
Abstract
Aims/hypothesis
Levels of uncoupling protein-2 (UCP2) are regulated in the pancreatic beta cells and an increase in the protein level has been associated with mitochondrial uncoupling and alteration in glucose-stimulated insulin secretion. However, it is not clear whether an increase in uncoupling protein-2 per se induces mitochondrial uncoupling and affects ATP generation and insulin secretion.
Materials and methods
Transgenic mice with beta cell-specific overexpression of the human UCP2 gene and INS-1 cells with doxycycline-inducible overproduction of the protein were generated and the consequences of increased levels of UCP2 on glucose-induced insulin secretion and on parameters reflecting mitochondrial uncoupling were determined.
Results
In transgenic mice, an increase in beta cell UCP2 protein concentration did not significantly modify plasma glucose and insulin levels. Glucose-induced insulin secretion and elevation in the ATP/ADP ratio were unaltered by an increase in UCP2 level. In INS-1 cells, a similar increase in UCP2 level did not modify glucose-induced insulin secretion, cytosolic ATP and ATP/ADP ratio, or glucose oxidation. Increased levels of UCP2 did not modify the mitochondrial membrane potential and oxygen consumption. Increased UCP2 levels decreased cytokine-induced production of reactive oxygen species.
Conclusion/interpretation
The results obtained in transgenic mice and in the beta cell line do not support the hypothesis that an increase in UCP2 protein per se uncouples the mitochondria and decreases glucose-induced insulin secretion. In contrast, the observation that increased UCP2 levels decrease cytokine-induced production of reactive oxygen species indicates a potential protective effect of the protein on beta cells, as observed in other cell types.
Similar content being viewed by others


Introduction
In the pancreatic beta cell, oxidative metabolism of glucose and mitochondrial ATP generation are key links between glucose sensing and insulin secretion. The production of ATP in the mitochondria, leading to elevation in the cytosolic ATP/ADP ratio, is the main trigger of glucose-stimulated insulin secretion by\(K^{ + }_{{{\text{ATP}}}} \)-dependent and -independent mechanisms (reviewed in [1]). Therefore, any modification in glucose-induced ATP formation will have important consequences for glucose-stimulated insulin secretion (GSIS).
In brown adipocytes, ATP generation and energy metabolism are modulated by the presence and activity of uncoupling protein-1 (UCP1). Based on their similarity of sequence, UCP2 and UCP3 were first suggested to play a role in regulated thermogenesis [2]. Later studies demonstrated that this may not be the case [3] and now evidence is accumulating in favour of a role in reducing the production of reactive oxygen species (ROS) by the mitochondria [4, 5].
Among the UCPs, UCP2 is the only uncoupling protein present in rodent and human pancreatic beta cells [6, 7]. As GSIS depends on ATP production, the uncoupling state that may be induced by increased UCP2 level and/or activity is predicted to impair insulin secretion. Indeed, several studies have associated an increase in UCP2 with inhibition of GSIS and indications of mitochondrial uncoupling [6–8]. An increase in UCP2 protein levels has been measured after long-term exposure of beta cells to high concentrations of fatty acids [9–12], with indications of mitochondrial uncoupling. Exposure of human islets to high glucose increases UCP2 expression, and alters GSIS [13]. Peroxisome proliferator-activated receptor (PPAR)α [14–16] and PPARγ [17, 18] agonists also increase UCP2 and decrease GSIS, except in a study in which a threefold to fourfold increase in UCP2 level, measured after overexpression of PPARα and RXR (retinoid X receptor), decreased neither the mitochondrial membrane potential nor GSIS [16].
In agreement with a negative role of UCP2 in GSIS, Ucp2 knock-out mice have an increased circulating insulin level, and in vitro their pancreatic islets secrete more insulin and show a higher ratio of ATP/ADP in response to glucose [19]. These effects are reversed by adenoviral production of Ucp2 [20]. In addition, islets from Ucp2 knock-out mice are protected from the long-term effects of fatty acids, suggesting that UCP2 may be an important mediator of lipotoxicity [20]. Most of these results favour the hypothesis that UCP2 negatively regulates glucose-induced changes in ATP/ADP and insulin secretion in pancreatic beta cells.
