

Abstract
Tissue acidosis and inflammatory mediators play critical roles in inflammatory pain. Extracellular acidosis activates acid-sensing ion channels (ASICs), which have emerged as key sensors for extracellular protons in the central and peripheral nervous systems and play key roles in pain sensation and transmission. Additionally, inflammatory mediators, such as serotonin (5-HT), are known to enhance pain sensation. However, functional interactions among protons, inflammatory mediators, and ASICs in pain sensation are poorly understood. In the present study, we show that 5-HT, a classical pro-inflammatory mediator, specifically enhances the proton-evoked sustained, but not transient, currents mediated by homomeric ASIC3 channels and heteromeric ASIC3/1a and ASIC3/1b channels. Unexpectedly, the effect of 5-HT on ASIC3 channels does not involve activation of 5-HT receptors, but is mediated via a functional interaction between 5-HT and ASIC3 channels. We further show that the effect of 5-HT on ASIC3 channels depends on the newly identified nonproton ligand sensing domain. Finally, coapplication of 5-HT and acid significantly increased pain-related behaviors as assayed by the paw-licking test in mice, which was largely attenuated in ASIC3 knock-out mice, and inhibited by the nonselective ASIC inhibitor amiloride. Together, these data identify ASIC3 channels as an unexpected molecular target for acute actions of 5-HT in inflammatory pain sensation and reveal an important role of ASIC3 channels in regulating inflammatory pain via coincident detection of extracellular protons and inflammatory mediators.
Introduction
Acid-sensing ion channels (ASICs) are members of the voltage-insensitive, amiloride-sensitive epithelial sodium channel/degenerin family of cation channels, which have emerged as key sensors for extracellular protons in central and peripheral neurons. To date, functional cloning studies have revealed four genes that give rise to at least six ASIC isoforms (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, and ASIC4) (Lingueglia et al., 1997; Waldmann et al., 1997a,b; Chen et al., 1998; Gründer et al., 2000). In brain neurons, calcium-permeable ASIC1a channels are required for acid-evoked currents and they contribute critically to acidosis-induced neuronal death (Xiong et al., 2008; Sluka et al., 2009; Gründer and Chen, 2010; Wang and Xu, 2011). In peripheral neurons, ASIC3 channels are important for sensing inflammatory pain (Mamet et al., 2002, 2003; Deval et al., 2008; Walder et al., 2011).
Tissue injury and inflammation cause acidosis, which is sufficient to activate ASIC3 channels and can trigger pain sensation (Steen and Reeh, 1993; Reeh and Steen, 1996; Frey Law et al., 2008). Interestingly, activation of ASIC3 by protons generates biphasic inward currents: a large transient current followed by a small sustained current with little or no desensitization during continued presence of the low extracellular pH (Lingueglia et al., 1997; Waldmann et al., 1997b; Babinski et al., 1999; Benson et al., 2002). The sustained current is particularly enhanced when ASIC3 is activated by protons in the presence of neuropeptides such as FMRF-amide and related products (Askwith et al., 2000), a property thought to be essential for sensing prolonged acidosis under pathological conditions. Previous studies have suggested that the sustained currents of ASIC3 channels have significant physiological or pathophysiological relevance, such as pain perception (Dubé et al., 2005b; Yagi et al., 2006).
In addition to acidosis, the inflamed tissues are also enriched with other factors, collectively called inflammatory mediators. Together, these factors confer nociception as shown by the continued discharge of nocifensive nerve fibers when superfused with a solution that contained bradykinin (BK), serotonin (5-HT), histamine (His), and prostaglandin E2 (PGE2) at pH 6.1, which mimics the inflammatory condition (Steen et al., 1995; Strassman et al., 1996; Kress et al., 1997). Importantly, a combined stimulation by the inflammatory mediators, BK, 5-HT, nerve growth factor, and glia-derived neurotrophic factor, was shown to increase ASIC3 expression and activity (Mamet et al., 2002, 2003). These observations suggest the prominent synergism between inflammatory mediators and protons, as well as their convergence on ASIC3 channels. However, whether the inflammatory mediators act directly at ASIC3 channels and the molecular mechanism(s) underlying the possible synergistic effect between inflammatory mediators and acidosis on these channels remain to be established.
We report here that 5-HT enhances the sustained currents of ASIC3 channels in a manner that depends on the recently identified nonproton ligand sensing domain (Yu et al., 2010). Moreover, 5-HT strongly augmented acid-induced nocifensive behavior in wild-type (WT) but not the ASIC3 knock-out (KO) mice and the effect was blocked by the nonselective ASIC inhibitor amiloride. We suggest that the classical inflammatory mediator 5-HT contributes to inflammatory pain sensation via enhancing acid-induced sustained ASIC3 activation by stimulating the nonproton ligand sensing domain.
Materials and Methods
Cell culture and transfection.
The cDNAs used for heterologous expression of ASIC channels and 5-HT2C receptors were as follows: rat ASIC1a, GenBank ID: 140970991; rat ASIC1b, 3445467; rat ASIC2a, 77404414; rat ASIC3, 27465600; and human 5-HT2C receptors, 377520136. All constructs were expressed in Chinese hamster ovary (CHO) cells as described previously (Yu et al., 2010). In brief, CHO cells were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% O2 and passaged twice a week. Transient transfection of CHO cells was performed using HilyMax liposome transfection reagent (Dojindo Laboratories). CHO cells were maintained in F-12 Nutrient Mixture (added 1.176 g of NaHCO3/L medium) supplemented with 10% fetal bovine serum (FBS) and 1% gluta-MAXTM-1 (100×; Invitrogen). When more than one ASIC subunit was cotransfected, the ratio of ASIC3 subunit and another ASIC subunit was kept at 1:3. All plasmids used contained, in addition to the desired ASIC cDNA, the coding sequence for enhanced green fluorescent protein (EGFP) to aid identification of transfected cells. Electrophysiological measurements were performed 24–48 h after transfection.
Acute dorsal root ganglion (DRG) neuron preparation was performed as previously described (Dubé et al., 2005a). Briefly, lumbar (L4–L5) DRG were dissected from the vertebral column of WT and ASIC3 KO littermates (6–8 weeks old in C57BL/6 background), and then cleaned with ice-cold Hank's balance salt solution (HBSS). The dissected samples were incubated in Ca2+/Mg2+-free HBSS containing 0.3% collagenase B plus 0.05% trypsin (Sigma) in a 1:1 ratio for 20 min at 37°C. After washing with fresh HBSS, the ganglia were dissociated by triturating and isolated cells were plated onto laminin-coated 8 mm glass coverslips. The neurons were cultured in DMEM supplemented with 1% gluta-MAXTM-1, 2% B27, and 10% FBS (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2 and 95% O2. Whole-cell patch-clamp recordings were performed on individual DRG neurons 4–12 h after isolation.
Site-directed mutagenesis.
The cDNA of rat ASIC3 was subcloned into the pEGFPC3 vector (Promega). Mutations were made using the Quik-Change mutagenesis kit (Stratagene) according to the manufacturer's protocol (Sigma-Genosys). All mutant constructs were verified by DNA sequencing and the predicted amino acid sequences were determined by computer analysis.
Electrophysiology.
Extracellular solutions contained the following (in mm): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, pH 6.0–7.4, or 10 MES hydrate, pH < 6.0, and 10 glucose, adjusted to the final pH with Tris-base. The pipette (3–4 MΩ for whole-cell and 6–8 MΩ for micropatch) solution contained the following (in mm): 120 KCl, 30 NaCl, 1 MgCl2, 0.5 CaCl2, 5 EGTA, 2 Mg-ATP, and 10 HEPES, adjusted to pH 7.2 with Tris-base. The osmolarity of all solutions were maintained at 300–320 mOsm (Advanced Instrument). Whole-cell voltage-clamp (with the holding potential of −60 mV) and current-clamp recordings were performed with an Axoclamp 700A amplifier and were sampled and analyzed using a Digidata 1320A interface (Molecular Devices); the micropatch recording with the outside-out configuration was performed with an Axopatch 200B amplifier (Molecular Devices). In most experiments, 70–90% of the series resistance was compensated. For current-clamp recordings, only DRG neurons with stable resting membrane potentials lower than −50 mV were included for analysis. All recordings were performed at room temperature (23 ± 2°C).
