1 were used to evaluate postinjury changes in excitatory input to ventral horn motorneurons (Gutierrez-Mecinas et al., 2016). VGlut1 labels intraspinal synaptic contacts formed mostly by excitatory propriospinal fibers, whereas VGlut2 is expressed in synaptic terminals of both supraspinal and intraspinal neurons. Consistent with labeling patterns described previously in rats (Oliveira et al., 2003; Landry et al., 2004; Persson et al., 2006), VGlut1 labeling was enriched in the deeper laminae of the gray matter dorsal horn and lamina VII of both WT and CX3CR1−/− mice, although significantly more excitatory presynaptic/postsynaptic puncta colocalized with laminae VII neurons in CX3CR1−/− mice (Fig. 7A–I). VGlut2 is more widely distributed throughout the spinal gray matter (Oliveira et al., 2003; Persson et al., 2006). Quantification of VGlut2/Homer-1 colocalization also revealed significantly more excitatory synapses in laminae IX neurons in CX3CR1−/− mice (Fig. 7J–R). Together, these data indicate that the relative magnitude of excitatory and inhibitory inputs on lumbar motor neurons is increased after SCI in CX3CR1−/− mice, presumably providing a more “normal” substrate for modulating motor neuron activities.

Plasticity of excitatory synapses is enhanced in spinal cord lumbar ventral horn after SCI in CX3CR1−/− mice. Immunolabeling for presynaptic VGlut1 (A, B, E, F) and VGlut2 (J, K, N, O) and (C, G, L, P) postsynaptic Homer-1 receptors at 56 dpi in CX3CR1+/+ and CX3CR1−/− mice. Merged image of double-labeling for VGlut1 or VGlut 2 and Homer-1 in lamina VII (D, H) and IX (M, Q), respectively. I, R, Schema indicate gray matter laminae where puncta were quantified. The number of excitatory synaptic puncta in CX3CR1−/− mice is increased in ventral horn lamina VII (I) and IX (R), respectively, relative to +/+ mice. Graphs are representative of a single experiment; n = 5 +/+ and n = 4 −/− mice/group. *p < 0.05 (Student's unpaired two-tailed t test). **p < 0.01 (Student's unpaired two-tailed t test). Scale bars: H, Q, 20 μm. Data are mean ± SEM.
CX3CR1 deletion impairs inflammatory signaling in spinal cord microglia remote from the injury epicenter
The remarkable changes in axonal and synaptic plasticity in remote (lumbar) regions of the injured spinal cord of CX3CR1−/− mice could result from the reduced inflammatory burden with neuroprotection at the injury epicenter (see above). However, thoracic SCI can elicit inflammatory signaling in microglia remote from the lesion site, and such changes have been shown to impair sensory and locomotor function and spinal learning (Detloff et al., 2008; Hansen et al., 2013; Grau et al., 2014; Shin et al., 2014). To determine whether lumbar spinal microglia respond differently after SCI in CX3CR1−/− compared with WT mice, we quantified the proportional area (PA) of Iba1+ microglia, a morphological index of activation (Blackbeard et al., 2007; Donnelly et al., 2009) and relative proportions of p38MAPK+ microglia.
After thoracic SCI in both WT and CX3CR1−/− mice, microglia adopt an “activated” phenotype at and well beyond the lesion epicenter, including lumbar spinal levels where activated microglia are distributed throughout white matter and also surrounding motor neuron soma (illustrated using tissue from a CX3CR1+/+ mouse in Fig. 8A,B). However, a comparison of microglia activation (Iba1 PA) between genotypes and as a function of distance from the lesion epicenter revealed a significant main effect of both genotype (F(1,13) = 12.04, p = 0.0042) and spinal level (F(1,13) = 4.87, p = 0.0459). Microglia activation was significantly reduced in CX3CR1−/− mice. These effects were most prominent in the lower lumbar (L4-L5) spinal cord (Fig. 8C–E). The (genotype × spinal level) interaction was not significant.