The studies mentioned above assume that, when present, UCP2 promotes proton leak in proportion to its level. However, in a cellular system, UCP1 has no uncoupling activity, unless adequately stimulated. Recent studies in mitochondria obtained from tissues with different levels of UCP2 or UCP3 show that these proteins exhibit uncoupling activity only in the presence of ROS, in particular superoxide of mitochondrial origin, with or without fatty acids [21]. As mitochondrial ROS production is dependent on the mitochondrial potential, mild uncoupling could effectively decrease ROS generation. Therefore, UCP2 activation by ROS could be considered as a protective feed-back loop, as suggested by studies in neurons or cardiomyocytes [22, 23].
To determine whether an increase in UCP2 per se induces mitochondrial uncoupling and affects GSIS, we generated transgenic mice and pancreatic beta cell lines with an increase in UCP2 and measured GSIS, changes in ATP/ADP and mitochondrial membrane potential. The data obtained in islets of transgenic mice and in pancreatic beta cell lines do not support the hypothesis that an increase in UCP2 protein concentration partly uncouples the mitochondria and alters glucose-stimulated insulin secretion. In agreement with the notion that the protein acts on ROS generation, we provide evidence that an increase in UCP2 protein concentration decreases cytokine-induced ROS production.
Materials and methods
Generation and analysis of transgenic mice
UCP2-transgenic mice were obtained by microinjection into fertilised eggs of an RIP-Ucp2 cDNA construct that comprised 600 bp of the rat insulin II promoter, the entire coding region of human UCP2 (GenBank Accession U82819) and a rabbit β-globin intron and polyadenylation signal. Founder animals were identified by Southern blot hybridisation and PCR analysis on DNA extracted from tail biopsies (DNAeasy tissue kit; Quiagen AG, Hilden, Germany). The sex distribution and percentage of transgenic mice was 30 ± 2.4, 26 ± 2.2, 20 ± 2.1, 25 ± 2.1 for control and transgenic males and control and transgenic females respectively (25 litters). Male mice aged 10–16 weeks were used for the experiments.
The mice were housed in the animal facility of the University of Geneva Medical School. They had free access to water and standard mouse diet. All animal procedures were approved by the Office Vétérinaire Cantonal.
Glucose tolerance tests
Glucose tolerance tests (GTTs) were performed in mice that had fasted overnight. A baseline blood sample was collected from the tail vein and glucose (1.0 or 1.5 mg/g body weight) injected i.p. Blood was collected at 15, 30, 60 and 120 min and centrifuged, and plasma glucose was measured using glucose oxidase (Roche Diagnostics AG, Rotkreuz, Switzerland). Plasma insulin was measured at 0 and 15 min using a kit for rat insulin radioimmunoassay (Linco Research, St Louis, MO, USA).
Pancreas perfusion
Pancreases were perfused at 1.5 ml/min with modified KRBH (121 mmol/l NaCl, 4.8 mmol/l KCl, 1.0 mmol/l CaCl2, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4, 24.6 mmol/l NaHCO3, 40 g/l dextran T-40 and 12.5 mmol/l HEPES, pH 7.4) containing 0.25% BSA and the additions mentioned, as described in [24]. Insulin was measured by radioimmunoassay.
Islet isolation and culture, measurements of insulin secretion and ATP levels
Pancreatic islets were isolated by collagenase digestion, hand-picked and cultured in RPMI 1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% FCS, 10 mmol/l HEPES, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. After 24 h of culture, islets were hand-picked again, preincubated and incubated in KRBH containing 0.5% fatty-acid-free BSA or BSA–oleate prepared as described [9], and used for measurement of insulin secretion and content, or ATP/ADP levels. Islet insulin was extracted with cold acid–ethanol (1.5% HCl, 75% ethanol) and measured by radioimmunoassay.
Total ATP and ADP were measured in groups of ten islets. After glucose addition (30 min), perchloric acid (0.6 mol/l final concentration) was added. After centrifugation, the supernatant fraction was neutralised with 2.7 mol/l K2CO3 and the precipitate was removed by centrifugation. ATP was assayed directly using a bioluminescence assay kit (HSII; Roche Diagnostics AG). For ADP measurements, ATP was first hydrolysed to AMP using ATP sulfurylase [25]. ADP was converted to ATP with pyruvate kinase and phosphoenolpyruvate and ATP was measured by the same bioluminescence assay. The same treatment was applied to the ADP standard curve.