Calcium imaging.
Cells were loaded with Fura-4 and imaged as described previously (Shigetomi et al., 2012). Briefly, cultured CHO cells grown on 8 × 8 mm glass coverslips were washed thrice with PBS and incubated with 2 μm Fluo-4 acetoxymethyl ester (Invitrogen) in HBSS for 10 min at 37°C. Pluronic F-127 (20%, v/v) solution in dimethylsulfoxide (Invitrogen) diluted at 1:2000 was added to facilitate the dye loading. Cells were again washed thrice and then incubated in the standard extracellular solution of pH 7.4 for 30 min at 37°C. The coverslip was transferred to a perfusion chamber placed on a laser scanning confocal microscope (LSM-710; Carl Zeiss), which is equipped with a 20× (NA 0.5) UV fluor oil-immersion objective lens. Images were typically taken every 2 s and all acquired images were processed by Carl Zeiss Zen software.
Radioligand binding assays.
CHO cells were washed with a preparation buffer containing the following (in mm): 50 Tris-HCl, 0.5 EDTA, 10 MgCl2, and 0.1 phenylmethanesulfonyl fluoride (trypsin and chymotrypsin inhibitor) (adjusted to pH 7.4 with Tris-base), detached with a cell scraper into 6 ml of the preparation buffer and homogenized using Pellet Pestle (Kontes; 10 s at 4°C). The homogenate was centrifuged (3000 g) for 15 min at 4°C (Beckman ultracentrifuge) and the resulting pellet was resuspended with the preparation buffer and centrifuged (27,000 g) for 15 min at 4°C (Beckman ultracentrifuge). Then the resulting pellet was resuspended with an analysis buffer containing: 50 mm Tris-HCl, 5 mm CaCl2, 0.1% (v/v) ascorbic acid (prevent indolamine oxidation) and 10 μm pargyline (a monoamine oxidase inhibitor) (adjusted to pH 7.4 with Tris-base). Binding assays were performed in a total volume of 400 μl for each concentration in tubes that contained increasing concentrations of [3H]-5-HT (0.1–3000 nm, PerkinElmer; specific activity: 80.8 Ci/mmol) in the absence or presence of an excess of unlabeled 5-HT (1 mm) to define nonspecific binding. All components of the assay were diluted in the analysis buffer and all results obtained as duplicates. The incubation was started by the addition of 200 μl of freshly isolated membrane protein. After 30 min at 37°C the reaction was terminated using a Brandel harvester by addition of an excess of deionized water (4°C) and rapid vacuum filtration through Whatman GF/C filters. Radioactivity was measured by liquid scintillation counting (Beckman LS 6500 multipurpose scintillation counter) using Optiphase HiSafe scintillant (PerkinElmer). A BCA Protein Assay Kit (Pierce Thermo Scientific) was used to determine protein concentrations in the membrane samples.
Animals and pain-related behavioral assays.
All behavioral measurements were performed using littermate WT and ASIC3 KO, or littermate WT and ASIC1a KO mice (male, 6–8 weeks old, and in C57BL/6 background). Three to four animals were housed per cage and maintained on a 12 h light/dark cycle with food and water ad libitum. Animals were acclimatized for 30 min before all behavioral experiments. A total volume of 20 μl of solution (in 0.9% NaCl) containing acetic acid (0.6%, pH 3.5–4.0), 5-HT (25 μm), 5-HT plus acetic acid (0.6%), or 5-HT plus acetic acid (0.6%) with a desired antagonist, was injected intraplantarly using a 30 G needle and paw-licking behavior was quantified for 30 min (Yu et al., 2010; Li et al., 2011). Behavioral and histological experiments were both conducted and scored with the experimenter blinded to the genotype. All experimental procedures were performed in strict compliance with the animal use and care guidelines of the Institute of Neuroscience, Chinese Academy of Sciences.
Immunohistochemistry.
For spinal cord c-Fos immunoreactivity, mice were deeply anesthetized with ether 90 min after hindpaw injection (as for behavioral experiments), and perfused transcardially with saline followed by 4% paraformaldehyde (PFA) in PBS, pH 7.4. The spinal cord was removed and fixed for 12 h in 4% PFA and stored for at least 48 h in 30% sucrose. Frozen spinal cord sections (15 μm) (obtained using Leica CM 1850 tissue slicer) were prepared from lumbar level L4/L5 and immunostained for c-Fos protein as described previously (Caterina et al., 2000; Lino-de-Oliveira et al., 2001). In brief, the tissue sections were successively washed and permeabilized with 0.3% Triton X-100 in PBS for 30 min at room temperature. After incubation in blocking solution (PBS containing 1% bovine serum albumin (BSA) and 0.3% Triton X-100) for 30 min at room temperature, sections were labeled for 24 h at 4°C with the primary rabbit monoclonal anti-c-Fos (1:1000; Cell Signaling Technology) in PBS supplemented with 1% BSA and 0.3% Triton X-100. The sections were washed three times with PBS (15 min each time) and then incubated with the secondary antibody (Alexa Fluor 488-conjugated goat anti-rabbit IgG, 1:1000; Invitrogen) in PBS supplemented with 1% BSA and 0.3% Triton X-100 for 2 h at 37°C. After washing twice with PBS (15 min each time), spinal cord sections were incubated with the nuclear counterstain 2-(4-amidinophenyl)-6-indole-carbamidine dihydrochloride (DAPI; Beyotime Institute of Biotechnology, 1:5000 in PBS). After staining is completed, the tissue sections were mounted with coverslips and sealed with clear nail polish. Fluorescence images were obtained using a laser scanning confocal microscope (LSM-710; Carl Zeiss).
Reagents and data analysis.
All drugs were purchased from Sigma unless otherwise mentioned. The sustained ASIC3 currents were measured at 10 s after the start of acid application. Results were expressed as means ± SEM. Statistical comparisons were performed using the Student's t test or one-way ANOVA where values of *p < 0.05, **p < 0.01, or ***p < 0.001 were considered significantly different.
Results
Acute effects of pro-inflammatory mediators on ASIC3 channel activation
Previous studies have shown that pro-inflammatory mediators increase the expression of ASICs (Mamet et al., 2002, 2003), resulting in enhanced nociceptor excitability. These studies represent a chronic modulation by increasing the availability of the ASIC channel numbers, but a direct interaction between ASICs and pro-inflammatory mediators has not been established. As numerous inflammatory mediators released from immune cells and fibroblasts are often associated with a high concentration of protons in inflamed tissues, we investigated the acute effects of pro-inflammatory mediators on ASIC3 channel activation. Whole-cell patch-clamp recordings were used to measure proton-gated currents in CHO cells expressing ASIC3. As previously reported (Waldmann et al., 1997b; Hesselager et al., 2004), lowering extracellular pH from 7.4 to 5.0 produced a rapidly activating current followed by fast inactivation and then a small, but significant, sustained current with no or very slow inactivation (Fig. 1A,B). All of the tested pro-inflammatory mediators, when applied alone, generated no current in CHO cells expressing ASIC3 channels (Fig. 1A). However, in the presence of 5-HT (50 μm), the sustained currents induced by pH 5.0 were dramatically enhanced in ASIC3-expressing cells (Fig. 1BII) but not in control cells (expressing the pEGFPC3 vector only; Fig. 1BI). The enhancement of pH 5.0-induced sustained currents did not occur to other pro-inflammatory mediators, such as His (50 μm), BK (50 μm), and PGE2 (50 μm). Substance P (SP; 50 μm) also significantly enhanced the pH 5.0-induced sustained currents but to a much less extent than 5-HT (Fig. 1B,C). Notably, the mixture (Mix) of these pro-inflammatory mediators had nearly the same effect on the pH 5.0-induced sustained currents of ASIC3 channels as 5-HT alone (Fig. 1B,C), suggesting the dominance of 5-HT in regulating the sustained activation of ASIC3 channels under acidosis.