Microglia activation is reduced in lumbar spinal cord of CX3CR1−/− mice after thoracic SCI. A, GFAP labeling (red) defines lesion margins, and activated microglia (GFP; green) are found within and several segments above and below the injury epicenter at 56 dpi (horizontal section centered on the injury site). B, Triple labeling for 5-HT (red), GFP (green), and ChAT (blue) shows microglia (GFP+) contacting motor neurons and 5-HT terminals in lumbar spinal cord. C, D, F, G, Activated microglia in lumbar spinal cord labeled with Iba-1 in CX3CR1+/+ (C, F) and CX3CR1−/− (D, G) mice. E, IBA-1 proportional area measurement at (L2-L3) and (L4-L5) spinal levels. F–H, High-power images of Iba-1+ microglia in white matter show genotype specific differences in morphology in lumbar ventral horn (shown in L2-L4). I–K, Genotype-specific differences exist for p38MAPK immunolabeling in lumbar spinal cords of CX3CR1+/+ (I) and CX3CR1−/− (J) mice. L–N, Double-labeling for p38MAPK (red, L–N), Iba-1 (green, L), MAC-1 (green, N), and GFAP (blue, M, N) reveals that p38MAPK is predominantly expressed by microglia (Iba1 and Mac1+ cells). E–K, Data were generated from a single experiment using n = 5 +/+ and n = 4 −/− mice/group. E, Data were analyzed using two-way ANOVA with Bonferroni post tests. H, K, Data were analyzed using Student's unpaired two-tailed t test. *p < 0.05. Scale bars: B, G, J, L–N, 20 μm. Data are mean ± SEM.
p38MAPK phosphorylation, an indicator of neurotoxic inflammatory signaling (Herlaar and Brown, 1999; Saklatvala, 2004), also was reduced in CX3CR1−/− microglia relative to WT microglia (Fig. 8I–K). Immunofluorescent double-labeling confirmed that p38MAPK is expressed mainly by microglia (and not astrocytes) (Fig. 8L–N). These data indicate that CX3CR1 regulates inflammatory signaling and cytoskeletal dynamics in remotely activated microglia.
Dendritic spine plasticity is enhanced in lumbar motor neurons of CX3CR1-deficient mice
In WT mice, postinjury activation of microglia with increased inflammatory signaling at remote spinal levels could indicate that these glia are responding to chronic axonal degeneration, motor neuron denervation, and synapse loss. To assess this, we quantified fine details of neuron morphology, including soma size, number of dendrites, and the number and length of dendritic spines on Golgi-stained motor neurons located throughout the ventral horn (lamina IX) of the L2-L5 spinal cord at 56 dpi. Surprisingly, no genotype-specific differences were found for any of these parameters (Fig. 9A–I).

Dendritic spine plasticity is enhanced in motor neurons of CX3CR1−/− mice. Golgi-Cox stain in lumbar spinal cord of CX3CR1+/+ (A, B) and CX3CR1−/− (C, D) mice at 56 dpi. B, Arrows indicate the presence of beaded dendrites (see also Fig. 10). E–I, Motor neuron perimeter, dendrite number, mean dendrite length, length of dendritic spines, and number of dendritic spines. J, Schematic showing morphology of mature and immature dendritic spines. K, High-power image showing filopodial (filo), mushroom (mush), and thin of dendritic spine morphologies. L–N, Quantification of the geometric characteristics of dendritic spines at L2-L3 and L4-L5 spinal levels. L, Immature (Filo) spines. M, Mature (Mush) spines width > 0.6 μm. N, Thin spines with a ratio of length/width >2 μm (immature). A significant switch of mature synapses to immature synapses occurs only in CX3CR1−/− mice. E–N, Data were generated from a single experiment using n = 5 +/+ and n = 4 −/− mice/group. L–N, Data were analyzed using two-way ANOVA with Bonferroni post tests. E–I, Data were analyzed using Student's unpaired two-tailed t test. *p < 0.05. Scale bars: A, C, 100 μm; B, D, 50 μm; K, 1 μm. Data are mean ± SEM.