Construction of plasmid and transfection of INS-1 cells
The parental INS-1 cell line expressing the reverse tetracycline-dependent transactivator (INS-1-r9 cells) was kindly provided by P. Iynedjian and H. Wang. The plasmid used for the second stable transfection was constructed by subcloning the UCP2 cDNA isolated from a human skeletal muscle cDNA library (GenBank accession number U82819) into the expression vector PUHD10-3. INS-1-r9 stable transfection, clone selection and screening were as described [26]. Four clones were obtained, and one of them was used in the present study.
Cell culture
Cells were plated (105 cells per well) into 24-well plates and cultured under standard cell culture conditions [9]. After 2–3 days, doxycycline (Sigma Chemical, St Louis, MO, USA) was added for 48 h to induce UCP2 production. Cells were incubated in the same medium at 5 mmol/l glucose without or with doxycycline 12 h before experiments.
Insulin secretion
Cells were washed, preincubated for 60 min in KRBH containing 2.8 mmol/l glucose and 0.5% BSA and incubated for 30 min with the additions mentioned, and the insulin concentration in the medium was determined. Total cellular insulin content, measured after cold acid–ethanol extraction, and insulin secretion were quantified by radioimmunoassay. DNA was measured according to Labarca and Paigen [27].
ATP/ADP and cytosolic ATP measurements
The cells were treated as for insulin secretion, except that 10 min after glucose addition the medium was rapidly removed and 0.75 ml of 0.6 mol/l perchloric acid was added to the cells. The extract was processed and ATP and ADP were measured as described previously [9].
Cytosolic ATP levels were monitored in cells infected with the ATP-sensitive luminescent probe luciferase [28]. Twenty-four hours after the induction of UCP2 production, the cells were infected with the specific AdRIP-Luc viral construct. Twenty-four hours later, cytosolic ATP was measured after the addition of 250 μmol/l beetle luciferin (AcrosOrganics, Geel, Belgium), the luciferase substrate, as described [28].
Western blotting
Mitochondria and cytosolic fractions were prepared from isolated islets and cultured cells using the Mitochondria/Cytosol fractionation kit (BioVision, Mountain View, CA, USA) according to the manufacturer’s instructions. Proteins were measured using the Bio-Rad protein assay (Bio-Rad Laboratories GmbH, München, Germany), with BSA as standard.
One hundred islets of similar size, obtained from one mouse, or the mitochondrial and cytosolic fractions of 100 islets, or 15 μg of mitochondrial proteins from INS cells, were subjected to electrophoresis on a 12% polyacrylamide gel, electrotransferred to Immobilon P membranes (Millipore Corporation, Bedford, MA, USA), blocked with 2.5% Top Block (VWR International AG, Luzern, Switzerland) in phosphate-buffered saline and 0.1% Tween, and incubated overnight at 4°C in the same buffer containing polyclonal antibody to UCP2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, the membranes were incubated with a horseradish peroxidase-coupled IgG antibody (Amersham Biosciences, Little Chalfont, UK) for 1 h at room temperature. Cytochrome oxidase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were detected on the same membrane, using a monoclonal antibody against subunit IV of cytochrome oxidase (Molecular Probes Inc. Eugene, OR, USA) or GAPDH (Chemicon International, Temecula, CA, USA), and horseradish peroxidase-coupled anti-mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA) as secondary antibody. The resolved bands were visualised by enhanced chemiluminescence detection reagents (ECL; Amersham) and exposed to Hyperfilm ECL (Amersham, UK) for 1–3 min.
Mitochondrial membrane potential and ROS formation
Mitochondrial membrane potential was measured using rhodamine 123 (Molecular Probes). Cells in 24-well plates were preincubated for 2 h in KRBH containing 1 mmol/l glucose, loaded with 10 μg/ml rhodamine 123 for 20 min, washed, and transferred to a thermostated plate reader (Fluostar Optima, BMG Labtechnologies, Offenburg, Germany). The mitochondrial membrane potential (Δψ m) was monitored with an excitation and emission set of 485 and 525 nm, respectively. Glucose 12 mmol/l and carbonylcyanide m-chlorophenylhydrazone (CCCP) 30 μmol/l were added successively as described [28].