Effects of pro-inflammatory mediators on proton-evoked ASIC3 currents. A, Representative traces showing currents in CHO cells expressing ASIC3 induced by an acidic solution, pH 5.0, or each of the pro-inflammatory mediators individually or in combination (Mix) as indicated at pH 7.4. 5-HT, serotonin; BK, bradykinin; His, histamine; SP, Substance P; PGE2, prostaglandin E2; Mix, a mixture of the above pro-inflammatory mediators. All mediators were at 50 μm. B, Representative traces showing currents in CHO cells expressing vector (pEGFPC3; BI) or ASIC3 (BII), induced by the acidic solution, pH 5.0, in the absence (black) or presence (red) of the pro-inflammatory mediators as indicated. C, Means ± SEM sustained currents at 10 s after the start of pH 5.0 application, normalized to that obtained with pH 5.0 solution alone (dashed line indicates control); n = 4–7. *p < 0.05 and **p < 0.01 versus control.
Dose dependence and specificity of 5-HT-mediated enhancement of sustained phase of proton-induced ASIC3 currents
As shown in Figure 2, A and B, 5-HT dose-dependently enhanced the pH 5.0-induced sustained ASIC3 currents, but had nearly no effect on the large initial transient currents. The EC50 of 5-HT-mediated enhancement of sustained ASIC3 currents was 41.2 ± 3.4 μm. The effective 5-HT concentrations used in this study were in the range of concentrations usually used in the literature or based on levels found in inflamed tissues (Mössner and Lesch, 1998; Mamet et al., 2002, 2003; Jitsuki et al., 2011). Strikingly, the enhancing effect of 5-HT on the sustained currents of ASIC3 channels was detectable at a concentration as low as 1 μm (p = 0.05 vs control), which can be commonly attained under multiple physiological and pathological conditions (Fig. 2A,B).

Effects of 5-HT and its analogs on proton-evoked ASIC3 currents. A, Representative current traces showing the effects of various concentrations of 5-HT on pH 5.0-induced currents in CHO cells expressing ASIC3. Dashed lines indicate the sustained current induced by pH 5.0 alone. B, Means ± SEM peak (open symbols) and sustained (filled symbols) currents normalized to that induced by pH 5.0 alone. n = 5–8. Solid line for sustained currents represents a fit to the Hill equation. C, Chemical structures of 5-HT and its precursors and metabolites. D, Sustained ASIC3 currents induced by pH 5.0 in the presence of 5-HT (50 μm) and its analogs, l-tryptophan (TRP; 50 μm), 5-hydroxy-l-tryptophan (5-HTP; 50 μm), or 5-hydroxyindoleacetic acid (5-HIAA; 50 μm). Data are means ± SEM sustained currents normalized to that induced by pH 5.0 alone (dashed line indicates control). n = 4–6. **p < 0.01 versus control.
As both TRPV1 and ASIC3 channels are major sensors for inflammatory pain associated with acid sensing presumably by coincident detection of proton and pro-inflammatory mediators, we tested the specificity of acute potentiation of ASIC3 currents by 5-HT. However, in CHO cells that expressed TRPV1 channels, neither capsaicin-induced (500 nm) or acid-induced (pH 5.0) TRPV1 currents were affected by acute application of 5-HT (10 μm) (data not shown). Therefore, as the vanguards of the sensory systems, ASIC3, but probably not TRPV1 channels, are responsible for the coincident detection of acidosis and 5-HT-associated inflammatory pain.
The acute modulation of ASIC3 channels by 5-HT prompted us to look for its structure–activity relationship by investigating the effects of 5-HT precursors and metabolites (Fig. 2C). As shown in Figure 2, when the 2-amino group of 5-HT (Fig. 2C) was replaced by a carboxyl group, the resulting compound 5-hydroxy-indoleacetic acid (5-HIAA; the major metabolite of 5-HT; Fig. 2C) lost the enhancement effect (Fig. 2D) on the sustained currents of ASIC3 channels. In addition, when the methylene (ortho position of 2-amino group) hydrogen was substituted with a carboxyl group, presumably by disrupting the polarity of the 2-amino group, the resulting compounds, both l-tryptophan (TRP; precursor of 5-HT; Fig. 2C) and 5-hydroxy-l-tryptophan (5-HTP; precursor of 5-HT; Fig. 2C) failed to enhance the ASIC3 currents (Fig. 2D). These results underscore a critical role of the 2-amino group of 5-HT in enhancing the sustained activation of ASIC3. The requirement of the 2-amino group in 5-HT for its enhancing effects raises a potential structural mechanism, which might involve interactions with acidic residues in the ASIC3 channel (see below).
pH dependence of 5-HT-mediated enhancement of the sustained ASIC3 currents
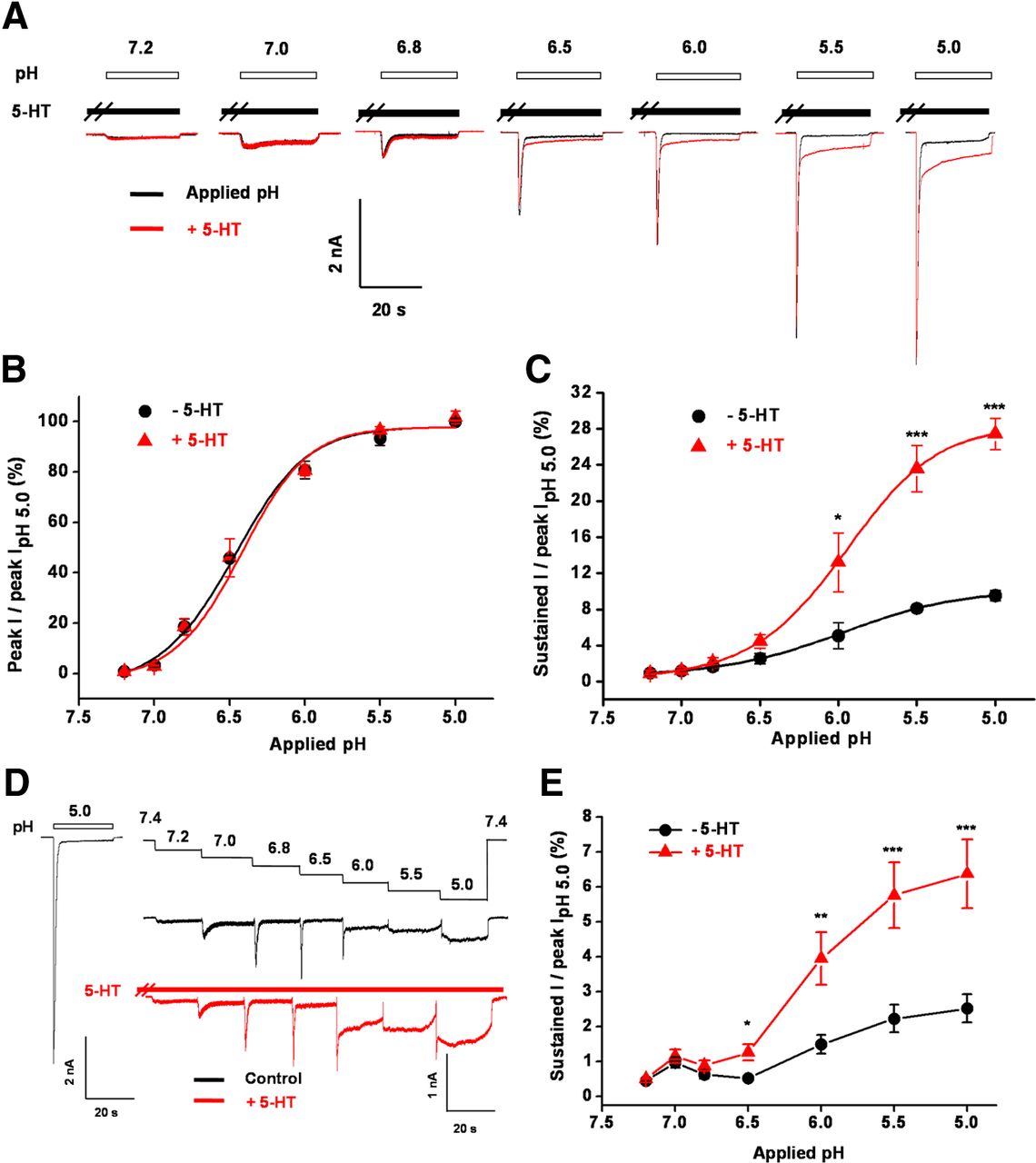
Previous studies have suggested that tissue acidosis plays a dominant role in inflammatory excitation and sensitization of nociceptors (Steen et al., 1995) and local proton concentrations gradually increase in inflamed or injured tissue. Hence, we examined the pH dependence of 5-HT modulation of ASIC3 using two different protocols. First, we used solutions with different pH values to stimulate ASIC3 channels expressed in CHO cells in the presence or absence of 5-HT (Fig. 3A). The interval between two adjacent stimulations was 2 min to allow recovery of the channels from desensitization. Noticeably, the enhancing effect of 5-HT (50 μm) on the sustained ASIC3 currents was pH dependent, being more pronounced at lower pH levels (Fig. 3A,C). Under mild acidosis, pH 6.8–7.2, the effect of 5-HT was negligible (Fig. 3A,C). As expected and in contrast to the sustained currents, 5-HT had no significant effects on the transient peak ASIC3 currents evoked by different levels of acidosis (Fig. 3A,B). Second, successive stepwise changes in pH mimic the slow acidification that accompanies muscle ischemia or inflammation (Yagi et al., 2006). Thus, we successively changed the extracellular pH between 7.4 and 5.0 in a stepwise manner in the presence or absence of 5-HT (50 μm) to stimulate ASIC3 channels expressed in CHO cells (Fig. 3D). Prior exposure to acidic solutions desensitized the ASIC3 channels and dramatically decreased the transient peak current (Fig. 3D). However, the sustained ASIC3 currents continuously increased as the pH dropped and the enhancing effect of 5-HT (50 μm) on the sustained ASIC3 currents was maintained under these conditions at lower pH (<6.5; Fig. 3D,E), similar to the results obtained with the first protocol (Fig. 3A–C). Gradual acidification is a common phenomenon that occurs under pathophysiological conditions, such as muscle ischemia and inflammation. These results thus suggest that 5-HT works particularly in enhancing the sustained ASIC3 currents under certain pathophysiological conditions, such as inflammation.

