
Abstract
The regulation of the striatum by the GPCR signaling through neuromodulators is essential for its physiology and physiopathology, so it is necessary to know all the compounds of these pathways. In this study, we identified a new important partner of the dopaminergic pathway: GPRIN3 (a member of the GPRIN family). GPRIN3 is highly expressed in the striatum but with undefined function. Cell sorting of medium spiny neurons (MSNs) in indirect MSNs and direct MSNs indicated the presence of the GPRIN3 gene in both populations with a preferential expression in indirect MSNs. This led us to generate GPRIN3 KO mice by CRISPR/Cas9 and test male animals to access possible alterations in morphological, electrophysiological, and behavioral parameters following its absence. 3D reconstruction analysis of MSNs revealed increased neuronal arborization in GPRIN3 KO and modified passive and active electrophysiological properties. These cellular alterations were coupled with increased motivation and cocaine-induced hyperlocomotion. Additionally, using a specific indirect MSN knockdown, we showed a preferential role for GPRIN3 in indirect MSNs related to the D2R signaling. Together, these results show that GPRIN3 is a mediator of D2R function in the striatum playing a major role in striatal physiology.
SIGNIFICANCE STATEMENT The striatum is the main input of the basal ganglia processing information from different brain regions through the combined actions of direct pathway neurons and indirect pathway neurons. Both neuronal populations are defined by the expression of dopamine D1R or D2R GPCRs, respectively. How these neurons signal to the respective G-protein is still debatable. Here we identified GPRIN3 as a putative selective controller of D2R function in the striatum playing a critical role in striatal-associated behaviors and cellular functions. This study represents the identification of a new target to tackle striatal dysfunction associated with the D2R, such as schizophrenia, Parkinson's disease, and drug addiction.
- Crispr/Cas9
- D2 receptors
- GPRIN3
- iMSNs
- striatum
Introduction
The striatum is the primary input nucleus of the basal ganglia integrating sensorial, emotional, and higher cognitive responses (Hassoun et al., 2005; Wen et al., 2016; Laurent et al., 2017; Maltese et al., 2018). It receives dense dopaminergic innervations from the substantia nigra and from the ventral tegmental area (VTA) (Watabe-Uchida et al., 2012). It is postulated that dopamine drives the striatal function through a combined balance between the activation of striatonigral (dMSNs, expressing D1 receptors, D1R) and striatopallidal (iMSNs, expressing D2 receptors, D2R) medium spiny neurons (MSNs). Together, MSNs comprise ∼95% of all cells in the striatum (Gerfen et al., 1990). Both, D1R and D2R are GPCRs; whereas D1R are coupled to Gs-olf, D2R act through Gαi/o proteins. The functionally opposite types of GPCRs operated by D1R and D2R suggest an antagonistic role for MSN populations in the striatum, but recently it has been shown that a timely ordered synergism of both populations is required for normal function (Jin et al., 2014; Tecuapetla et al., 2016).
Considering the multitude of functions played by the striatum, MSNs are the target not only of dopamine, but also other endogenous G-protein-regulating neuromodulators, such as serotonin, cannabinoids, adenosine, and opioids, since the control of many functions is dependent on multiple synchronized ligand actions (Augusto et al., 2013; Araque et al., 2017; Cavaccini et al., 2018; Nummenmaa et al., 2018). The malfunction of this intricate machinery translates into several striatum-related pathologies, such as addiction, Huntington's disease, Parkinson's disease, depression, and schizophrenia, among others (Li et al., 2015; Florio et al., 2018; Rivas-Grajales et al., 2018). It is still debatable how GPCRs fine-tune the signaling between the binding of certain neuromodulators and the subsequent molecular mechanisms.
The G-protein-regulated inducer of neurite outgrowth (GPRIN) family of proteins (GPRIN1, GPRIN2, and GPRIN3) are Gαi/o-regulated proteins suggested to intermediate the communication between GPCRs and the sequential intracellular targets (Iida and Kozasa, 2004). As an example of such functional “bridge,” the first identified member of the GPRIN family (GPRIN1) is known to intermediate the function of μ-opioid receptors (Ge et al., 2009). GPRIN1 binds to Gαo-protein-activating Cdc42, resulting in remarkable cellular morphological changes (Nakata and Kozasa, 2005). Similar observations were made concerning GPRIN2 (Masuho et al., 2008). Indeed, GPRIN1 and GPRIN2 have been described as alternative (to adenylyl cyclase) mediators of GPCRs signaling (Iida and Kozasa, 2004).
GPRIN3, on the other hand, has a much less defined role but is highly expressed in the brain and has recently been tagged as a partner of β-arrestin-2, regulating dopamine receptor desensitization with impact on selective behavioral tasks (Mototani et al., 2018). Here, we show the preferential expression of the GPRIN3 in iMSNs, and its presence in much higher levels in the striatum compared with the PFC, hippocampus, or cerebellum. The higher expression of GPRIN3 in iMSNs might be related to the fact that D2R are Gαi/o protein-coupled receptors, making them more likely (but maybe not uniquely) functional partners of GPRINs, whereas D1R, expressed in dMSNs, are Gs-olf protein-coupled receptors.
We therefore formulate the hypothesis that the KO of GPRIN3 would destabilize D2R function, having deep impact on the striatal function. Moreover, some functional overlapping hints on the potential partnership between GPRINs and the D2R. For instance, fluctuations in the D2R availability in the striatum impact the cellular morphology inducing a profound reorganization of the basal ganglia as the overexpression of the D2R is sufficient to reduce the dendritic arborization of MSNs inducing the shrinkage of the striatum (Cazorla et al., 2012).
In taking with the evidence that GPRINs intermediate the signaling between the Gαi/o and the adjacent intracellular mechanisms, here our aim is to untangle the relative role of GPRIN3 in morphological and electrophysiological properties of MSNs as well as the impact of GPRIN3 manipulation on behaviors relevant to dopamine in dMSNs and iMSNs.
Materials and Methods
Animals.
All procedures were performed in accordance with the guidelines provided by the Institutional Animal Care Committee and were approved by the local ethics committees (Université Libre de Bruxelles Pôle Santé 551-N). Unless stated, 2–4 mice were housed together, with ad libitum access to food and tap water. Housing conditions were as follow: temperature: 21°C; humidity: 50%; light/dark cycle: 12 h/12 h. Only males were used for the experiments.
Generation of GPRIN3-KO mice.
sgRNAs targeting N-terminal (sgRNA-a) or C-terminal (sgRNA-b) regions of GPRIN3 gene were designed using Crispr Design software (http://crispr.mit.edu/) (Cong et al., 2013). The two selected sgRNA target sequences (sgRNA-a: 5′-GGTGACTAAAGCTTCGATAG-3′, sgRNA-b: 5′-AGAAGAAATGATCGTCACTG-3′) were used to design dsDNA templates containing a T7 RNA polymerase promoter, the target sequence followed by the sgRNA scaffold (Chen et al., 2013). The sequences were synthesized as linear dsDNA gBlock fragments (IDT), and the corresponding sgRNAs were in vitro transcribed using the T7 RNA polymerase (New England Biolabs). The inactivation of GPRIN3 sequence by Cas9/CRISPR was performed directly in fertilized mouse oocytes. Briefly, C57BL/6J female mice underwent ovulation induction by intraperitoneal injection of 5 IU equine chorionic gonadotrophin (PMSG; Folligon, InterVet), followed by intraperitoneal injection of 5 IU human chorionic gonadotropin (Pregnyl Essex Chemie) 48 h later. For the recovery of zygotes, C57BL/6J females were mated with males of the same strain immediately after the administration of human chorionic gonadotropin. All zygotes were collected from oviducts 24 h after the human chorionic gonadotropin injection, and were then freed from any remaining cumulus cells by a 1–2 min treatment of 0.1% hyaluronidase (Sigma-Aldrich) dissolved in M2 medium (Sigma-Aldrich). Mouse embryos were cultured in M16 (Sigma-Aldrich) medium at 37°C and 5% CO2. For micromanipulation, embryos were transferred into M2 medium. All microinjections were performed using a microinjection system comprised of an inverted microscope equipped with Nomarski optics (Nikon), a set of micromanipulators (Narishige), and a FemtoJet microinjection unit (Eppendorf). Injection solution containing both sgRNAs (150 ng/μl), Cas9 mRNA and Cas9 protein (12.5 ng/μl) (Toolgen) was microinjected into the male pronuclei of fertilized mouse oocytes until 20%–30% distension of the organelle was observed. Embryos that survived the microinjection were transferred on the same day into the oviducts of 8- to 16-week-old pseudopregnant Crl:CD1(ICR) females (0.5 d used after coitus) that had been mated with sterile genetically vasectomized males (Haueter et al., 2010) the day before embryo transfer. Pregnant females were allowed to deliver and raise their pups until weaning age. sgRNA preparation and microinjection were performed by Pawel Pelczar (Center for Transgenic Model, Basel, Switzerland). Founder screening was performed by genomic PCR using 3 couples of primers: Primer_a_F (5′-GGGACTGTACCTGACCCTCT-3′) and Primer_a_R (5′-ACTGGCTCTCCCTCACTGAA-3′) (amplification of sgRNA-a site), Primer_b_F (5′-GGTGTTCTGCAGTCCATGCT-3′) and Primer_b_R (5′-GCATCCTCTTGTGGTACTTGC-3′) (amplification of sgRNA-b site) or Primer_a_F and Primer_b_R (amplification of deletion band). PCR products were then purified using Qiaquick PCR purification kit (QIAGEN) and sent to Eurofins for sequencing. Founder 11 (male) was bred with C57BL6 females, and the offspring heterozygous for the 11-c mutation (GPRIN3+/−) were selected. This mutation consists of an insertion of 1 bp, which creates a frameshift and a premature STOP codon in GPRIN3 coding sequence. This mutation can be detected by BstBI restriction after PCR amplification. GPRIN3+/− mice were backcrossed on C57BL6 background for at least two generations. GPRIN3−/− mice were then obtained from GPRIN3+/− × GPRIN3+/− breedings and GPRIN3+/+ littermates were used as controls. All genotypes were confirmed after the end of experiments.