Production of ROS was measured with the ROS-sensitive fluorescent dye CM-H2DCFDA (Molecular Probes) after addition of recombinant human IL1β (PeproTech, Rocky Hill, NJ, USA). Cells were washed and incubated in RPMI without indicator, containing 5 μmol/l CM-H2DCFDA, for 20 min and transferred to the thermostated plate reader. ROS production was monitored during 30 min with an excitation and emission set of 485 and 525 nm, respectively. ROS production was linear during the 30 min of measurement. The slope of the curve was taken as an index of ROS production and expressed as arbitrary units/min.
Oxygen consumption
Cells were detached by incubation with PBS, washed, resuspended in KRBH medium containing 10% FCS and 5 mmol/l glucose, and incubated for 15 min at 37°C. After centrifugation, they were resuspended in the same medium containing 1% FCS and transferred into the stirred thermostated cuvette of a Clark-type electrode. Oxygen consumption was measured at 12 mmol/l glucose, and 10 μmol/l oligomycin and 30 μmol/l CCCP (Sigma Chemical) were added successively. The results were standardised to DNA concentration.
Glucose oxidation measurement
Cells were detached and preincubated with KRBH containing 2.8 mmol/l glucose and 0.5% BSA for 60 min. Cells in suspension were incubated with 2.8 or 12 mmol/l glucose and radiolabelled glucose for 60 min. Glucose oxidation was measured as 14CO2 production from [U-14C]glucose (0.1 μCi/μmol) (Amersham). Carbon dioxide was trapped and measured as described [9].
Statistical analysis
Results are expressed as mean ± SE. ANOVA and two-tailed paired or unpaired t tests or the Mann–Whitney test were used for statistical analysis.
Results
Transgenic mice with beta cell specific overexpression of UCP2
Three independent founders with the RIP-hUCP2 transgene were obtained. Two of them were backcrossed for nine or ten generations on a C57Bl/6J background and one was used in the present study, but similar results (shown in the Electronic supplementary material) were obtained in the offspring of the other founder, suggesting that the phenotype was independent of the site of integration of the DNA in the genome. Transgenic mice had a sixfold increase in UCP2 production in pancreatic islets (Fig. 1a,b). Figure 1a shows that UCP2 was found exclusively in the mitochondria. No significant differences were found between control and UCP2-overproducing mice for body weight fed and fasting glycaemia, insulinaemia, and insulin/glucose ratio. Plasma NEFA were slightly increased in fed but not in fasted transgenic mice (Table 1). Pancreatic insulin content and islet ATP and ADP contents were not significantly different between the two groups (Table 1).

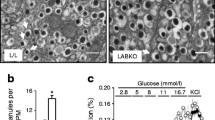
Levels of UCP2 protein in pancreatic islets and INS-1 cells. a UCP2 levels in pancreatic islets of control and RIP-UCP2 mice and in the cytosolic and mitochondrial fraction of islets from transgenic mice. Whole islets or cytosolic and mitochondrial proteins were separated by SDS-PAGE and UCP2 and GAPDH were measured by western blotting as described in the Materials and methods section. b UCP2 was quantified by densitometric scanning and normalised to the level of GAPDH. Mean ± SE of four independent measurements; p < 0.005 between control and RIP-UCP2. c Concentration-dependent effect of doxycycline (Doxy) on the level of UCP2 in isolated mitochondria of INS-1 cells. For comparison, 15, 7.5, 3.75 and 1.9 ng of recombinant UCP2 (Rec) are shown. d UCP2 was quantified by densitometric scanning and normalised to the level of cytochrome oxidase (cyt ox). Mean ± SE of three measurements; p < 0.005 between mitochondria from non-induced cells and cells induced with 250 ng/ml doxycycline
Transgenic mice with beta-cell-specific overproduction of UCP2 had normal glucose tolerance and glucose-stimulated insulin secretion. Intraperitoneal GTT was performed in overnight-fasted control and transgenic mice. Figure 2a and b shows no significant difference in the glucose profile between the two groups, in insulin levels at 0 and 15 min, or in change in insulin concentration. To further analyse the possible role of UCP2 overproduction in GSIS, pancreases from control and transgenic mice were perfused with glucose and tolbutamide. As shown in Fig. 3, increased levels of UCP2 did not alter the insulin secretion profiles in response to glucose and tolbutamide, while basal insulin secretion was slightly decreased in pancreases from transgenic mice. This decrease did not reach statistical significance, even when using the nonparametric Mann–Whitney test.