pH dependence of 5-HT potentiation. A, Representative current traces elicited by application of different pH in the absence (black) or presence of (red) 5-HT (50 μm) to CHO cells expressing ASIC3 channels. B, Means ± SEM peak currents evoked by different pH in the absence (black) or presence (red) of 5-HT, normalized to that by pH 5.0 alone. n = 4–7. C, Means ± SEM sustained currents evoked by different pH in the absence (black) or presence (red) of 5-HT, normalized to the peak current induced by pH 5.0 alone. n = 4–7. *p < 0.05 and ***p < 0.001, different from those in the absence of 5-HT. D, Representative current traces elicited by successive stepwise decreases of pH from 7.4 to 5.0 as indicated in the absence (upper black) or presence (lower red) of 5-HT (50 μm) from a CHO cell expressing ASIC3 channels. Current trace on the left represents pH 5.0-induced current in the absence of 5-HT from the same cell, serving as a measure for the maximal peak current. E, Means ± SEM sustained components of pH 5.0-induced currents as shown in D, normalized to the peak current induced by pH 5.0 alone. n = 4–6. *p < 0.05, **p < 0.01, and ***p < 0.001, different from those in the absence of 5-HT.
Isoform selectivity of 5-HT-mediated enhancement among ASICs
Almost all ASIC isoforms are present in primary sensory neurons of the trigeminal, vagal, and dorsal root ganglia (Alvarez de la Rosa et al., 2002; Wemmie et al., 2006; Lingueglia, 2007; Sluka et al., 2009; Deval et al., 2010). ASIC1a/1b, ASIC2a/2b, and ASIC3 are expressed abundantly in the small- and medium-sized nociceptive sensory neurons that are able to detect noxious chemical, thermal, and high-threshold mechanical stimuli (Lingueglia et al., 1997; Waldmann et al., 1997a,b; Chen et al., 1998, 2002; Price et al., 2001; Benson et al., 2002). To investigate subunit selectivity of 5-HT modulation, we tested the effects of 5-HT in CHO cells expressing different ASIC isoforms (Fig. 4A). We found that 5-HT (50 μm) did not potentiate homomeric ASIC1a, ASIC1b, or ASIC2a channels (Fig. 4Aa,B). Furthermore, we tested whether 5-HT could enhance acid-evoked currents mediated by heteromeric channels containing ASIC3. To minimize the formation of ASIC3 homomers, ASIC3 and another ASIC subunit were coexpressed at the 1:3 ratio. Interestingly, 5-HT (50 μm) significantly enhanced the sustained currents of heteromeric ASIC3–ASIC1a (3 + 1a) and ASIC3–ASIC1b (3 + 1b) channels but not heteromeric ASIC3–ASIC2a (3 + 2a) or ASIC3–ASIC2b (3 + 2b) channels (Fig. 4Ab,B). These results show that 5-HT specifically enhances sustained activation of the homomeric ASIC3 channels and heteromeric ASIC3 subunit-containing 3 + 1a and 3 + 1b channels.

Subunit selectivity of 5-HT potentiation on ASIC activation. A, Representative current traces induced by pH 5.0 in the absence (black) or presence (red) of 5-HT (50 μm) from CHO cells expressing homomeric (a) ASIC3, ASIC1a, ASIC1b, and ASIC2a channels, or heteromeric (b) ASIC3 plus 1a, 1b, 2a, or 2b, channels. B, Means ± SEM sustained components of pH 5.0-induced currents in the presence of 5-HT (50 μm) normalized to that induced by pH 5.0 alone (dashed line). n = 4–7. *p < 0.05 and **p < 0.01, different from control (pH 5.0 alone); #p < 0.05 and ##p < 0.01, different from homomeric ASIC3 as indicated.
The effect of 5-HT on ASIC3 is not dependent on serotonin receptors
CHO cells have been reported to be devoid of 5-HT receptors except for 5-HT1B (Giles et al., 1996) and have frequently been used as a host for expressing cloned 5-HT receptors. Therefore, it is unlikely that the enhancement of sustained currents by 5-HT of ASIC3 channels expressed in CHO cells is dependent on activation of endogenous 5-HT receptors present in these cells. If that was the case, then 5-HT could act directly on the ASIC3 channel.
To verify if classical 5-HT receptors were indeed not involved in the enhancing effect of 5-HT on sustained ASIC3 activation, we treated the cells with Y-25130 (Tocris Bioscience) and methysergide to inhibit 5-HT3 and 5-HT1,2,7 subtype receptors, respectively. The DRG neurons, where the natural interaction between 5-HT and ASIC3 channels most likely occurs, express the mRNAs for 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2C, 5-HT3, and 5-HT7 receptors (Pierce et al., 1997). Therefore, these blockers are of relevance to potential regulation of native ASIC3 by 5-HT receptors in sensory neurons responsible for inflammatory pain. We confirmed the effectiveness of Y-25130 (200 nm) by showing the block of 5-HT (100 μm)-induced 5-HT3 currents in mouse DRG neurons (Fig. 5A,B), similar to the published observations (Yakushiji and Akaike, 1992). We also validated that methysergide (40 μm) effectively decreased the elevation of intracellular Ca2+ concentration induced by 5-HT (50 μm) on CHO cells that heterologously expressed 5-HT2c receptors (Fig. 5C). In contrast, neither Y-25130 (200 nm) nor methysergide (40 μm) significantly affected the effect of 5-HT on enhancing the proton-evoked sustained currents in ASIC3-transfected CHO cells (Fig. 5D,E), demonstrating that the enhancement of sustained ASIC3 current by 5-HT is independent of classical 5-HT receptors.