Protein extraction from striatum and mass spectrometry analysis.
Striatum tissue was microdissected from GPRIN3+/+ and GPRIN3−/− mice before homogenization in 250 μl lysis buffer containing 62.5 mM Tris-HCl, pH 6.8, and 2% SDS. The homogenates were centrifuged at 16,000 × g for 15 min at 4°C. Protein concentration was assayed in the supernatants with DC Protein Assay (Bio-Rad); 300 μg of crude protein extracts was mixed with 4× Laemmli buffer and incubated during 5 min at 95°C. The samples were applied to 9% SDS-PAGE and stained by PageBlue Protein Staining (Thermo Fisher Scientific).
The gel bands were excised from the stained gel (64 bands per lane) and subjected to tryptic digestion. Briefly, the bands were incubated in 30 μl of 25 mM NH4HCO3, then reduced with 10 mM dl-DTT during 30 min at 56°C, and alkylated with 55 mM iodoacetamide during 20 min at room temperature. Proteins were digested overnight with 75 ng of trypsin (Promega) at 37°C. After gel shrinking with acetonitrile (ACN), formic acid was added to 1% (v/v) and peptides were purified using StageTips C18 (Thermo Fisher Scientific) according to the manufacturer's instructions. The samples were evaporated to dryness in a vacuum centrifuge and resuspended in 10 μl of 0.1% HCOOH. Aliquots (8 μl) of digested proteins were injected for mass spectrometric analysis.
Mass spectra were acquired using a 5600 Triple TOF mass spectrometer (AB Sciex) interfaced to a 425 microLC system (Eksigent). Peptides were separated on a separation column (ChromXP C18 CL, 150 × 0.3 mm, 3 μm) using a two-step ACN gradient (5%–25% ACN/0.1% HCOOH in 48 min, then 25%–60% ACN/0.1% HCOOH in 20 min) and were sprayed online in the mass spectrometer. MS1 spectra were collected in the range 400–1250 m/z for 250 ms. The 20 most intense precursors with a 2–4 charge state were selected for fragmentation, and MS2 spectra were collected in the range 200–1500 m/z for 100 ms. Precursor ions were excluded for reselection for 12 s.
Experimental ablation and D2-eGFP strain.
Adora2a-Cre+/− iDTR+/− and D1-Cre+/− iDTR+/− mice were obtained by crossing Adora2a+/− mice (Durieux et al., 2009) or D1-Cre+/− mice (Gong et al., 2007) with iDTR+/+ mice (Buch et al., 2005). Specific ablation of iMSNs or dMSNs was performed as previously described (Durieux et al., 2009, 2012).
The two Cre strains (Adora2a-Cre+/− and D1-Cre+/−) were bred with the Rosa26-tdTomato reporter strain (Madisen et al., 2010). Resulting Adora2a-Cre+/− tdTomato+/+ and D1-Cre+/− tdTomato+/+ mice allowed iMSN or dMSN identification and were used for FACS experiments. GPRIN3 KO strain was bred with D2-eGFP strain (Gong et al., 2003) to obtain D2-eGFP+/− GPRIN3+/+ (control group) and D2-eGFP+/− GPRIN3−/− (experimental group) mice. All strains were of C57BL6 background, except D2-eGFP mice, which were kept on CD1 background. Consequently, double-transgenic mice D2-eGFP GPRIN3 were of mixed background.
Striatal cell dissociation and FACS.
Ten- to 16-week-old Adora2a-Cre+/− tdTomato+/+, D1-Cre+/− tdTomato+/+, and tdTomato+/+ mice (control) were used for FACS experiments, performed as previously described (Ena et al., 2013) with small modifications. Striata were dissected from acute brain slices (see Brain slice preparation), dissociated enzymatically (Protease Type XIII 1.5 mg/ml; Sigma-Aldrich), and then triturated to obtain dissociated cells. To remove excessive debris, the cell suspension was subjected to 3 successive cycles of centrifugation (2 min at 2000 rpm) and resuspended in L-15 medium (Invitrogen) supplemented with 10 μm HEPES and 5% (m/v) trehalose (Saxena et al., 2012). Cell suspension was then filtered through a 40 μm cell strainer (Falcon; BD Biosciences), and DAPI (1:5000, D9564, Sigma-Aldrich) was added to label dead cells. Cells were sorted on a FACS Aria cell sorter (BD Biosciences) for PE signal (detecting tdTomato) and Fl3 (detecting Hoechst). Sorted cells were collected directly in RLT lysis buffer (QIAGEN) to proceed with the RNA extraction. Each sample was a collection of cells pooled from 2–4 animals.
RNA extraction and qRT-PCR.
Total RNA extraction from brain structures (striatum, cortex, hippocampus, or cerebellum) was performed using Qiazol lysis reagent (QIAGEN) and RNeasy Minikit (QIAGEN) as recommended by the manufacturer. Samples were treated with RNase Free DNase Set (QIAGEN) and quantified by measuring the optical density (ND-1000 Spectrophotometer, NanoDrop Technologies); 1 μg of total RNA was used to synthesize a first-strand cDNA using an MMLV-RT kit (Invitrogen) and random hexamer (Roche Diagnostics). For total RNA extraction from sorted cells, RNeasy Microkit (QIAGEN) was used. In both cases, qRT-PCR analysis was performed with Power SYBR Green (Applied Biosystems) on the ABI 7500 Fast Real Time PCR system (Applied Biosystems) using the following primers: GPRIN3: Mm_Gprin3_1_SG QuantiTect Primer Assay (QIAGEN). RER-1 (5′-CCACCTAAACCTTTTCATTGCG-3′ and 5′-TTTGTAGCTGCGTGCCAAAAT-3′) or RPL13 (5′-CCCGTGGCGATTGTGAA-3′ and 5′-TCATTGTCCTTCTGTGCAGGTT-3′) were used as housekeeping gene. Ct (threshold cycle) values for each gene were normalized to the Ct value for RER-1 or RPL13 to obtain relative expression level. The samples obtained from cortex or from D1-Cre+/− tdTomato+/+ mice were used as reference and set at an arbitrary value of 100.
In situ hybridization.
The 20-μm-thick striatal coronal slices were produced from fresh-frozen brain and mounted onto glass slides. In situ hybridization and subsequent quantification were performed as described previously (Durieux et al., 2009). The following probes were used: GPRIN3: 5′-CGACTCCCCTCGAGACTCTTCCTTTCCAGAAGCTGCCACTQTCGA-3′, enkephalin (enk): 5′-GGCCAAGGTGTCTTCCTCATCTGCATCCTTCTTCATGAAGCCGCC-3′, substance P: 5′-GCGTCACTGCCAAGCTTGGACAGCTCCTTCATCGTGCTTTGC-3′.
Stereological analysis.
The striatal volume was accessed using the Cavalieri method through the StereoInvestigator software (MBF Bioscience). Brains of 2- to 3-month-old GPRIN3+/+ or GPRIN3−/− male mice were quickly removed after decapitation and incubated overnight in formol. They were then transferred to 70% ethanol bath before inclusion in paraffin. Serial coronal slices (15 μm thick) were produced using an apotome and Nissl staining performed. Slices were observed at 10× magnification on an AxioImager M1 (Carl Zeiss) equipped with a stage controller (MBF Bioscience) and a camera Axioplan (Carl Zeiss). A 30 μm grid was superposed with the slice, and the striatal surface was manually delimited.
Brain slice preparation and patch clamping.
All the procedures were similar to those described previously (De Backer et al., 2018). Animals were deeply anesthetized using halothane and killed by dislocation and decapitation. Brains were rapidly removed and immersed in ice-cold cutting solution (in mM as follows: 140 choline, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, and 14 glucose) gassed with a 95% O2 and 5% CO2 mixture; 220 μm corticostriatal coronal slices in the cutting solution were prepared using a Vibratome VT 1000 S (Leica Microsystems). Slices were then transferred to a chamber filled with a standard aCSF (in mM as follows: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose, pH 7.2–7.4, 330 mOsm/L) also gassed with 95% O2 and 5% CO2. Slices were left in the chamber at 34°C to recover for at least 1 h before recording.