An increase in UCP2 protein concentration in pancreatic beta cells does not affect glucose tolerance. Glucose (1.5 g/kg body weight) was injected i.p. in overnight-fasted 12- to 14-week-old control (open squares and bars) and RIP-UCP2 transgenic mice (filled squares and bars). a Plasma glucose measured at 0, 15, 30, 60 and 120 min. b Plasma insulin measured at 0 and 15 min and change in insulin concentration (Δ). Results are mean ± SE of nine animals per group

An increase in UCP2 protein concentration does not affect insulin secretion by the perfused pancreas. Insulin release in response to glucose (Glu 2.8, 7 and 16.7 mmol/l) and tolbutamide (100 μmol/l) was measured in perfused pancreas from control (open squares) (n = 6) and RIP-UCP2 (filled squares) (n = 4) mice, as described in the Materials and methods section
The possibility that UCP2 has to be activated by fatty acids to exhibit uncoupling properties was investigated in isolated islets from control and transgenic mice by addition of 0.4 mmol/l BSA–oleate (Fig. 4a). Basal and GSIS were similar in the two groups. As demonstrated by others, short-term exposure of islets to oleate greatly amplifies GSIS (Fig. 4a). Similar amplification was measured in islets from control and transgenic mice, indicating that fatty acids did not activate UCP2 or that the potent effect of fatty acids on the fatty acid receptor GPR 40 [29] or of fatty acyl CoA on the secretory process [30] overcame possible changes in glucose-induced ATP production.

Glucose-stimulated insulin secretion and changes in ATP/ADP ratio are not affected by increased UCP2 levels. a Insulin secretion was measured in response to glucose (Glu 4, 8, and 16 mmol/l), without or with 0.4 mmol/l oleate (ol), and to 30 mmol/l KCl, in batch of three isolated islets from control (open bars) and RIP-UCP2 (filled bars) mice. Results are mean ± SE of four to six measurements. b ATP/ADP ratio was measured in isolated islets 30 min after exposure to 2 mmol/l (open bars) and 20 mmol/l (filled bars) glucose, as described in the Materials and methods section. Results are mean ± SE of seven or eight measurements representing four or five mice in each group. *p < 0.05 compared with glucose 2 mmol/l
An increase in mitochondrial uncoupling is also expected to result in a decreased ATP/ADP ratio. Figure 4b shows that the ATP/ADP ratios are comparable in control and transgenic islets at low glucose concentration, and increase similarly after glucose addition.
Inducible overproduction of UCP2 in INS-1 cells
To further study the effects of increased levels of UCP2, the beta cell clone INS1-r9, stably expressing the reverse tetracycline-dependent transactivator, was transfected with the pTRE plasmid containing UCP2 cDNA. Increasing the concentration of doxycycline from 50 to 1,000 ng/ml produced a two- to 15-fold increase in UCP2 protein levels (Fig. 1c). Addition of 250 ng/ml doxycycline produced an increase in UCP2 production similar to that observed in the islets of transgenic mice (Fig. 1d). This concentration was used in subsequent experiments.
This increase in UCP2 protein levels did not affect cell proliferation, as measured by DNA after 3 and 6 days of saline or doxycycline addition (data not shown).
Overproduction of UCP2 in INS-1 cells does not alter glucose-induced insulin secretion and ATP formation
Insulin secretion was measured in response to secretagogues acting via an increase in ATP (glucose, leucine) or in a manner independent of ATP generation (tolbutamide). Increasing UCP2 did not alter insulin secretion in response to any of the stimulants (Fig. 5a). Higher levels of UCP2 (more than 12-fold increase) resulted in a 27% decrease in glucose-stimulated insulin secretion, but also a 27 and 19% decrease in secretion stimulated by tolbutamide and KCl respectively (p < 0.01 by paired t test, five independent experiments), suggesting a non-specific effect.