The enhancement by 5-HT on sustained ASIC3 currents is independent of 5-HT receptors. A, Representative traces showing that the 5-HT3 receptor antagonist, Y-25130 (200 nm), blocked 5-HT-evoked currents in isolated mouse DRG neurons. B, Means ± SEM 5-HT-induced currents in the presence and after washout of Y-25130, normalized to that before the Y-25130 treatment (dashed line, control); n = 10. ***p < 0.001, compared with control. C, Time courses of fluorescence changes in Flou-4 loaded CHO cells expressing the human 5-HT2C receptor or a control vector. Cells were stimulated with 5-HT (50 μm) and then treated with methysergide (Methy; 40 μm), a 5-HT1,2,7 receptor antagonist, as indicated. Data are means ± SEM (n = 23, 16, for 5-HT2C and control, respectively). D, Representative traces showing the effects of Methy (40 μm) and Y-25130 (200 nm) on proton-evoked currents and 5-HT-mediated enhancement of proton-evoked sustained currents in CHO cells expressing ASIC3. E, Means ± SEM sustained currents under conditions as indicated, normalized to that evoked by pH 5.0 alone; n = 4–8. **p < 0.01 versus as indicated. F, Effect of 5-HT on pH 5.0-induced currents in excised outside-out patches from CHO cells expressing ASIC3. Left, Representative current traces without and with 5-HT treatment. Right, Means ± SEM sustained currents normalized to that evoked by pH 5.0 alone (control); n = 4. **p < 0.01, compared with control.
Next, we examined whether the acute potentiation of ASIC3 channels by 5-HT involved any intracellular signaling pathway. For this purpose, we tested the effect of 5-HT on proton-evoked currents from ASIC3-expressing CHO cells using excised outside-out patches, in which diffusible intracellular signaling components should have been mostly lost. Similar to that seen with the whole-cell recordings, 5-HT (50 μm) significantly enhanced the sustained ASIC3 currents in the outside-out configuration (Fig. 5F), suggestive of a negligible role of intracellular signaling cascades. Collectively, these data are consistent with the notion that 5-HT preferentially interacts with ASIC3 channels to enhance the sustained channel activation and thereby plays a novel role in mediating pain sensation and transmission during inflammation.
Crucial role of the nonproton sensing domain in the enhancement of sustained ASIC3 currents by 5-HT
Recent studies have identified a nonproton ligand sensing domain in ASIC3 that mediates the sustained channel activation, an effect that is independent of an abrupt decrease in extracellular pH (Yu et al., 2010; Li et al., 2011). We reasoned that the sustained activation of ASIC3 channels enhanced by 5-HT in the presence of acidosis might share similar mechanisms to that caused by nonproton ligands in the absence of acidosis. As expected, we found that as for the activation by nonproton ligands, glutamate 79 (E79) and glutamate 423 (E423) were essential for the enhancement of sustained ASIC3 currents by 5-HT (Fig. 6). When E79 was substituted with serine (S), cysteine (C), or glutamine (Q), the mutated channel became largely insensitive to 5-HT (Fig. 6A,B). Similar results were obtained when E423 was replaced with alanine (A), C, or Q (Fig. 6A,B). We did not test the E79A mutant because it resulted in strong steady-state desensitization (Cushman et al., 2007; Yu et al., 2010). The crucial role of these acidic residues was reminiscent of the essential requirement of the 2-amino group of 5-HT in enhancing sustained ASIC3 activation (Fig. 2C,D). Thus a potential interaction between either E79 or E423, or both, and the 2-amino group of 5-HT might constitute a molecular mechanism underlying the acute sensitization of ASIC3 channels by 5-HT.

5-HT-mediated enhancement involves the nonproton ligand sensing domain of the ASIC3 channel. A, Representative current traces illustrating mutations of key residues (E79 and E423) in the nonproton ligand sensing domain of the ASIC3 channel abolished the enhancing effect of 5-HT (100 μm). ASIC3 mutants were expressed in CHO cells. B, Means ± SEM pH 5.0-induced sustained currents in the presence of various concentrations of 5-HT, normalized to that in the absence of 5-HT (dashed line). CHO cells expressed WT or mutant ASIC3 constructs as indicated. n = 4–7. WT data are the same as that shown in Figure 2B, re-graphed for comparison. **p < 0.01, compared with WT. C, Representative current traces illustrating effects of 5-HT pretreatment (100 μm, 30 s) on currents induced by GMQ (100 μm) at different pH conditions in CHO cells expressing WT ASIC3. Black, currents evoked by pH change alone; pink, GMQ-induced currents at different extracellular pH levels; blue, GMQ-induced currents at different extracellular pH levels after pretreatment with 5-HT. D, Means ± SEM sustained currents normalized to peak current induced by pH 5.0 alone, activated at different pH levels in the absence and presence of GMQ and 5-HT as indicated. n = 6. *p < 0.05 and **p < 0.01, different from those with 5-HT pretreatment. E, F, Typical recordings showing the effect of 5-HT (1 mm) on ASIC3E79C activation induced by DTNB (0.5 mm, pH 7.4) with a different drug application sequence. Coadministration of 5-HT with DTNB slowed down the development of DTNB-induced currents because of steric competition. The reducing reagent DTT completely reversed the DTNB-induced currents. Inset shows the structure of DTNB and the resultant covalent link of 5-thio-2-nitrobenzoic acid (TNB) to E79C via a mechanism of Ellman's reaction. G, Pooled data from the combination of experiments in E and F. n = 7. **p < 0.01, compared with DTNB alone.
In addition, we examined if 5-HT could compete with 2-guanidine-4- methylquinazoline (GMQ), which activates ASIC3 at the physiological pH (7.4) via binding to the nonproton ligand sensing domain. Consistent with the previous study (Yu et al., 2010), GMQ (100 μm) strongly sensitized the response of ASIC3 channel to mild extracellular acidosis (pH 7.0, 6.8, and 6.5), but this effect was gradually decreased as the extracellular pH was lowered, because of the pH-dependent increase of the sustained current in the absence of GMQ (Fig. 6C,D). Pretreatment with 5-HT (100 μm, 30 s) significantly attenuated the enhancing effect of GMQ under mild acidosis, but at more extreme acidosis (pH ≤ 6.0), the enhancing effect of 5-HT on sustained ASIC3 current dominated (Fig. 6C,D), giving rise to similar levels of acid-evoked sustained currents as with 5-HT alone (compare with Fig. 3C). Plausibly, these results can be interpreted as preoccupation by 5-HT prevented the binding of GMQ to the nonproton ligand sensing domain, inhibiting its action on the ASIC3 channel, supporting the idea that 5-HT and GMQ bind to the same channel domain.
To further verify the possible interaction of 5-HT with the nonproton ligand sensing domain of ASIC3, we tested if 5-HT could interfere with the activation of E79C mutant by the thiol probe, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), which covalently modifies the nonproton sensing domain of ASIC3 (Yu et al., 2010; Li et al., 2011). As shown previously, bath application of DTNB (0.5 mm, pH 7.4) slowly activated the ASIC3E79C channel, presumably reflecting the time-dependent modification of the E79C site by the thiol-reactive probe (Fig. 6E,F, inset). We reasoned that if 5-HT binds to the nonproton ligand sensing domain, it should perturb the interaction and hence covalent bond formation between 5-thio-2-nitrobenzoic acid and C79 because of steric effects. As expected, 5-HT significantly slowed down the development of DTNB-induced ASIC3E79C channel activation, regardless of the sequence of 5-HT and DTNB applications (Fig. 6E,F). Furthermore, a reducing reagent, 1,4-dithiothreitol (DTT) blocked the DTNB-induced channel activation (Fig. 6E,F). To quantify the effect of 5-HT, we measured the rate (pA/s) of DTNB-induced activation of ASIC3E79C, defined as the ratio of maximal current (pA) and the duration (s) of DTNB application. The rate of DTNB-induced activation of ASIC3E79C was significantly higher in the absence than in the presence of 5-HT (Fig. 6G), demonstrating an inhibitory effect of 5-HT on the DTNB-dependent modification of the E79C site, which supports the notion that 5-HT directly interacts with the nonproton ligand sensing domain of ASIC3.