Because microglia affect synaptic plasticity and because indices of microglia activation differ between genotypes with respect to lumbar spinal level (Fig. 8), we next analyzed dendritic spine morphology taking care to bin data by lumbar segment (L2-L3 vs L4-L5). The maturation status of newly formed dendritic spines and the functional state of ongoing synaptic transmission correlate with dendritic spine morphology (Risher et al., 2014). Fully mature synapses are associated with bulbous or mushroom-shaped spines containing many neurotransmitter receptors. Conversely, longer filopodial-like spines that help initiate synaptic contact are predictive of immature synapses (Yuste and Bonhoeffer, 2001; Hayashi and Majewska, 2005). A rapid and unbiased Golgi analysis technique was used to measure the length and diameter of dendritic spines, which were then classified as filopodial or thin (immature) or mushroom (mature) (Risher et al., 2014). Immature synapses were more frequent at caudal lumbar levels (L3-L4), whereas mature synapses were more frequent at the L2-L3 level. Importantly, in CX3CR1−/− mice, in which microglia activation and inflammatory signaling are reduced relative to WT microglia, these spinal level-dependent differences were magnified (Fig. 9L–N). The percentage of each spine classification was analyzed using two-way ANOVA with genotype and spinal level as independent variables. There was a significant main effect of spinal level for both mushroom (F(1,14) = 13.76, p = 0.0023) and thin spines (F(1,14) = 6.03, p = 0.0278). There also was a main effect of genotype for thin spines (F(1,14) = 6.03, p = 0.0278). The (genotype × spinal level) interaction was not significant. The presence of more immature dendritic spines in caudal spinal cord of CX3CR1−/− mice, together with the data in Figures 56–7 showing enhanced synaptic plasticity at similar levels in these mice, indicates that CX3CR1 signaling may normally inhibit microglia-dependent synapse formation.
CX3CR1 deficiency enhances synapse formation in part by preserving dendritic arbors
When evaluating Golgi-stained neurons, varicosities were detected in a subset of dendritic arbors (Figs. 9B, 10A,B). These may represent early stages of cytoskeletal dysfunction and neuron degeneration. Microglia can release glutamate, and excessive release of glutamate into the extracellular space can cause structural collapse in neurons, including the formation of dendritic varicosities (Ikegaya et al., 2001; Greenwood and Connolly, 2007; Greenwood et al., 2007). An unbiased quantitative comparison of the size and numbers of varicosities in neurons from Figure 9 revealed smaller and fewer dendritic varicosites in CX3CR1−/− mice relative to WT mice (Fig. 10C,D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Microglia-dependent CX3CR1 signaling causes dendrite pathology after SCI. A, B, Dendritic varicosities form on a subset of ventral horn lumbar motor neurons (lamina IX) after SCI. C, D, Greater numbers and larger varicosities are found on motor neurons in CX3CR1+/+ compared with CX3CR1−/− mice. Representative images of neurons in neuron-microglia cocultures (E–G) or neurons exposed to microglia-conditioned media (H–J). K, L, CX3CR1−/− microglia cause less dendrite pathology than CX3CR1+/+ microglia; the ratio of dendrite beads/axon length is decreased on neurons from both coculture (K) and media transfer experiments (L). Graphs are representative mean data from one of three independent replicate experiments. C, D, n = 5 +/+ and n = 4 −/− mice/group. *p < 0.05 (Student's unpaired two-tailed t test). **p < 0.01 (Student's unpaired two-tailed t test). K, L, Student's unpaired two-tailed t test. **p < 0.01. Scale bars: A, 10 μm; G, J, 50 μm. Data are mean ± SEM.
To determine whether microglia cause these varicosities to form, microglia from WT or CX3CR1−/− mice were cocultured with primary cortical neurons (Fig. 10E–G). Although we found varicosities on dendrites cocultured either with WT and CX3CR1−/− microglia, more varicosities were found formed on dendrites when cocultured with WT microglia (Fig. 10F,K). To determine whether these effects were caused by cell contact or soluble factors released by microglia, a separate culture of cortical neurons was exposed to medium conditioned by microglia isolated from WT or CX3CR1−/− mice (Fig. 10H–J). Again, more dendritic varicosities formed when neurons were exposed to WT microglia-conditioned media (Fig. 10I,L). These data are consistent with the in vivo data (Fig. 10A,B) and indicate that microglia adversely affect dendrite structure via release of toxic soluble factors produced downstream from CX3CR1 signaling. Whether these varicosities indicate imminent degeneration or loss of function is not known. Moreover, the precise nature of the soluble factor(s) has not been determined.
Discussion
Data in this report indicate that deletion of CX3CR1 in microglia and macrophages is neuroprotective (spares myelinated axons) and improves indices of endogenous repair, both at the lesion epicenter and in spared white and gray matter (e.g., serotonergic axon growth/sprouting, NG2 cell proliferation), both proximal and distal to the epicenter.