Glucose-induced insulin secretion and changes in ATP/ADP ratio and cytosolic ATP are not affected by high levels of UCP2 in INS-1 cells. a Insulin secretion (expressed as percent insulin content) was measured in response to glucose (Glu 4, and 12 mmol/l), 10 mmol/l leucine and 100 μmol/l tolbutamide (Tolb) in control (open bars) and UCP2-overproducing cells (filled bars). The insulin content was similar in the two groups: 25.0 ± 2.4 and 25.1 ± 2.6 ng/μg DNA for control and UCP2-overproducing cells, respectively. b ATP/ADP ratios were measured before (open bars) and 10 min after addition of 12 mmol/l glucose (final concentration, cross-hatched bars) as described in the Materials and methods section. All results are mean ± SE of four independent experiments performed in triplicate. c Cytosolic ATP levels were monitored in control cells (open squares and circles) and in cells overproducing UCP2 (filled squares and circles). After 8 min in the baseline condition, cells were unstimulated (squares) or stimulated with 12 mmol/l glucose for 10 min (circles), then ATP generation was suppressed by the addition of 2 mmol/l NaN3. Values are mean ± SE of two representative experiments performed in quadruplicate
Increasing UCP2 did not modify basal and glucose-stimulated changes in ATP/ADP ratio (Fig. 5b), nor did it modify the absolute values of ATP (200 ± 19 and 189 ± 21 pmol/μg DNA, not significant) and ADP (27 ± 4 and 25 ± 2 pmol/μg DNA, not significant) in control and UCP2-overproducing cells, respectively. The glucose-induced increase in cytosolic ATP, which is more directly related to closure of the KATP channels, was increased similarly in control and UCP2-overproducing cells (Fig. 5c).
Increasing UCP2 does not affect oxygen consumption, glucose oxidation and mitochondrial membrane potential \( {\left( {\Delta \Psi _{\mu } } \right)} \), meaning that no uncoupling activity is induced
Oxygen consumption and glucose oxidation were measured to test whether increased metabolism may mask putative uncoupling activity. Control and UCP2-overproducing cells showed no difference in oxygen consumption (Table 2). The rate of oxygen consumption remaining after addition of an inhibitor of ATP synthase represents proton leak by the mitochondria, and is expected to increase with mitochondrial uncoupling. Table 2 shows that the addition of oligomycin decreased oxygen consumption to the same extent in control and UCP2-overproducing cells. The two groups increased their O2 consumption equally after addition of the uncoupler CCCP.
Basal glucose oxidation was similar in the control and UCP2-overproducing cells (0.126 ± 0.010 and 0.131 ± 0.020 nmol glucose/μg DNAxh for control and UCP2-overproducing cells respectively, not significant) and increased similarly when raising the glucose concentration to 12 mmol/l (0.314 ± 0.022 and 0.320 ± 0.030 nmol glucose/μg DNA per hour). To determine whether increased levels of UCP2 would induce changes in \( {\left( {\Delta \Psi _{\mu } } \right)} \), the latter was measured before and after glucose addition. The hyperpolarisation induced by high glucose concentration was similar in the two groups, as was the depolarisation induced by 30 μmol/l CCCP (Fig. 6). Taken together with the oxygen consumption and glucose oxidation results, this last observation indicates that increasing UCP2 level does not result in any sign of mitochondrial uncoupling.

The mitochondrial membrane potential is not modified by UCP2 overproduction in INS-1 cells. Mitochondrial membrane potential was monitored in control cells (squares) and in cells overproducing UCP2 (triangles), loaded with rhodamine 123, and left without stimulation (closed symbols) or stimulated with glucose (open symbols) and CCCP added where indicated. The results are means of quadruplicate measurements from one representative experiment performed four times. Insert shows detail of glucose-induced hyperpolarisation of control and UCP2-overproducing cells. The SEs have been omitted for clarity (they were between 1.5 and 5%)
Increasing UCP2 levels decrease ROS production
Several studies have suggested that UCPs may decrease ROS generation, in particular when the production of free radicals is increased and ‘activates’ the uncoupling protein. Increased ROS production has been described in pancreatic beta cells after exposure to high glucose concentrations [31, 32]. However, our control cells did not exhibit any increase in ROS generation after short (60 min) or long (24 h) exposure to 20 mmol/l glucose (data not shown). Mitochondrial ROS production in the pancreatic beta cell is also known to increase after administration of cytokines [33]. For this reason, the possibility that increased expression of UCP2 may act on IL1β-induced ROS production was tested. Control and UCP2-overproducing cells were exposed to various concentrations of IL1β for 5 h and ROS generation was measured. Increasing UCP2 levels resulted in a significant decrease in IL1β-induced ROS production (Fig. 7).