After the recovery period, slices were placed in a submersion-type chamber at room temperature with 2 ml/min rate of aCSF saturated with 95% O2 and 5% CO2. Striatal neurons were recorded at room temperature. Whole-cell patch-clamp recordings were obtained from striatal neurons (GPRIN3−/− and GPRIN3+/+), iMSNs (eGFP+ neurons from D2-eGFP+/− GPRIN3−/− brain slices or eGFP+ neurons from pSicoR-injected D1-Cre+/− mice), and dMSNs (eGFP− neurons from D2-eGFP+/− GPRIN3−/− brain slices). MSNs were identified with a 63× water-immersion objective of an upright microscope (Axioskop 2FS Plus; Carl Zeiss), and somatic eGFP expression was verified routinely using epifluorescence from a source composed of a LED array (OptoLED; Cairn Research) emitting at 488 nm and coupled to the microscope that was equipped with a dichroic mirror and a high-pass emission filter centered at 505 and 507 nm, respectively. Whole-cell recordings were made under infrared differential interference contrast optics with a back-illuminated CCD camera (iXon+; Andor Technology) with borosilicate-glass patch electrodes (Hilgenberg) obtained with a vertical two-stage puller (PIP5; HEKA). Recordings started 2 min after obtaining the whole-cell configuration to allow diffusion of the intracellular solution (in mM as follows: 125 potassium methylsulfonate, 4 MgCl2, 10 HEPES, 0.1 EGTA, 5 Na2-phosphocreatine, 0.5 Na2-GTP, 4 Mg2ATP, and 0.022 CaCl2). Biocytin (0.5%) was added when 3D reconstruction of patched-neurons was planned. Current- and voltage-clamp recordings were obtained with a Dual EPC-10 operational amplifier (Molecular Devices) interfaced to an OS X iMac running PatchMaster software (HEKA). The signal for voltage-clamp recordings was filtered at 3 kHz and digitized at 20 kHz, and that for current-clamp recordings was filtered at 3 kHz and digitized at 10 kHz using the built-in EPC-10 filter (HEKA). Access resistance, membrane resistance, and membrane capacitance were extracted from voltage-clamp recordings by analyzing current relaxation induced by a 10 mV step change from a holding potential of −80 mV as described previously (D'Angelo et al., 1995). Neuronal excitability was investigated in current clamp by setting the resting membrane potential (RMP) at −80 mV and injecting 1 s depolarizing steps (from 0 to 250 pA in 10 pA increments). Action potential (AP) frequency was measured by dividing the number of interspike intervals (ISIs + 1) by the time interval between the first and the last spike. Rheobase current was defined as the first current step within a series of 10 pA steps beginning at 0 pA that was capable of eliciting at least one AP (Gertler et al., 2008). Quinpirole (20 μm; Sigma-Aldrich) was infused into the aCSF solution, and the recordings were performed 10 min after. All recordings were analyzed using the IgorPro 6.3 software (Wavemetrics).
Immunohistochemistry and 3D reconstruction.
We used a slightly modified version of the one used by Lambot et al. (2016) from our laboratory. When added in the internal solution during patch experiments, biocytin allows the labeling of patched neurons. Slices were incubated overnight at 4°C in PFA solution (4% in 0.01 m PBS). After fixation, the slices were washed 3 times in PBS and incubated in PBS-0.1% Triton X-100 (Sigma-Aldrich) for 2 h under gentle shaking to permeabilize cellular membranes. Slices were then transferred in solution containing streptavidin-coupled AlexaFluor-488 (1/200 in PBS-0.1% Triton X-100 solution, Jackson ImmunoResearch Laboratories) or NL557 (1/3000 in PBS-0.1% Triton X-100 solution, R&D Systems) for 1 h under shaking. Cellular nuclei were finally stained by incubation in Hoechst solution (1/5000 in PBS, H3570, Thermo Fisher Scientific) for 5 min. After several washes in PBS, slices were mounted between slide and coverslip (no. 1.5, VWR) with Fluorsave mounting medium (34–578-920ML, Thermo Fisher Scientific).
Confocal microscopy and 3D reconstruction.
Serial optical sections (z stacks) were imaged on a laser-scanning confocal system (LSM 780; Carl Zeiss), mounted on an Axiovert 200M inverted microscope (Carl Zeiss), equipped with a c-Achroplan NIR 40×/0.8W objective (Carl Zeiss). The excitation beam of an argon laser (488 nm) and bandpass emission filter (500–550 nm) were used for the detection of the Alexa-488 fluorophore, whereas an HeNe laser (543 nm) was used for detection of NL557 and fluorescence was measured between 585 and 700 nm.
After the acquisition, the image was deconvoluted (Huygens-II deconvolution software; Scientific Volume Imaging) and the 3D image rendered. During the deconvolution step, an algorithm (Classic Maximum Likelihood Estimation) determines a point spread function, which is used to calculate, for each pixel of each acquired image in a z series, the statistical likelihood of the exact origin of the photons emitted by its fluorescent source. Reconstruction of the dendritic arbor of the neurons was done by the computer-aided filament tracing function in the 3D image analysis software Imaris (Bitplane). A complementary manual tracing strategy adjusted the automatic reconstruction. Total dendritic arborization reconstruction allowed Sholl, dendrite branching, and dendrite segment length analysis.
Lentivirus-mediated knockdown of GPRIN3.
The design of shRNA targeting GPRIN3 mRNA, lentiviral production, and titration were performed as previously described (Ena et al., 2013). Oligonucleotides containing the shRNA sequence 5′-GAACTGCACTAGTGTACGA-3′ were cloned into HpaI and XhoI restriction sites in pSicoR lentiviral vector as described previously (Ventura et al., 2004). Lentiviral stock was produced by transient cotransfection of pSicoR, psPAX2, and pMDG2G into HEK293T cells with the calcium phosphate method. After harvest and titration, lentiviral stock was diluted in D-PBS (Invitrogen) to reach 5 × 108 TU/ml. pSicoR-mediated GPRIN3 knockdown was first evaluated in C57BL6 male mice, by performing unilateral striatal injection and comparing in situ hybridization signal for GPRIN3 mRNA in both hemispheres. After validation of GPRIN3 knockdown, intrastriatal injections were performed as previously described (Ena et al., 2013). Briefly, 5 μl of diluted lentiviral stock was bilaterally injected into striatum of 8- to 10-week-old D1-Cre+/− male mice. The patch-clamp analysis was performed 1 month after stereotaxic injection.
Behavioral tests.
Behavioral experiments were conducted on 9- to 16-week-old male GPRIN3 KO (GPRIN3−/−) and control mice (GPRIN3+/+), during the light phase of the day. When needed, mice were video-tracked using Ethovision XT equipment (Noldus Technology).
Open field.
Locomotor activity was measured during 1 h in square open field boxes (40 × 40 cm) in dark light. A central zone (20 × 20 cm) was defined in each box. The total distance traveled and the time spent in the center zone were measured.
Rotarod.
The rotarod apparatus (Ugo Basile) consisted of 4 plastic rollers (3 cm diameter) with small grooves running along its turning axis. The protocol consisted of 4 trials per day for 4 consecutive days. In each trial, the animals were placed on the rotating rod at a constant speed (4 rpm), and then the rod was accelerated continuously from 4 to 40 rpm over a time period of 300 s. The latency to fall off the rotarod was measured for each session. Animals that did not fall during the entire experiment were scored as 300 s (Bearzatto et al., 2006).
Elevated plus maze.
The elevated plus maze was used on the basis of its documented ability to detect both anxiolytic- and anxiogenic-like drug effects in mice (Lister, 1987). Briefly, the apparatus consisted of four arms (30 cm long and 7 cm wide) made of wood covered with impermeable Formica, and placed 40 cm above the floor. Two opposite arms were surrounded by walls (17 cm high, closed arms) while the other two were devoid of enclosing walls (open arms). The four arms were connected by a central platform (7 × 7 cm). Each mouse was placed in the center of the maze facing a closed arm, and their behavior was monitored for 5 min: anxiogenic-like effects were defined as a decrease in the proportion of open arm entries divided by the total number of arm entries, and the time spent on open arms relative to the total time spent on both arms. Whenever a mouse placed all four paws onto an arm, an entry was recorded. The total number of closed arm entries was used as a measure of locomotor activity.
Light-dark box.
Anxiety-like behavior was next evaluated using the light/dark apparatus, which was composed of two rectangular compartments (20 × 20 × 14 cm) separated by a tunnel (5 × 7 × 10 cm). One compartment is constituted of a black floor and walls dimly lit (5 lux), whereas the other is constituted of a white floor and walls intensely lit (1000 lux). The apparatus is equipped with infrared beams and sensors. Mice were placed in the dark compartment, and behavior was automatically recorded for 5 min.
Marble-burying test.
Defensive burying was measured using the marble-burying test, performed with 20 small glass marbles (15 mm) evenly spaced in a transparent single cage (21 × 11 × 17 cm) over 4 cm sawdust bedding. The cage was covered by a plastic lid in a room illuminated at 40 lux. The mice were left in the cage for 10 min, and the number of marbles buried more than halfway in the sawdust was counted.
Tail suspension.
The tail suspension test is based on the fact that animals subjected to the short-term inescapable stress of being suspended by their tail will develop an immobile posture. The total duration of immobility induced by tail suspension test was measured according to the method previously described (Steru et al., 1985). Briefly, mice were suspended 50 cm above the floor with an adhesive tape placed ∼1 cm from the tip of their tail. Immobility time was recorded during a 6 min period. Mice were considered immobile only when they hung passively and completely motionless.
Sucrose preference.