The benefits of CX3CR1 deletion may be explained by functional changes in microglia and macrophages. WT microglia and macrophages activated with inflammatory stimuli become neurotoxic and less supportive of endogenous repair (Kigerl et al., 2009; Miron et al., 2013; Kroner et al., 2014; Gensel et al., 2015). However, the same inflammatory stimuli cause CX3CR1−/− macrophages to increase expression of FGF-2, TGF-β1, and IGF1 mRNA. Proteins encoded by these genes are known to be secreted by macrophages and can promote neuroprotection (Diemel et al., 2003; Frost et al., 2003; Suh et al., 2013; Cohen et al., 2014). Some of these proteins also are likely responsible for increasing survival, differentiation, and proliferation of NG2 cells (McTigue et al., 2000, 2001; Levine, 2016).
After SCI, NG2+ cell numbers increase inside the lesion and at the interface between injured and spared tissue (Lytle et al., 2006, 2009; Tripathi and McTigue, 2007; Wu et al., 2010). This stereotypical response is augmented in CX3CR1−/− mice. Even though NG2 is designated as an axon-growth inhibitory proteoglycan, it also can act as a substrate for growing axons (Tan et al., 2005, 2006; Petrosyan et al., 2013; Levine, 2016), especially serotonergic axons. In NG2 null mice, spontaneous regrowth of serotonergic axons is impaired after SCI (de Castro et al., 2005). Moreover, NG2 cells stabilize degenerating axons at the lesion border, limiting axon “die-back” (McTigue et al., 2006; Yang et al., 2006; Busch et al., 2010). Thus, the enhanced NG2 cell response in/around the contusion lesion in CX3CR1−/− mice could be responsible for the enhanced axonal density and sprouting that we observed (Figs. 3, 4).
Data in this report also show that, in remote lumbar spinal segments, inflammatory signaling is reduced in CX3CR1−/− microglia and that these cells colocalize to regions where axon and synaptic plasticity are enhanced and motor neuron dendrite pathology is attenuated. Importantly, these changes occur on/around motor neurons located within lumbar spinal segments containing the central pattern generator (CPG).
Within the CPG, serotonergic (5-HT) axons play a key role in activating and modulating lumbar motor circuitry (Grillner and Wallén, 1985; Jacobs and Fornal, 1997; Schmidt and Jordan, 2000; Murray et al., 2010; Gackière and Vinay, 2014; Leech et al., 2014). After SCI, 5-HT axons have a propensity to sprout and grow, bypassing the lesion and repopulating gray matter distal to the injury site. This vigorous growth response of 5-HT axons has been implicated in spontaneous recovery of locomotor function after SCI (Schmidt and Jordan, 2000; Murray et al., 2010; Gackière and Vinay, 2014; Leech et al., 2014). Despite this dynamic spontaneous growth, the overall magnitude of growth and lamina-specific distribution of 5-HT axons can be enhanced further using various genetic, pharmacologic or cell-based therapies (Müllner et al., 2008; Boato et al., 2010; Leech et al., 2014). These data indicate that optimal growth and guidance of 5-HT axons require the presence of specific molecular cues, some of which may be blocked or suppressed by CX3CR1-dependent signaling in microglia or macrophages. Indeed, data in the current report indicate that the density of 5-HT+ axons that grow into or around the lesion epicenter increased in mice deficient in CX3CR1 signaling. Also, the number of 5-HT+ axons that spontaneously repopulate lumbar spinal cord gray matter increases in CX3CR1−/− mice, and the preferential growth of 5-HT axons around lumbar motor neurons is topographically appropriate. From these data, we can conclude that local guidance cues that regulate lamina-specific regrowth of 5-HT axons after SCI are inhibited by factors released downstream of CX3CR1 signaling in microglia.
Enhanced serotonergic axon plasticity and greater locomotor function in CX3CR1−/− mice could facilitate synaptic reorganization and reformation of appropriate sensorimotor networks below the level of injury. After SCI, dennervated neuronal circuitry tries to maintain function by modulating the axon sprouting and synaptic densities on newly formed circuits. Several reports have documented a progressive increase in synapse and axon density in segments distal to the lesion (Müllner et al., 2008; Tan et al., 2008; Bandaru et al., 2015). However, these newly formed circuits, unless “sculpted” or modulated by appropriate functional activity (e.g., rehabilitation), can promote aberrant neuronal functions, including pathological reflex control and neuronal exhaustion (Tan et al., 2012; Beauparlant et al., 2013; Bandaru et al., 2015). Postinjury changes in the relative magnitude of excitatory and inhibitory synapses on lumbar motor neurons contribute to such functional deficits.