5-HT binding to ASIC3 channels
We then tested whether 5-HT binds to the ASIC3 channels using [3H]-5-HT. Consistent with a previous report (Giles et al., 1996), significant difference between the total and nonspecific binding (determined in the presence of 1 mm unlabeled 5-HT) was only detectable at [3H]-5-HT concentrations >0.2 μm in samples prepared from CHO cells (Fig. 7A). Remarkably, at 3 μm [3H]-5-HT, both total and specific bindings were significantly higher in samples prepared from cells transfected with WT ASIC3 than that with the vector (pEGFPC3, control) or a mutated channel with dramatically reduced sensitivity to 5-HT as shown in Figure 6A and B (ASIC3E423A) (Fig. 7A,C). In contrast, nonspecific bindings were about the same among these samples (Fig. 7B). Thus, 5-HT can bind to the ASIC3 channel (presumably through interactions with E79 and/or E423) and the enhanced sustained activation depends on the integrity of the nonproton ligand sensing domain (Li et al., 2010, 2011; Yu et al., 2010).

[3H]-5-HT binding to ASIC3 channels. A, Total [3H]-5-HT binding to membranes from CHO cells expressing pEGFPC3 vector (control), WT, and E423A mutated ASIC3 channels (n = 3). B, Nonspecific [3H]-5-HT binding determined in the presence of 1 mm unlabeled 5-HT. C, Specific [3H]-5-HT binding determined from the data in A and B. *p < 0.05, compared with WT.
5-HT enhances pH 5.0-induced currents and excitability of DRG neurons
ASICs expressed in primary sensory neurons respond to local acidosis with membrane depolarization, which is thought to be the initial trigger for pain sensation (Mamet et al., 2002; Sugiura et al., 2005; Deval et al., 2008). To gain insights into the pathophysiological function of ASIC3-dependent synergism of 5-HT and protons, we first tested the effect of 5-HT on acid-evoked currents in acutely isolated DRG neurons from WT mice by patch-clamp recording. To exclude the interference by classical 5-HT receptors, we included Y-25130 (200 nm) and methysergide (40 μm) in the recording solution to block 5-HT3 and 5-HT1,2,7 receptors (Yakushiji and Akaike, 1992; Dahlöf and Maassen Van Den Brink, 2012), respectively. We also included AMG9810 (10 μm), a TRPV1 selective antagonist, to block proton-induced TRPV1 activation (Gavva et al., 2005). Under these conditions, the prevalent type of currents induced by the pH 5.0 solution among small- and medium-sized (15–30 μm) WT mouse DRG neurons were ASIC-like (rapidly inactivating in response to acid; 79.4%, 27 of 34 total tested neurons) (Fig. 8A,C), which is consistent with previous studies (Dubé et al., 2005b; Deval et al., 2008). Among these 27 neurons, 5-HT dose-dependently (with an EC50 = 62.2 ± 6.1 μm) enhanced the sustained currents induced by the pH 5.0 solution in 19 of them (70.4%, Fig. 8A,B). These effects were completely absent in DRG neurons from ASIC3 KO mice (Fig. 8B). Only 30.8% (4 of 13 total) tested ASIC3 KO neurons showed ASIC-like current and none of them displayed sustained component. In addition, the potentiation by 5-HT, as well as the peak acid-induced current, in WT neurons were largely inhibited by the nonselective ASIC antagonist amiloride (200 μm) (Fig. 8A; peak current was 18.1 ± 0.02% and sustained current was 21.1 ± 0.08% of that induced by pH 5.0 in the presence of 200 μm 5-HT, n = 19).

Effects of 5-HT on pH 5.0-induced currents and excitability of mouse DRG neurons. A, Representative traces illustrating pH 5.0-induced ASIC-like currents in the absence and presence of various concentrations of 5-HT in isolated mouse DRG neurons. The extracellular bath solution contained methysergide (Methy; 40 μm) and Y-25130 (200 nm) to block 5-HT receptors and AMG9810 (10 μm) to block TRPV1 channels. Amiloride (200 μm) was used to block all ASIC channels (rightmost trace). B, Means ± SEM peak and sustained currents normalized to that induced by pH 5.0 alone. For WT, n = 19 (out of 34); for KO, n = 4 (out of 13). Solid line for sustained currents in WT represents a fit to the Hill equation. C, Distributions of acid- and 5-HT-enhanced currents in all tested DRG neurons from WT mice. D, Original voltage- and current-clamp recordings from DRG neurons isolated from WT (left) and ASIC3 KO (right) mice in the presence of Methy (40 μm), Y-25130 (200 nm), and AMG9810 (10 μm). Top row, ASIC3-like current induced by a pH drop from 7.4 to 5.0 in the absence or presence of 5-HT (100 μm), or 5-HT (200 μm) and amiloride (200 μm), with neurons held at −60 mV. Middle row, Membrane depolarization recorded by current-clamp (I = 0 pA) induced by the pH drop for corresponding neurons as in the top row under the same conditions. Bottom row, Regions marked with blue rectangles in the middle row shown in expanded time scale. E, F, Means ± SEM for number of APs (E) and level of membrane depolarization at the plateau (F) evoked by pH 5.0 from WT and ASIC3 KO mice under conditions shown in D. *p < 0.05 and **p < 0.01, versus as indicated; #p < 0.05 and ##p < 0.01 versus WT.
A small proportion of DRG neurons (23.5%, 8 of 34 total neurons tested) (Fig. 8C) was acid sensitive but unresponsive to 5-HT (100 μm). These could represent 5-HT-insensitive ASIC subtypes (Fig. 4). In addition, 20.6% (7 of 34) of the tested WT neurons (Fig. 8C) manifested a small sustained current (smaller than 50 pA) in response to pH 5.0, which was unaffected by amiloride (200 μm) (data not shown), indicative of a more complex acid response pattern in DRG neurons. Together, these data demonstrate that 5-HT not only enhanced ASIC3 currents in CHO cells expressing ASIC3 channels (Fig. 2A,B), but also enhanced ASIC3-like current in native neurons.
To investigate whether the 5-HT-mediated enhancement of ASIC3 activation is sufficient to increase neuronal excitability, we recorded the small- and medium-sized (15–30 μm) DRG neurons in current-clamp mode in the presence of methysergide (40 μm), Y-25130 (200 nm), and AMG9810 (10 μm). In acid-responsive neurons, a pH drop from 7.4 to 5.0 induced a significant depolarization, which consisted of an initial burst of action potentials (APs) followed by a plateau (Fig. 8D). Importantly, in 81.8% tested neurons (18 of 22 total), acute treatment with 5-HT (100 μm) dramatically increased the number of APs as well as the degree of plateau depolarization (Fig. 8D–F), indicating that 5-HT potentiation of ASIC3 response indeed enhances the excitability of DRG neurons, a view supported by the voltage-clamp results in the same neurons that 5-HT enhanced pH 5.0-induced sustained currents (Fig. 8D). In contrast, 5-HT (100 μm) was ineffective on acid-induced currents and acid-triggered excitation in neurons from ASIC3 KO mice (n = 9; Fig. 8D–F). Consistent with the importance of ASIC3 in mediating 5-HT-regulated neuronal excitability, amiloride (200 μm) significantly attenuated 5-HT-potentiated currents and excitability induced by pH 5.0 in WT neurons (Fig. 8D–F). Collectively, these results clearly show that 5-HT-mediated enhancement of the sustained ASIC3 activation is to increase the excitability of DRG neurons, an effect that may lead to enhanced nocifensive responses.