Sucrose preference was assessed using a two-bottle choice protocol. Mice were transferred from their home cages to individual cages with access to two bottles containing tap water and were allowed to habituate to the housing and drinking conditions for 3 d before the initiation of the test. During the choice phase, one of the two bottles was filled with 2% sucrose solution and the other with tap water. Mice had access to both bottles for a period of 24 h. Bottles were weight before and after the test. The sucrose intake (g/kg/d), water intake (g/kg/d), total fluid consumption (g/kg/d), and the sucrose preference ratio (defined as sucrose intake normalized by total fluid consumption) were posteriorly calculated.
Operant training and progressive ratio task.
The task was conducted in six identical automated operant chambers (Imetronic), each set in a ventilated, sound-isolated cubicle. Test cages were equipped with a grid floor, a pellet dispenser, and a retractable lever. Each pellet dispenser is coupled with a light cue above the food magazine. First, mice were placed on a food restriction schedule and kept at 80%–85% of their free feeding weight. Mice were then submitted to a magazine training phase, when food pellets (Dustless Precision chocolate flavor pellets #F05301, Bio-Serv) are provided at random time intervals (one daily session for a minimum of 3 d). Each single session was considered finished after 30 min or after 40 pellets dispensed. Subsequently, mice were submitted to a continuous reinforcement phase (CRF, 5 d), where they were trained to press a lever to obtain food pellets (each lever press was rewarded during this session). The lever was retracted after each press, while the food magazine light was switched on and a food pellet was dispensed. The light above the food magazine was switched off, and the lever reextended after the food magazine was revisited by the mouse or after 3 s. The CRF phase was followed by random ratio (RR) phases (ranging from; RR5, where the reward was delivered on average every 5 lever presses, to RR25, where the reward was delivered on average every 25 lever presses). Similar to the food magazine training, each trial of CRF and RR phases ended after 30 min or when 40 food pellets were earned. The number of lever presses was then normalized by the duration of each session.
After the instrumental learning, mice were submitted to a progressive ratio task to assess their motivation to have access to food. During this test, the number of lever presses required to earn a food pellet progressively increased as follows: 1, 2, 3, 5, 12, 18, 27, 40, 60, 90, 135, 200, 300, 450, 675, and 1000. The maximum duration of each session was 2 h but ended automatically if no lever press was computed for 3 min. Recorded parameters were the number of rewards earned, the number of lever presses, the session duration, and the breakpoint (defined as the ratio at which the animals stop pressing the lever).
Finally, mice were submitted to a devaluation task, to detect potential alterations in reward value. After the progressive ratio task, mice were retrained on an RR25 schedule for 2 consecutive days, and then the food pellet reward was devalued by reinstalling ad libitum access for food. Mice were submitted to the same RR25 protocol, and effect of devaluation on operant behavior was assessed by comparing the normalized number of lever presses before and after the instauration of ad libitum access to food.
Cocaine-induced sensitization.
Cocaine response was tested in arena (20 × 40 cm) under light intensity of 70 lux. Mice were first injected intraperitoneal daily with saline (0.9% NaCl) during 3 consecutive days. After this habituation phase, mice received subeffective doses of cocaine (1 and 5 mg/kg) on daily basis, to detect a potential increase in sensitivity to cocaine (Bello et al., 2011). This was followed by a classical sensitization protocol, consisting of daily intraperitoneal administration of 20 mg/kg cocaine during 5 consecutive days. The total distance traveled was recorded after cocaine injection for a period of 1 h.
Western blot.
Western blot analyses were performed using proteins lysates of dorsal and ventral striata as described previously (Rial et al., 2014). Acute brain slices from cocaine and saline-treated animals were obtained as previously described (see Brain slice preparation). Striata were then manually dissected out, cut into dorsal and ventral part, and stored at −80°C until utilization. Tissue was homogenized in lysis solution (in mM: 50 Tris-HCl, pH 7.5, 150 NaCl, 1 EDTA, 1% Triton X-100, 0.1% SDS) supplemented with Halt protease and phosphatase inhibitors (Thermo Fisher Scientific). After incubation on ice for 30 min, samples are clarified by centrifugation and proteins content is quantified using Bradford protein assay (Bio-Rad); 80 μg of total proteins and PageRuler Prestained Protein ladder (Thermo Fisher Scientific) were loaded on a denaturing 10% polyacrylamide gel and separated by electrophoresis at constant current (40 mA). After migration, proteins were transferred to a nitrocellulose membrane using Trans-Blot Turbo Transfer kit (Bio-Rad), as recommended by the manufacturer. Nonspecific sites were blocked at room temperature in blocking solution: PBS-Tween 0.1% (PBS-T)/Odyssey blocking buffer (Li-COR, #P/N 927-40000) 50/50 v/v for 1 h, and then membrane was incubated overnight with primary antibody (phospho-Thr34 DARPP-32 (AF2874, R&D Systems) 1/200, phospho-Thr75 DARPP-32 (2301, Cell Signaling Technology) 1/2500, and total DARPP-32 (sc-271111, Santa Cruz Biotechnology, 1/750) in blocking dilution. After several washes in PBS-T, proteins of interest were revealed by incubating the membrane with fluorophore-coupled adequate secondary antibody (P/N 925-68073 or P/N 925-32212 1/10000, Li-COR) in blocking solution for 1 h. Finally, membranes were washed in PBS-T and PBS and dried before revelation using c400 (Azure Biosystems). Quantification was done using Image Lab (Bio-Rad) software. Signals for phospho-Thr34 and phospho-Thr75 were normalized by the signal detected for total DARPP-32.
Statistical analysis.
All data are expressed as mean ± SEM, and the choice of the statistical tests was guided by the Shapiro–Wilk's W normality test. All tests were performed using the Prism 6 software package (GraphPad). Two-tailed Student's t test for independent samples for comparison of two groups (usually genotype); and two-way ANOVA was used for multifactorial comparison, provided that they passed the normality test. Following significant analyses of variance, multiple post hoc comparisons were performed using the Tukey's or Sidak's post hoc tests when appropriate. When not normal, the data were evaluated using the Mann–Whitney Test. Survival curves were analyzed by the Mantel–Cox test. The accepted level of significance for the tests was p ≤ 0.05.
Results
GPRIN3 mRNA is unevenly expressed in dMSNs and iMSNs
By cell sorting of iMSNS and dMSNs as described previously (Ena et al., 2013), we identified GPRIN3 in both MSN populations but more expressed in iMSNs. The expression of GPRIN3 in both MSN populations was investigated by inducing unilateral specific ablation of iMSNs or dMSNs injecting diphtheria toxin (DT) in the right striatum of Adora2a-Cre+/− iDTR+/− or D1-Cre+/− iDTR+/− mice (Durieux et al., 2009, 2012), respectively, followed by in situ hybridization. Enkephalin and substance P signals were also quantified to control iMSN- or dMSN-specific ablations. We found 42% decrease in GPRIN3 mRNA level and 50% decrease in GPRIN3 mRNA (Fig. 1B,C) (one-way ANOVA, F(3,18) = 4.68, p < 0.05; n = 2 of 8 mice; 3 of 12 mice), respectively.

GPRIN3 is expressed in both MSN subpopulations and is enriched in iMSNs. A, Strategy for generation of GPRIN3−/− strain using CRISPR/Cas9 (top) and comparison of GPRIN3+/+ and GPRIN3−/− sequences (bottom). GPRIN3−/− strain presents an insertion of 1 bp, creating an early frameshift and a premature STOP codon in GPRIN3 coding sequence. Amino acid residues and their position are given below the sequence. B, C, Unilateral injection of DT in the right striatum of Adora2a-Cre+/− iDTR+/− (B) or D1-Cre+/− iDTR+/− (C) mice allowed specific ablation of iMSNs or dMSNs, respectively, as a significant decrease in in situ hybridization signals detected for enkephalin (Enk) and substance P (SP) was observed. In both cases, specific ablation of one MSN population also resulted in significant decrease in GPRIN3 signal, showing that GPRIN3 is expressed in both MSN populations (n = 2 or 3). *p < 0.05 (two-way ANOVA followed by Sidak's test). D, Example of iMSN or dMSN sorting. Sorting from tdTomato+/− (negative control, first row), Adora2a-Cre+/− tdTomato+/+ (second row), and D1-Cre+/− tdTomato+/+ (third row). After discarding cell debris (first column), dead cells were discarded on base of DAPI staining (second column). Among DAPI cells, tdTomato+ cells were sorted (third column). E, After cell sorting, GPRIN3 mRNA level was assessed by qRT-PCR, showing higher expression of GPRIN3 mRNA in iMSNs than in dMSNs (n = 2). *p < 0.05 (unilateral t test). F, Among the brain structures analyzed, the striatum exhibited the highest GPRIN3 mRNA level, as shown by qRT-PCR (n = 4 or 5). ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc test). Error bar indicates SEM.
While it confirmed that GPRIN3 is expressed in both MSN populations, this semiquantitative approach does not provide direct and accurate quantification of GPRIN3 mRNA in each MSN population. Using FACS and subsequent qRT-PCR, we then purified iMSNs and dMSNs from Adora2a-Cre+/− tdTomato+/+ or D1-Cre+/− tdTomato+/+ mice, respectively, and quantified the relative GPRIN3 mRNA level between both MSN populations. This confirmed the preferential expression of GPRIN3 mRNA in iMSNs (almost twofold) (Fig. 1D,E).