Serotonin regulates neuronal expression of KCC2, a potassium-chloride cotransporter that regulates intraneuronal chloride homeostasis and the maturation and function of excitatory and inhibitory neurotransmission (Chamma et al., 2012; Gackière and Vinay, 2015). After SCI, loss of serotonergic input is associated with an increase in excitatory synapses and a decrease in inhibitory synapses on lumbar motor neurons. Loss of inhibitory input increases motor neuron excitability. Regrowth or sprouting of serotonergic axons or pharmacological interventions that increase signaling via 5-HT receptors can restore KCC2 expression and normalize motor neuron excitability (Bos et al., 2013). In the present study, increased 5-HT fiber density in the lumbar spinal cord of CX3CR1−/− mice was associated with a significant increase in inhibitory synapses. Likewise, a notable increase of excitatory synapses was observed in the same group. These data could indicate that microglia, via CX3CR1-dependent signaling, normally inhibit postinjury remodeling of dendritic spines below the level of injury.
Published data show that dynamic remodeling of dendritic spines occurs throughout gray matter on neurons distal to the injury site (Bandaru et al., 2015). This compensatory response of neurons to denervation is enhanced in CX3CR1−/− microglia. In addition to improved recovery of locomotor function, it is possible that microglia CX3CR1-dependent changes in synapse formation also will affect other functional indices. For example, activating CX3CR1 can enhance synaptic strength and neurotransmission in nociceptive pathways (Clark et al., 2015). Thus, CX3CL1-CX3CR1 signaling may normally contribute to the onset and propagation of hyperreflexia and neuropathic pain after SCI (Tan et al., 2008; Bandaru et al., 2015).
Microglia may also regulate neuronal excitability by modulating sensitivity of neurons within CPG circuitry. Microglia associate with the axon hillock, specifically with the axon initial segment (Baalman et al., 2015) and CX3CL1-mediated activation of microglia (via CX3CR1) facilitates excitatory synaptic transmission (Clark et al., 2015). Thus, inhibiting CX3CR1 signaling in microglia could alter microglia-neuron cross talk to reverse the hyperexcitable state that is characteristic of neuronal networks in the CPG of SCI mammals.
Inflammatory signaling in microglia stimulates the synthesis and release of potentially dangerous cytokines and enzymes that can damage microtubules causing dendritic beading and synapse loss (Khairova et al., 2009; Maezawa and Jin, 2010; Hardingham et al., 2013; Yang et al., 2013). Thus, reduced inflammatory signaling in microglia could limit damage to axons and dendrites, yielding a circuitry that is better able to support recovery of function. Notably, in CX3CR1−/− mice, where inflammatory signaling is reduced in microglia, we found fewer dendritic varicosities on neurons (Fig. 9), and an identical phenotype was observed in vitro on neurons incubated with media conditioned by CX3CR1−/− microglia (Fig. 10).
Together, data in this report indicate that, in mice with deficient CX3CR1 signaling, the lesion milieu created by inflammatory microglia/macrophages is less toxic and more favorable to cell survival, repair, and plasticity. Future studies are needed to identify the complex molecular mechanisms that control CX3CR1-dependent signaling and whether manipulating CX3CR1, either through use of blocking antibodies or small molecule inhibitors, can augment structural and functional plasticity alone or as an adjunct to facilitate rehabilitation.
Footnotes
- Received September 9, 2016.
- Revision received February 21, 2017.
- Accepted February 22, 2017.
This work was supported by Cnpq-Brazil (Science without borders) Fellowship to C.M.F. and the Ray W. Poppleton Endowment to P.G.P. We thank Dr. Alexandre L. R. Oliveira for assistance and access to the Transmission Electron Microscopy, State University of Campinas (UNICAMP), São Paulo, Brazil; Randy J. Nelson (Ohio State University) for access to the Neurolucida MBF Bioscience system; and Taryn Aubrecht for assisting with Neurolucida software.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Phillip G. Popovich, Ohio State University, 460 West 12th Avenue, 694 Biomedical Research Tower, Columbus, OH 43210. phillip.popovich{at}osumc.edu.
- Copyright © 2017 the authors 0270-6474/17/373568-20$15.00/0