5-HT potentiates acid-induced ASIC3-dependent nocifensive behaviors
To ascertain whether 5-HT facilitates pain-related behaviors through interacting with ASIC3 channels, we injected 5-HT (25 μm) and acetic acid (0.6%) into the right hindpaws of WT and ASIC3 KO mice and measured the total time that the animal spent licking the injected paw during a 30 min period. As shown in Figure 9A, ASIC3 WT mice showed a dramatic increase in paw-licking time after coinjection of 5-HT and acetic acid when compared with injection of acetic acid or 5-HT alone. This effect was attenuated by inclusion of the nonselective ASIC inhibitor amiloride (200 μm) (Fig. 9A), but not the TRPV1 antagonist, AMG9810 (10 μm) (Fig. 9B), demonstrating the involvement of ASIC channels. Furthermore, the enhanced pain-related behavior in response to coapplication of acetic acid and 5-HT was not detected in ASIC3 KO mice (Fig. 9A), providing critical evidence that ASIC3 channels mediate the synergistic action of acid and 5-HT on pain responses. In support of a specific role of ASIC3, the pain responses induced by the coadministration of 5-HT and acetic acid were unaffected in ASIC1a KO mice, compared with the WT littermates (Fig. 9C). These data collectively indicate that 5-HT potentiates acidic pain behavior specifically via ASIC3 channels.

5-HT potentiates acid-induced pain-related behavior in mouse via ASIC3 channels. A, Pain-related behavior as determined by the time spent on paw licking following saline or chemical injection (20 μl) as indicated. Means ± SEM. n = 9–14, *p < 0.05, **p < 0.01, and ***p < 0.01, versus as indicated. ##p < 0.01, WT mice versus their KO littermates. Acetic acid, 0.6%, pH 3.5–4.0; 5-HT, 25 μm; amiloride, 200 μm. B, Effects of methysergide (Methy; 40 μm), Y-25130 (200 nm), and AMG9810 (10 μm) on pain behaviors evoked by coapplication of 5-HT (25 μm) and 0.6% acetic acid. n = 8–10. *p < 0.05, compared with coinjection of 5-HT and 0.6% acetic acid. C, Effects of 5-HT (25 μm) on 0.6% acetic acid-induced pain behaviors in ASIC1a KO mice and their WT littermates. Data are means ± SEM n = 8–10. ***p < 0.001 versus as indicated. D, Lack of effect of MK-212 (5-HT2 receptor agonist, 50 μm) and trazodone (Tra; 5-HT2 receptor agonist, 50 μm) on pH 5.0-induced currents in mouse DRG neurons. Ip and Is represent peak current and sustained current, respectively. E, Lack of effect of MK-212 (50 μm) and Tra (50 μm) on potentiating pain behaviors induced by 0.6% acetic acid in ASIC3 KO mice and their WT littermates. *p < 0.05, versus as indicated, indicating the effects of MK-212 or Tra alone. n = 8. F, Lack of effect of BK (50 μm) on pain behaviors induced by 0.6% acetic acid in ASIC3 KO mice and their WT littermates. n = 8–10.
To further explore the mechanism underlying the pain sensitization, we tested the effects of two 5-HT derivatives, MK-212 and trazodone (Tra; both are 5-HT2 receptor agonists), on pH 5.0-induced currents in mouse DRG neurons. Neither MK-212 (50 μm) nor Tra (50 μm) (pretreatment 30 s) enhanced ASIC3 currents (Fig. 9D). Consistently, neither MK-212 nor Tra potentiated the pain responses induced by 0.6% acetic acid in these mice (Fig. 9E). Importantly, injection of MK-212 (50 μm) or Tra (50 μm) alone induced marked increase of pain responses compared with saline but there was no significant difference between ASIC3 KO mice and their WT littermates (Fig. 9E). Moreover, consistent with the findings in DRG neurons that 5-HT-mediated enhancement of sustained ASIC3 currents was independent of 5-HT receptors (Fig. 8A,B), methysergide (40 μm) and Y-25130 (200 nm) failed to eliminate the effect of 5-HT on sensitization of pain responses induced by acetic acid (Fig. 9B). To further validate the unique role of 5-HT among pro-inflammatory mediators in enhancing acidosis-induced pain responses via ASIC3 channels, we tested BK, a pro-inflammatory mediator that is unrelated to 5-HT and had no effect on pH 5.0-induced ASIC3 response (Fig. 1B,C). The pain responses to BK (50 μm), as well as their potentiation by BK which was marginal, were similar in ASIC3 KO mice and their WT littermates (Fig. 9F). Therefore, the ASIC3-mediated pain responses are specifically enhanced by 5-HT.
To confirm the activation of pain signals at the cellular level in mice injected with 5-HT and acid, we examined the expression of neuronal activity marker, c-Fos, for indications of engagement of nociceptive pathways. After injection of 5-HT (25 μm) and acetic acid (0.6%) into the hindpaws of WT mice, which produced a robust nocifensive behavior characterized by licking (Fig. 9A), we observed abundant c-Fos protein expression in superficial laminae of dorsal spinal cord of the same animals (Fig. 10). These responses were greatly reduced in ASIC3 KO mice, demonstrating an important contribution of ASIC3 channels to the evoked pain-like responses.

c-Fos expression in superficial dorsal horn area of spinal cord. A, Representative images of c-Fos immunostaining in superficial laminae of spinal cord sections from WT and ASIC3 KO mice injected with saline or acetic acid plus 5-HT from the same group in Figure 9A. Images obtained from ipsilateral (Ipsi.) to the injection site. DAPI labels the nuclei. White rectangles label the superficial dorsal horn. Scale bar, 200 μm. B, Enlarged images for regions marked by white rectangles in A. Scale bar, 200 μm. C, Means ± SEM the numbers of c-Fos immunoreactivity cells in superficial dorsal horn (laminae I–IV) Ipsi. and contralateral (Contra.) to the injected site for mice injected with acetic acid, 5-HT, and/or amiloride as in Figure 9A. *p < 0.05, **p < 0.01, and ***p < 0.001, versus as indicated; and ##p < 0.01, WT versus KO. n = 4–8.
Discussion
Extracellular acidosis is a key factor in inflammatory pain and hyperalgesia (Issberner et al., 1996; Reeh and Steen, 1996). ASIC3 channels have emerged as critical pH sensors predominantly expressed in nociceptors (Chen et al., 1998; Price et al., 2001; Voilley et al., 2001; Deval et al., 2008), which respond to extracellular acidification with a sustained (noninactivating) component (Waldmann et al., 1997b). The sustained currents mediated by ASIC3 channels have implications in pain perception (Sutherland et al., 2001; Dubé et al., 2005b; Yagi et al., 2006). We present evidence here that the pro-inflammatory mediator 5-HT and protons synergistically enhance the sustained ASIC3 currents through interaction with the recently identified nonproton ligand sensing domain (Yu et al., 2010), revealing a novel mechanism for nociceptive sensitization to inflammation.
5-HT enhances the sustained activation of ASIC3-containing channels independent of 5-HT receptors
5-HT is an important inflammatory mediator in pain processing and modulation. In peripheral neurons, 5-HT sensitizes afferent nerve fibers, thus contributing to hyperalgesia in inflammation and nerve injury (Sommer, 2004, 2006) probably by sensitizing nociceptors. Coincidentally, tissue acidosis is an important feature of inflammation in which extracellular pH can drop to values as low as 5.4 (Jacobus et al., 1977). The accumulating protons are sufficient to activate nociceptors (Steen et al., 1992) possibly gating acid-sensitive ion channels such as ASICs and TRPV1 channels. The coexistence of both protons and inflammatory mediators under these pathophysiological conditions suggests that functional interactions among different agents may occur in nociceptors. Notably, the enhancing effect of 5-HT on ASIC3 activation is more pronounced at lower pH, suggesting that 5-HT is particularly prominent in potentiation and extending the acid response under pathophysiological conditions when extreme acidosis occurs, thereby more effectively enhancing the body's response to injury.