Still using qRT-PCR, we compared GPRIN3 mRNA levels in the cortex, striatum, hippocampus, and cerebellum. This experiment indicated that the highest level of GPRIN3 mRNA is detected in the striatum (Fig. 1F; one-way ANOVA, F3,14 = 16.34, p < 0.0001; n = 4 or 5).
Inactivation of GPRIN3 in mice using CRISPR/Cas9
GPRIN3 coding sequence is included in the second exon of GPRIN3; therefore, our initial strategy aimed to induce the deletion of the main part of exon-2 using two sgRNAs (Fig. 1A). Similar strategies have previously been used to generate large genetic deletions (Brandl et al., 2015; Zhang et al., 2015). Comicroinjection of sgRNA-a, sgRNA-b, and Cas9 protein in one-cell zygote (from C57BL6 mice) was performed, and the offspring was analyzed by PCR. Although the combination of sgRNA-a and sgRNA-b was able to induce the deletion of the exon-2 of GPRIN3 in Neuro2A cells (data not shown), the offspring did not exhibit the same deletion. PCR amplification analysis and sequencing of each sgRNA target site highlighted mutations in 14 (35.9%,) pups at sgRNA-a and in 1 (2.6%) pup at sgRNA-b targeting sites, suggesting that sgRNA-b is less efficient in vivo than in vitro. Nevertheless, indels at sgRNA-a target site could be adequate to KO the GPRIN3 gene if sufficient in the induction of a frameshift in the coding sequence and/or a premature STOP codon. Founder 11 (male) was bred with C57BL6 females, and the offspring presenting the 11-c mutation were selected because it generates an early frameshift in GPRIN3 coding sequence and a premature STOP codon (Fig. 1A). These animals were backcrossed on C57BL6 background for two generations, and then homozygotes for GPRIN3 11-c mutation were generated. Homozygous mice with 11-c mutation will be referred as GPRIN3 KO (GPRIN3−/−) mice.
We validated the efficiency of gene inactivation in these mice by assessing the loss of GPRIN3 protein by mass spectrometry analysis. This permitted to detect GPRIN3 by microsequencing of 9 distinct peptides in 7 adjacent gel bands stretching from ∼90 to 110 kDa after SDS-PAGE on microdissected striatum tissue from GPRIN3+/+ mouse, whereas no GPRIN3 peptides were detected on microdissected striatum tissue from GPRIN3−/− animal (Table 1). It is noteworthy that peptides corresponding to GPRIN1 protein (∼110 kDa) were equally detected in wild type (WT) and KO mice.
List of the GPRIN3 peptide sequences detected during the mass spectrometry analysisa
Striatal neurons lacking GPRIN3 show increased dendritic arborization and decreased neuronal excitability
Previous studies reported GPRIN1 as a modulator of neurite outgrowth (Iida and Kozasa, 2004; Masuho et al., 2008). To clarify the possible role exerted by GPRIN3 on the striatal function, we addressed the effects of the genetic inactivation of GPRIN3 on the striatal volume by performing stereological analysis. We found that GPRIN3 KO did not alter striatal volume (Fig. 2A) (Mann–Whitney Test, p > 0.9999; n = 2). Nevertheless, we also addressed the effect of GPRIN3 KO on MSNs morphology by performing the 3D reconstruction of striatal dendritic arbors, followed by Sholl analysis as previously described (Lambot et al., 2016). MSNs from GPRIN3−/− mice (Fig. 2C) present increased dendritic arborization (two-way ANOVA, F(1,294) = 70.03, p < 0.0001; n = 6–10) (Fig. 2D) compared with MSNs from GPRIN3+/+ mice (Fig. 2B). Increased number of dendritic segments was observed in neurons from GPRIN3−/− mice compared with controls (two-way ANOVA, F(1,70) = 5.495, p = 0.0219; n = 6–10) (Fig. 2E), whereas the dendritic length was similar in both groups (two-way ANOVA, F(1,70) = 0.567, p = 0.454; n = 6–10) (Fig. 2F), suggesting that the increase in dendritic arborization in GPRIN3 KOs is the consequence of the increased dendritic branching. Moreover, our whole-cell voltage-clamp recordings revealed increased cell capacitance in MSNs from GPRIN3−/− mice (Fig. 2G) (Mann–Whitney Test, p = 0.0048; n = 13–16), without alterations in membrane resistance (Fig. 2H) (Mann–Whitney Test, p = 0.5890; n = 13–16), indicating that GPRIN3-deficient MSNs had a larger surface area, hence confirming our morphological measurements of dendritic arborization.

Striatal neurons lacking GPRIN3 show increased dendritic arborization and decreased neuronal excitability. A, Similar striatal volume in GPRIN3 KO mice (n = 2, Mann–Whitney Test, p > 0.9999). B, C, Representative maximal intensity projections of GPRIN3+/+ (B) and GPRIN3−/− (C) MSN. Scale bar, 50 μm. D, GPRIN3−/− MSNs exhibited an increase in dendritic arborization, as shown by 3D Sholl analysis (n = 6–10, two-way ANOVA, p < 0.0001). E, F, The number of dendrites in function of branch order is increased in MSNs lacking GPRIN3 (E, n = 6–10, two-way ANOVA, p < 0.05), whereas the dendritic length remained unchanged (F, n = 6–10, two-way ANOVA, p > 0.05). G–I, According to whole-cell current-clamp recordings, GPRIN3−/− MSNs showed increased cell capacitance (G, n = 13–16, Mann–Whitney Test, p < 0.01), unchanged membrane resistance (H, n = 14–18, Mann–Whitney Test, p > 0.05), and increased RMP (I, n = 13–16, Mann–Whitney Test, p < 0.01). J, Voltage-clamp recordings showed that the rheobase is not modified by GPRIN3 KO (n = 13–16, Mann–Whitney Test, p > 0.05). K, Representation of AP frequency in function of injected current highlighted a decrease in cell excitability in GPRIN3−/− MSNs (n = 13–16, p < 0.0001). *p < 0.05, **p < 0.01, ***p < 0.0001. Error bar indicates SEM.
Additionally our electrophysiological data indicated increase in the RMP in MSNs from GPRIN3−/− mice (Fig. 2I) (Mann–Whitney Test, p = 0.0330; n = 13–16) but similar rheobase (Fig. 2J) (Mann–Whitney Test, test, p = 0.2008; n = 13–16). GPRIN3 gene inactivation results in decreased MSN excitability (as shown in the excitability curves; Fig. 2K) (two-way ANOVA, F(1,432) = 16.43, p < 0.0001; n = 13–16). These changes in neuronal morphology and excitability may indicate striatal reorganization with possible impact on behavior.
Loss of GPRIN3 increases motivation and decreases response to cocaine, but does not change locomotion, motor learning, anxiety, anhedonia, and helplessness
We have previously demonstrated the involvement of MSN populations in locomotor, exploratory, and operant behaviors (Durieux et al., 2009, 2012; Laurent et al., 2017). Therefore, the impact of GPRIN3 KO was first evaluated on behaviors, such as locomotion (open field test), motor coordination and motor learning (accelerated rotarod), and also anxiety-like (elevated plus maze, light-dark box, and marble-burying tests) and depressive-like behaviors (tail suspension and sucrose preference), given that all these behaviors are related to the striatum. Our data indicate no changes in those tests (results summarized in Table 2).
Locomotor and emotional tests in GPRIN3−/− and control mice
As the striatum is also involved in reward-directed behavior and motivation (for review, see Volkow et al., 2017); we evaluated how the loss of GPRIN3 would impact on operant behavior. Although both groups increased their response rate along time (Fig. 3A) (two-way ANOVA, F(time)15,225 = 88.94, p < 0.0001; n = 8–10), GPRIN3−/− mice tend to press the lever for reward more frequently than the control group during the instrumental learning phase (Fig. 3A) (two-way ANOVA, F(genotype)15,225 = 3.424, p = 0.0841; n = 8–10). Afterward, mice were tested in a progressive ratio task. We found that GPRIN3−/− mice reached a higher breakpoint than the control group (Fig. 3B) (Mann–Whitney Test, p = 0.0250; n = 8–10), indicating a higher motivation for reward. GPRIN3−/− mice also responded for longer periods (Fig. 3C) (Mantel–Cox test, p = 0.0037; n = 8–10). Goal-directed behaviors are sensitive to changes in reward value. This was assessed in GPRIN3−/− and WT mice by comparing their performance before and after the reestablishment of ad libitum food access. We demonstrated that both groups decreased similarly their performance after reward devaluation (Fig. 3D) (two-way ANOVA, F(time)1,16 = 114.1, p < 0.0001; n = 8–10).

GPRIN3 KO mice exhibit increase in motivation and decrease in cocaine-induced hyperlocomotion. A, GPRIN3 KO mice were trained under an instrumental protocol consisting of a CRF phase followed by RR phases with increasing press requirement. Globally, GPRIN3 deficiency induced a tendency to facilitate instrumental learning (n = 8–10, two-way ANOVA, p = 0.0841). B, C, The instrumental learning was followed by a progressive ratio task, in which GPRIN3 KO mice raised a higher breakpoint (B, n = 8–10, Mann–Whitney Test, p < 0.05) and responded for longer periods (C, n = 8–10, Mantel–Cox, p < 0.01). D, Devaluation of the reward was then achieved by allowing ad libitum food access overnight. Comparison of performance before and after the reestablishment of food access under a RR25 protocol showed no difference in devaluation process between experimental and control groups (n = 8–10, two-way ANOVA, pgenotype > 0.05). E, Cocaine effects on the locomotor activity indicated similar sensitivity to cocaine in lower doses (1 and 5 mg/kg) but decreased effects in the high (20 mg/kg) and sensitization in GPRIN3 KO mice (n = 6 or 7, F(genotype), p = 0.0022). F, Saline-normalized locomotor effect of cocaine on locomotor activity of control and GPRIN3 KO mice (n = 6 or 7, t test p = 0.03). *p < 0.05, **p < 0.01, ***p < 0.001. Error bar indicates SEM.
Considering the observed modifications in reward, we decided to check for possible changes in the impact of drugs of abuse induced by the invalidation of GPRIN3. We next accessed the impact of GPRIN3 absence on cocaine-induced locomotor activity. We did not observe differences between the groups following administration of subthreshold doses of cocaine (1 and 5 mg/kg). Mice were then submitted to a cocaine-sensitization procedure, where a time response was observed for both genotypes (Fig. 3E) (two-way ANOVA, F(time)4,44 = 3.055, p = 0.0263; n = 6 or 7). Still, as can be seen in Figure 3E, the ability of cocaine to increase the locomotor response is significantly reduced in GPRIN3−/− mice (two-way ANOVA, F(genotype)1,11 = 15.73, p = 0.0022; n = 6 or 7). Thus, GPRIN3 is critically involved in motivation and sensitization to drugs of abuse, but not in gross locomotor and emotional behavior.
GPRIN3 KO modifies the phosphorylation pattern of DARPP-32
DARPP-32 plays a crucial role in the integration of dopaminergic signaling in MSNs (Svenningsson et al., 2004). Moreover, its activity is tightly regulated by its phosphorylation pattern. We therefore investigated whether this pattern is affected by the loss of GPRIN3 by performing Western blot analyses aiming the phospho-Thr34, phospho-Thr75, and total DARPP-32 in dorsal and ventral striatum (Fig. 4A). This highlighted a decrease in phospho-Thr34 DARPP-32 in dorsal and ventral striatum samples from GPRIN3−/− mice (Fig. 4B) (unilateral t test, pdorsal = 0.0279 and pventral = 0.0196; n = 5), whereas the phospho-Thr75 DARPP-32 (Fig. 4C) (unilateral t test, pdorsal = 0.8718 and pventral = 0.6890; n = 5) and total DARPP-32 (Fig. 4D) (unilateral t test, pdorsal = 0.0793 and pventral = 0.9853; n = 5) remained unchanged.

Modification of DARPP-32 phosphorylation pattern in GPRIN3 KO mice. A, Protein lysates from dorsal and ventral striata were used in Western blot experiments aiming phospho-Thr34, phospho-Thr75, and total DARPP-32 forms. Similar experiments were also conducted after repeated administrations of cocaine. B, Phospho-Thr34 DARPP-32 level was decreased in dorsal and ventral striatum of GPRIN3 KO mice (n = 5, unilateral t test, pdorsal < 0.05, pventral < 0.05). Repeated injections of cocaine also induced a significant decrease in phospho-Thr34 DARPP-32 level in dorsal and ventral striata of control mice (n = 5, unilateral t test, pdorsal < 0.01, pventral < 0.05), while the same effect was observed only in ventral striatum of GPRIN3 KO mice (n = 5, unilateral t test, pdorsal > 0.05, pventral = 0.0556). C, D, GPRIN3 KO did not induce significant modifications in phospho-Thr75 and total DARPP-32 levels under these conditions. *p < 0.05, **p < 0.01. Error bar indicates SEM.
Cocaine administration has been shown to modify the pattern of phosphorylation of DARPP-32 (Svenningsson et al., 2005), so we also collected dorsal and ventral striata after repeated administration of cocaine and performed similar experiments. Chronic cocaine treatment decreased the expression of phospho-Thr34 DARPP-32 (Fig. 4B) (unilateral t test, pdorsal = 0.0035 and pventral = 0.0357; n = 5) in samples from GPRIN3+/+ mice, whereas no effect was found in dorsal striatum of GPRIN3−/− mice and a trending similar effect was observed in ventral striatum (Fig. 4B) (Mann–Whitney Test, pdorsal = 0.3095 and pventral = 0.0556; n = 5). As described previously (Svenningsson et al., 2005), no significant effect of chronic administration of cocaine on phospho-Thr75 (Fig. 4C) (GPRIN3+/+: unilateral t test, pdorsal = 0.4524 and pventral = 0.3719; n = 5) or total DARPP-32 (Fig. 4D) (GPRIN3+/+: unilateral t test, pdorsal = 0.2694 and pventral = 0.5900; n = 5) was observed in WT mice, and this was similarly observed in GPRIN3−/− mice (Fig. 4C; Mann–Whitney Test, pdorsal = 0.5476 and pventral = 0.4206; n = 5) (Fig. 4D; Mann–Whitney Test, pdorsal > 0.9999 and pventral = 0.8413; n = 5). These observations add the notion that the inactivation of GPRIN3 results in changes in the acute effects of cocaine (through DARPP32-Thr34) and not on its chronic effects (Thr75).
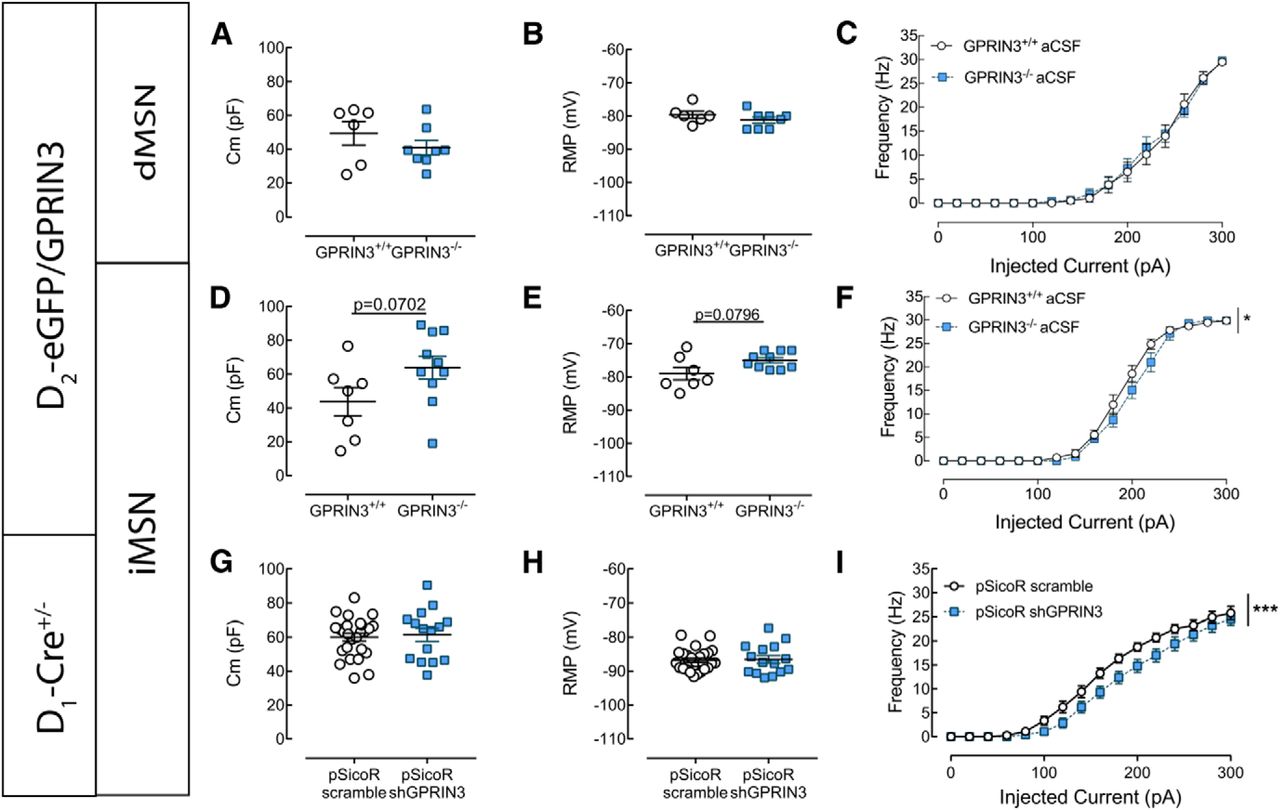
Loss of GPRIN3 differentially affects the morphology of dMSNs and iMSNs and electrophysiological properties of iMSNs
Next, we investigated whether iMSNs and dMSNs are equally affected by GPRIN3 deficit. D2-eGFP+/−GPRIN+/+ and D2-eGFP+/−GPRIN−/− double-transgenic mice were generated, in which eGFP allowed us to discriminate between iMSNs and dMSNs.