Nearly all ASIC subunits are expressed in the nervous system, with ASIC3 and ASIC1b (Waldmann et al., 1997a; Chen et al., 1998; Bässler et al., 2001; Price et al., 2001) showing exclusive expression in nociceptors. Due to a lack of specific pharmacological tools, the exact subunit composition and stoichiometry of ASICs in native neurons remain largely unknown. Deletion of any one of ASIC1, ASIC2, and ASIC3 subunits did not abolish acid-evoked currents, but altered current kinetics in a manner consistent with the heteromultimerization of remaining subunits (Benson et al., 2002; Hesselager et al., 2004). We show here that acidosis and 5-HT synergistically enhance the sustained activation of homomeric ASIC3 channels and heteromeric ASIC3/1a and ASIC3/1b channels, which likely contributes to inflammation pain processing. Hence, the 5-HT-mediated enhancement of sustained ASIC3 activation mostly occurs in vivo in sensory neurons expressing homomeric ASIC3 and/or heteromeric ASIC3-containing channels (Xie et al., 2003; Yagi et al., 2006; Deval et al., 2008). Interestingly, it was recently shown that a specific 5-HT2 receptor agonist, α-methyl-5-HT, upregulated the activity of ASICs via a 5-HT2 receptor and protein kinase C-dependent signaling pathway in rat DRG neurons (Qiu et al., 2012). However, this represents a slow and indirect modulation of ASICs. Alternatively, since α-methyl-5-HT is structurally similar to 5-HT, it may also interact directly with ASIC3 channels.
The finding that 5-HT enhances the sustained ASIC3 activation independent of classical 5-HT receptors is reminiscent of the recent identification of a nonproton ASIC3 ligand (i.e., GMQ) and its sensing domain on the ASIC3 channel. GMQ was shown to directly activate ASIC3 channels at the physiological pH (7.4) by targeting the nonproton ligand sensing domain, and sensitize the channel response to extracellular mild pH (∼7.0), but not extreme pH (∼5.0) acidosis (Yu et al., 2010). GMQ was shown to activate sensory neurons and cause pain-related behaviors in an ASIC3-dependent manner (Yu et al., 2010). Recently, an endogenous substance, agmatine, was found to interact synergistically with multiple inflammatory signals to sensitize the pain perception by targeting the nonproton sensing domain of ASIC3 channels (Li et al., 2010). Both GMQ and agmatine potentiate the responses predominantly to mild acidosis, which is associated with the “window” current through ASIC3 channels at modest pH changes presumably contributing to myocardial ischemia (Yagi et al., 2006). Here, we extended the functional implication of the nonproton ligand sensing domain with the demonstration that 5-HT, a substance that has a clear role in inflammatory responses, also interacts with this domain to enhance sustained ASIC3 channel activation, preferentially under extreme acidosis. We favor a direct and specific interaction between 5-HT and the nonproton ligand sensing domain of ASIC3 channels for the following reasons. First, pretreatment with 5-HT reduced the potentiation effect of GMQ on ASIC3 activation induced by mild acidosis. Second, 5-HT application reduced the rate of covalent modification-induced ASIC3E79C channel activation. Third, mutations at the key residues of the nonproton ligand sensing domain dramatically decreased the affinity and efficacy of 5-HT on potentiation of ASIC3 currents. Finally, radioligand binding experiments clearly demonstrated that 5-HT binds to membrane samples prepared from cells that expressed WT but not a mutated ASIC3 channel with the critical Glu residue in the nonproton ligand sensing domain substituted by an Ala. Together, our data suggest that 5-HT serves as an endogenous modulator for sustained ASIC3 activation by targeting the nonproton sensing domain of the channel. However, in contrast to GMQ/agmatine, the enhancement of 5-HT on ASIC3-sustained activation is more prominent at lower pH. The reasons for this may lie in the subtle differences in the interactions of these ligands with amino acid residues in the nonproton ligand sensing domain, causing differential degrees of modulation of proton sensitivity of the ASIC3 channel. The differential modulation of ASIC3 channels by GMQ/agmatine and 5-HT highlights the multifunctional roles of the nonproton ligand sensing domain in ASIC3 gating and regulation.
Physiological implications
Acidic solutions evoke nociceptor excitation and significant nociceptive behavioral sensitization to mechanical stimulation (Steen et al., 1992). Previous studies have demonstrated that continuous intracutaneous infiltration of low pH buffer, pH 5.2, into human skin induces sustained, localized pain (Steen and Reeh, 1993). Thus, it has been proposed that local acidosis plays a major role in cutaneous pain and hyperalgesia (Issberner et al., 1996; Reeh and Steen, 1996).
ASIC3 channels are expressed almost exclusively in sensory neurons (Waldmann et al., 1997b; Waldmann and Lazdunski, 1998; Krishtal, 2003) and predominantly in nocifensive sensory neurons that innervate cardiac and skeletal muscles (Benson et al., 1999; Sutherland et al., 2001; Molliver et al., 2005). Previous studies suggest that ASIC3 channels mediate the proton-gated currents in cardiac ischemia-sensing neurons and the sustained currents through ASIC3-containing channels play an important role in sensing extracellular acidosis during myocardial ischemia and mediate angina (Yagi et al., 2006). ASIC3 mRNA and protein levels are significantly increased by plantar incision 24 h after surgery, implicating a role of ASIC3-containing channels in postoperative pain (Deval et al., 2011). Pharmacological inhibition of ASIC3 channels with the toxin APETx2 or in vivo knockdown of the ASIC3 protein significantly reduced the postoperative spontaneous, thermal, and postural pain behaviors (Deval et al., 2011).
ASIC3 protein levels in DRG neurons are upregulated, which contribute to hyperalgesia in several inflammation models (Ohtori et al., 2006; Hori et al., 2010; Chen et al., 2011). Incubating DRG neurons with a mixture of nerve growth factor, 5-HT, interleukin-1, BK, and brain-derived neurotrophic factor for 8 h or 24 h stimulated ASIC, particularly ASIC3, gene expression, which may contribute to the pain-enhancing effects of these mediators (Mamet et al., 2002, 2003). Thus, the ASIC3 channel has emerged as an attractive candidate for inflammatory pain processing. Indeed, previous studies have shown that pain induced by cutaneous injection of acid appears to be largely mediated through ASICs because the pain sensation was significantly attenuated by the nonselective ASIC inhibitor amiloride in human volunteers (Steen et al., 2001; Ugawa et al., 2002). During extracellular acidosis, 5-HT may be released simultaneously from platelets and mast cells after tissue injury or inflammation (Lehtosalo et al., 1984; Dray, 1995) and sensitize the nociceptors. TRPV1 channel is also sensitive to protons and plays an important role in inflammatory pain (Caterina et al., 1997; Tominaga et al., 1998; Davis et al., 2000). However, the TRPV1 antagonist capsazepine failed to influence the C-fiber excitability induced by an acidic solution that contained inflammatory mediators (Habelt et al., 2000). We found that the TRPV1 antagonist AMG9810 failed to block 5-HT-facilitated hyperalgesia in WT mice, implying the specific interaction between 5-HT and the ASIC3 channel. Furthermore, 5-HT facilitated peripheral pain sensitivity to the same degree between ASIC1a KO mice and their WT littermates, but the facilitation was completely lacking in ASIC3 KO mice. Therefore, ASIC3-containing channels are likely the sole targets of the combined stimulation by 5-HT and protons and as such they constitute a potential target for the treatment of inflammatory pain. We conclude that 5-HT and protons synergistically potentiate the sustained activation of ASIC3-containing channels, an effect that alerts the body of tissue damage/injury to initiate protective responses. Previous investigations have also shown that arachidonic acid (Gerdsen et al., 2000; Smith et al., 2007) and ATP (Birdsong et al., 2010) enhance ASIC activity. Together, these studies suggest that the ASIC3 channel may serve as a coincident detector, which integrates the signals from multiple inflammatory mediators to cause inflammatory pain. Thus the present results reinforce the ASIC3 channels as a promising therapeutic target for relieving inflammatory pain.
Footnotes
This study was supported by grants from the National Natural Science Foundation of China (Nos. 31230028, 91132303, 91213306, and 31170787) and Key National S&T Program “Major New Drug Development” Grant 2012ZX09504001-003. We thank Dr. M. J. Welsh (Howard Hughes Medical Institute, University of Iowa, Iowa City, IA) for providing ASIC3 and ASIC1a knock-out mice.
- Correspondence should be addressed to Dr. Tian-Le Xu, Neuroscience Division, Department of Biochemistry and Molecular Cell Biology, Institute of Medical Science, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Road, Shanghai 200025, China. xu-happiness{at}shsmu.edu.cn





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}