Inactivation of GPRIN3 induced the reorganization of the dendritic tree in dMSNs (e.g., in Fig. 5A,B), as shown by the Sholl analysis (Fig. 5C) (two-way ANOVA, F(genotype)1,147 = 22.82, p < 0.0001; n = 5 or 6). The modifications are discrete since we could not detect decrease in the length of dendritic segments (Fig. 5D) (two-way ANOVA, F(genotype)1,35 = 0.7913 p = 0.3798; n = 5 or 6) or in their number (Fig. 5E) (two-way ANOVA, F(genotype)1,35 = 0.00008.839, p = 0.9926; n = 5 or 6). In iMSNs, we found that the inactivation of GPRIN3 results in increased dendritic arborization compared with controls (e.g., in Fig. 5F,G) (quantified by the Sholl analysis in Fig. 5H) (two-way ANOVA, F(genotype)1,210 = 16.8, p < 0.001; n = 4–8), accompanied by a tendency to increase the number of dendritic segments (Fig. 5J) (two-way ANOVA, F(genotype)1,49 = 3.102, p = 0.0844; n = 4–8,) without modifying their length (Fig. 5I) (two-way ANOVA, F(genotype)1,49 = 1.521, p = 0.2233; n = 4–8).

Modification of morphological properties of GPRIN3−/− dMSNs and iMSNs. A, B, Representative maximal intensity projections of GPRIN3+/+ (A) and GPRIN3−/− (B) dMSNs. Scale bar, 50 μm. C–E, GPRIN3−/− dMSNs exhibited modification of dendritic arborization, as shown by 3D Sholl analysis (C, n = 5 or 6, two-way ANOVA, p < 0.0001), although no significant modification in the dendritic length (D, n = 5 or 6, two-way ANOVA, p > 0.05) or in the number (E, n = 5 or 6, two-way ANOVA, p > 0.05) of dendritic segments was observed. F, G, Representative maximal intensity projections of GPRIN3+/+ (F) and GPRIN3−/− (G) iMSNs. Scale bar, 50 μm. H, GPRIN3−/− iMSNs exhibited an increase in dendritic arborization, as shown by 3D Sholl analysis (n = 4–8, two-way ANOVA, p < 0.0001). I, J, The dendritic length remained unchanged in iMSNs lacking GPRIN3 (I, n = 4–8, two-way ANOVA, p > 0.05), whereas the number of dendrites in function of branch order tended to increase (E, n = 4–8, two-way ANOVA, p = 0.0844). ***p < 0.0001. Error bar indicates SEM.
Electrophysiological recordings from the same animals indicated lack of modifications in cell capacitance (Fig. 6A) (Mann–Whitney Test, p = 0.5728; n = 6–8) or in RMP (Fig. 6B) (Mann–Whitney Test, p = 0.2987; n = 6–8) in dMSNs from GPRIN3−/− mice. Cell excitability remained unchanged in GPRIN3−/− dMSNs (Fig. 6C) (two-way ANOVA, F(genotype)1,192 = 0.1221, p = 0.5839; n = 6–8). Interestingly, GPRIN3 inactivation in iMSNs resulted in a tendency to increase the cell capacitance (Fig. 6D) (Mann–Whitney Test, p = 0.0702; n = 7–10) and higher RMP (Fig. 6E) (Mann–Whitney Test, p = 0.0796; n = 7–10) and overall decreased neuronal excitability (Fig. 6F) (two-way ANOVA, F(genotype)1,256 = 4.389, p = 0.0236; n = 7–10). Together, these results suggest that the loss of GPRIN3 affect the electrophysiological properties of iMSNs.

Modification of electrophysiological properties of GPRIN3−/− MSNs. A–F, Whole-cell patch-clamp recordings were performed using D2-eGFP+/− GPRIN3+/+ and D2-eGFP+/− GPRIN3−/−, allowing the discrimination of iMSNs and dMSNs. dMSNs lacking GPRIN3 exhibited unchanged membrane capacitance (A, n = 6–8, Mann–Whitney Test, p > 0.05) and RMP (B, 6–8, Mann–Whitney Test, p > 0.05). In addition, representation of AP frequency in function of injected current (C) showed no modification of cell excitability in GPRIN3−/− dMSNs (n = 6–8, two-way ANOVA, p > 0.05). On the other hand, in iMSNs of GPRIN3 KO, we observed a tendency to higher membrane capacitance (D, n = 7–10, Mann–Whitney Test, p = 0.0702) and RMP (E, n = 7–10, Mann–Whitney Test, p = 0.0796) and induced a decrease in cell excitability (F, n = 7–10, two-way ANOVA, p < 0.05). G–I, Whole-cell patch-clamp recordings were performed using D1-Cre+/− mice 1 month after intrastriatal injection of pSicoR-based lentivirus expressing a scramble shRNA (scramble, control group) or a GPRIN3-targeting shRNA (shGPRIN3). eGFP+ cells (i.e., iMSNs) were used in these experiments. Repression of GPRIN3 in iMSNs did not alter membrane capacitance (G, n = 25–30, unpaired t test, p > 0.05) nor RMP (H, n = 25–30, unpaired t test, p > 0.05). Nevertheless, expression of shGPRIN3 in iMSNs decreased cell excitability, as shown by the representation of AP frequency in function of injected current (I, n = 25–30). p < 0.0001 (two-way ANOVA). *p < 0.05, ***p < 0.0001. Error bar indicates SEM.
Specific iMSN knockdown of GPRIN3 in adult mice
To discriminate developmental and nondevelopmental functions of GPRIN3, we generated an iMSN-specific knockdown of GPRIN3 using pSicoR-based lentiviruses (Ventura et al., 2004; Ena et al., 2013). First, we performed the validation of the viral injections. Our results indicate a reduction of 45% in the GPRIN3 mRNA levels in the injected striatum following pSicoR shGPRIN3 (data not shown). Then we injected those viruses in the striatum of adult D1-Cre mice. This resulted in simultaneous expression of both eGFP and GPRIN3-targeting shRNA in Cre recombinase negative neurons (i.e., iMSNs), while the expression of Cre recombinase in D1 MSNs (i.e., dMSNs) induced the excision of eGFP and shRNA. eGFP+ cells that were not MSNs were discarded from our electrophysiological recordings. We found that GPRIN3 knockdown in iMSNs did not alter the membrane capacitance (Fig. 6G) (unpaired t test, p = 0.7933; n = 25–30) or the RMP (Fig. 6H) (unpaired t test, p = 0.4971; n = 25–30). Nevertheless, the cell excitability was decreased by GPRIN3 repression (Fig. 6I) (two-way ANOVA, F(genotype)1,838 = 32.7, p < 0.0001; n = 25–30), as it was in iMSNs from KO mice, indicating that the lack of GPRIN3 tonically modifies the firing pattern of iMSNs.
Lack of GPRIN3 modifies the effects of the activation of D2R on iMSN excitability
To address a potential mechanism to explain the prominent role of GPRIN3 in iMSNs, we hypothesize that the lack of GPRIN3 would disrupt D2R function. Thus, we patched eGFP+ neurons of D2-eGFP+/− GPRIN3+/+ and D2-eGFP+/− GPRIN3−/− mice in the absence or presence of quinpirole (20 μm), a D2R agonist. Passive electrophysiological properties were maintained in striatal slices from GPRIN3+/+ mice. No changes in capacitance (Fig. 7A) (Mann–Whitney Test, p = 0.5338; n = 6 or 7), RMP (Fig. 7B) (Mann–Whitney Test, p = 0.8572; n = 6 or 7) were observed. As expected (Perez et al., 2006; Janssen et al., 2009), in D2-eGFP+/− GPRIN3+/+ iMSNs, the activation of the D2R induced a decrease in cell excitability (Fig. 7C) (two-way ANOVA, F(1,176) = 12.81, p = 0.0005; n = 6 or 7). Conversely, quinpirole effects on D2-eGFP+/− GPRIN3−/− iMSNs did not modify cell excitability (Fig. 7F) (two-way ANOVA, F(1,288) = 1.596, p = 0.2051; n = 6 or 7) or membrane capacitance (Fig. 7D) (Mann–Whitney Test, p = 0.8534; n = 10 per group), but decreased RMP (Fig. 7E) (Mann–Whitney Test, p = 0.0154; n = 10 per group).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
GPRIN3 inactivation and specific iMSN knockdown modify the effects of D2R activation on cell excitability in iMSNs. A–F, Effect of quinpirole (20 μm) in the presence or absence of GPRIN3 was assessed by whole-cell patch-clamp recordings using D2-eGFP+/− GPRIN3+/+ and D2-eGFP+/− GPRIN3−/−. In the control group, quinpirole-induced activation of D2R had no effect on membrane capacitance (A, n = 6 or 7, Mann–Whitney Test, p > 0.05) nor on RMP (B, n = 6 or 7, Mann–Whitney Test, p > 0.05), although it induced a decrease in cell excitability, as shown by the representation of AP frequency in function of injected current (C, n = 6 or 7, two-way ANOVA, p < 0.001). In the opposite, D2-eGFP+/− GPRIN3−/− neurons exhibited no change in membrane capacitance (D, n = 10 per group, Mann–Whitney Test, p > 0.05), decrease in RMP (E, n = 10 per group, Mann–Whitney Test, p < 0.01), and no modification in cell excitability (F, n = 10 per group, two-way ANOVA, p > 0.05). G–L, To assess effects of GPRIN3 repression on quinpirole-induced D2R activation, similar experiments were performed using D1-Cre+/− mice 1 month after intrastriatal injection of pSicoR-based lentivirus expressing a scramble shRNA (scramble, control group) or a GPRIN3-targeting shRNA (shGPRIN3). Only eGFP+ cells (i.e., iMSNs) were used in these experiments. In the control group, quinpirole application (20 μm) did not change RMP (H, n = 12, Wilcoxon test, p > 0.05) but induced a decrease in cell excitability (I, n = 12, two-way ANOVA, p < 0.0001). On the other hand, quinpirole application did not modify RMP in iMSNs expressing shGPRIN3 (K, n = 8, p > 0.05) but induced an expected increase in cell excitability (L, n = 8, two-way ANOVA, p < 0.0001). *p < 0.05, ***p < 0.0001. Error bar indicates SEM.
Similar experiments were also performed in the specific iMSNs GPRIN3 knockdown mice (see above). We showed that in the scramble shRNA-expressing iMSNs, quinpirole does not alter RMP (Fig. 7H) (Wilcoxon test, p = 0.1455; n = 12 per group), whereas it induced an expected decrease in excitability (Fig. 7I) (two-way ANOVA, F(1,221) = 19.08, p < 0.0001, n = 12 per group). Remarkably, in iMSNs expressing the GPRIN3 shRNA, quinpirole application did not result in decrease in cell excitability as expected but on the contrary in an increase in this parameter (Fig. 7L) (two-way ANOVA, F(1,107) = 28.23, p < 0.0001; n = 8 per group), although no modification in RMP (Fig. 7K) (Wilcoxon test, p = 0.7422; n = 8 per group) was found, showing that GPRIN3 knockdown modifies cellular response to the activation of the D2R. Our double approach of this question and the convergence of the results shows the importance of GPRIN3 for the D2R signaling in iMSNs that is not related to the development.
Discussion
The present study provides first evidence that GPRIN3 partners with D2R at the GPCR signaling level in the striatum. The expression of GPRIN3 is significantly higher in the striatum compared with other brain areas, such as the cerebellum, cortex, and the hippocampus, and we highlight its paramount role in the control of MSNs morphological and electrophysiological properties and of the dopaminergic signaling in the striatum, specifically through the control of D2R function in iMSNs.
Considering the role of GPRIN1 on neural development (Masuho et al., 2008) and of GPRIN3 on the desensitization of Gαi/o-coupled receptors, we first addressed whether the KO of GPRIN3 could change the total volume of the striatum. Our stereological approach indicated similar striatal volume of GPRIN3+/+ and GPRIN3−/− mice, suggesting that GPRIN3 does not play a role in the gross striatal development.
The expression of GPRIN1 and GPRIN2 in cell cultures, conjunctly with their subcellular localization, is able to induce the growth of neurites, depending on the presence and function of Gαi/o (Chen et al., 1999; Iida and Kozasa, 2004). We here observed that the KO of GPRIN3 is able, in iMSNs, to increase the number of intersections and dendrites, without remarkable changes in their length. Together with the increase in the cell capacitance (proportional to the cell-membrane surface) in GPRIN3 KO cells, this suggests a qualitatively differential role for GPRIN3 compared with the other members of the GPRIN family, where it regulates the neuronal ramification other than the cell size. Indeed, GPRIN3 seems to negatively regulate neuronal dendritic arborization.
The differences found between dMSNs and iMSNs in the morphological changes caused by the inactivation of GPRIN3 are fundamental for the understanding of our electrophysiological and behavioral data. Distal parts of dendritic branching are known to be the points of convergence between dopamine and glutamate inputs in MSNs (Smith and Bolam, 1990) and primary targets of cocaine, which itself promotes increase in distal branching in the NAc of mice (Robinson and Kolb, 1999). The selective increase in distal branching in iMSNs after the inactivation of GPRIN3 suggests a “presensitization” process, able to change dynamically the targets of cocaine and therefore altering its effects (Pierce and Kalivas, 1997), as a lower hyperlocomotor effect can be observed in GPRIN3 KO mice. This is heralded by the decrease in the phosphorylation of Thr34 residue of DARPP-32 after repeated cocaine administration, meaning lower effects of cocaine (Svenningsson et al., 2005).
The increase in excitability properties of MSNs could be related to differential effects of cocaine (Larson et al., 2015). Interestingly, the KO of GPRIN3 increased the RMP of the MSNs but conversely shifted the excitability curve to the right, indicating decreased excitability. Phenotypically, a similar response, which might be connected to our results, can be achieved by the blockade of D2Rs with raclopride, whereas opposite effects can be mimicked by the overexpression of D2R (Cazorla et al., 2012; Chan et al., 2012), both related to decreased and increased responses to cocaine, respectively.
Indeed, the decrease in excitability promoted by quinpirole (a D2R agonist) described previously (Cazorla et al., 2012; Lalchandani et al., 2013) was blunted in D2-eGFP/GPRIN3 KO cells and reversed by the viral inactivation of GPRIN3 (with pSicoR shGPRIN3), indicating profound changes in D2R function, which are in line with the purported role of the GPRIN family in mediating the Gαi/o signaling. However, on all the results discussed above, it cannot be ruled out the possibility that our observations are caused by the loss of control over the D2R desensitization by inactivating GPRIN3 as previously described (Mototani et al., 2018).
The above results and the indication of a hierarchically prevalent role of GPRIN3 in iMSNs led us to anticipate a functional interaction between GPRIN3 and the D2R. Motivation and coding for reward take place in the striatum (Maia and Frank, 2011). It is known that the selective upregulation of D2R in the striatum enhances the willingness to work for food (Gallo et al., 2018). The differential role of iMSNs and dMSNs on this subject indicates that, whereas iMSNs code for motivation and punishment, dMSNs relate to reward (Kravitz et al., 2012). As discussed by Salamone et al. (2007), the role of dopamine in motivation (specifically in the NAc) is not related to the modification of the hedonic value of the reward but controlling the effort-related processes. Similarly, we noticed an increase in motivation, where the D2R is preponderant, when the GPRIN3 is inactivated, without changes in sucrose consumption (where the hedonic component is dominant) and more likely D1R-dependent.
In view of our results, a new layer to the already complex physiology of the striatum is added with normal functional partners of dopamine and of the D2R. For instance, the presence of receptors heteromers and fundamentally their dynamics, such as the D2R-A2AR (A2AR; adenosine A2A receptor), may gain new prospects (Ferré et al., 2008) since their antagonistic nature may be orchestrated by GPRIN3. Moreover, through the control of D2R fine-tuning, GPRIN3 might influence the striatal function of the glutamatergic (Escobar et al., 2015), GABAergic (Muñoz-Arenas et al., 2015), endocannabinoid (Pan et al., 2008), and adenosinergic (Higley and Sabatini, 2010) systems.
In keeping with the actions of cocaine on the dopaminergic system, it is known that the correct D2R signaling in iMSNs is essential for multiple effects of cocaine. The observation of a decreased cocaine-induced locomotor activation in GPRIN3 KO mice might be related with the incorrect signaling of D2R leading to higher activation of lateral inhibition pathways to short-projecting dMSNs, resulting in reduced locomotor response (Dobbs et al., 2016). This hypothesis gains more significance noticing that, in the absence of GPRIN3, the DARPP-32 phosphorylation pattern (Thr34) is decreased compared with the control group, indicating a downstream change in the D2R signaling properties. Since MSNs arborization is positively regulated by dopamine (Meredith et al., 1995), the higher neuronal arborization observed in GPRIN3 KO mice suggests more dopamine inputs compared with control mice. These suggest a morphological state resembling chronic exposure to cocaine (MacAskill et al., 2014; Spencer et al., 2017; Dos Santos et al., 2018), corroborating our hypothesis that the lack of GPRIN3 induces a “presensitization process.”
Together, our results indicate a prevalent role of GPRIN3 in dopamine signaling in the striatum and more precisely in iMSNs by controlling cellular arborization, maintenance of excitability, and D2R-dependent behaviors. It is still to be determined how the manipulation of GPRIN3 would impact on pathological situations, but the notion that it interferes with D2R signaling paves new ways of thinking about striatal-related dysfunctions and treatments. For instance, diseases in need of dopamine replacement, such as Parkinson's disease, or those where the decrease of dopaminergic tonus is essential (e.g., schizophrenia), could benefit from a fine-tuning in the D2R signaling exerted by GPRIN3.
Footnotes
- Received September 22, 2018.
- Revision received July 16, 2019.
- Accepted July 19, 2019.
This work was supported by FRS-FNRS (Belgium), Fondation Simone et Pierre Clerdent, Action de Recherche Concertée (FWB), and PAI 7/10 (Federal Science Policy). D.K. was supported by FRS-FNRS Aspirant PhD fellowship. D.R. is a BELSPO Fellowship recipient. J.-F.D.B. was supported by FRS-FNRS Aspirant PhD fellowship and Van Buren fund. A.d.K.d. is a Research Director of the FRS-FNRS and an investigator of WELBIO. D.C. is a Senior Research Associate of the FRS-FNRS. We thank Souad Laghmari and Virginie Imbaut for expert technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Alban de Kerchove d'Exaerde at adekerch{at}ulb.ac.be or Serge N. Schiffmann at sschiffm{at}ulb.ac.be.
- Copyright © 2019 the authors