Cytokine-induced ROS production is decreased by UCP2 overproduction. ROS production was assessed in control (open bars) and UCP2-overproducing cells (filled bars) using the fluorescent marker CM-H2-DCFDA. ROS production was measured 5 h after addition of 0, 62, 125 or 250 pg/ml IL1β, as described in the Materials and methods section. Mean ± SE of triplicate measurements in one representative experiment performed three times. AU arbitrary units. *p < 0.05 compared with control cells
Discussion
The aim of the present study was to investigate whether an increase in UCP2 in beta cells could per se induce mitochondrial uncoupling and alter glucose-induced changes in ATP/ADP ratio and insulin secretion. For the first time, transgenic mice with pancreatic beta-cell-specific increase in UCP2 protein were generated. These mice had no particular phenotype; their plasma insulin and glucose levels were not significantly modified in the fed or in the fasted states or during a GTT. Glucose-induced ATP generation and insulin secretion were comparable in control and transgenic mice, suggesting that an increase in UCP2 per se is not sufficient to uncouple beta cells and decrease ATP production and insulin secretion.
Our results in transgenic mice are in contrast to those obtained in UCP2−/− mice, which exhibit a clear increase in basal and glucose-induced ATP level and insulin secretion [19]. The difference in ATP/ADP ratio and mitochondrial membrane potential measured in islets of wild-type and UCP2−/− mice implies that even in the basal state, i.e. in the absence of activator, UCP2 is active and promotes proton leak in control mice. This is in contrast with a general consensus that, in a cell, a UCP has an uncoupling activity only in the presence of specific activators [34].
It is difficult to explain why UCP2 ablation has such important consequences for insulin secretion, whereas its increase has no effect. One important feature of overall ablation of UCP2 in mice is a slightly elevated plasma level of inflammatory cytokines such as IFN-γ and TNF-α and an increased generation of nitric oxide and cytokines upon antigenic challenge [35]. One would expect that this constant low inflammatory state reduces beta cell viability. However, recent data obtained in rat beta cells demonstrate that a transient and moderate increase in NF-κB (nuclear factor-κB) activity improves beta cell function and insulin secretion [36]. Furthermore, attenuation of NF-κB activation in transgenic mice impairs GSIS and decreases the expression of genes involved in glucose metabolism and insulin exocytosis [37]. Taken together, these data indicate that moderate activity of NF-κB could be beneficial to the beta cell. An alternative explanation for the lack of phenotype of mice with a beta cell specific increase in UCP2 could be that the ‘basal’ level of UCP2 present in wild-type mice may be sufficient to trigger a full effect of the protein. Indeed, in Fig. 7 in [20], overproduction of UCP2 in pancreatic islets of UCP2−/− mice resulted in a level of UCP2 above that observed in wild-type mice, but GSIS was similar to that measured in islets from wild-type mice, suggesting that the excess UCP2 had no further effect compared with the control situation. Alternatively, the genetic background could play a role, although the studies using UCP2-/- mice, as the present study, used C57Bl/6J mice [38].
The consequences of increased levels of UCP2 in the beta cell line INS-1 confirmed and extended the results obtained in islets. Increased levels of UCP2 similar to those measured in transgenic mice did not alter GSIS or induce any sign of uncoupling. The data are in agreement with the observation that the threefold to fourfold increase in UCP2 measured in INS-1E cells with high levels of PPARα and RXR did not modify \( \Delta \Psi _{\mu } \) or GSIS [16], and with the notion that UCPs have to be activated to exhibit uncoupling activities [34]. The possibility remains, however, that increasing expression of UCP2 induced slight changes in mitochondrial potential that cannot be detected by the methods used.
In previous studies, the role of UCP2 in pancreatic beta cells was mostly studied after supraphysiological production of the protein. The consequences of such high levels of UCP2 may, however, reflect an uncoupling artefact caused by disruption of the mitochondrial membrane integrity, and not the physiological role of the protein [3]. In the present study, overproduction of UCP2 by more than tenfold also decreased insulin secretion. Indeed, there is now evidence in several cell types that a moderate increase in UCP2 [34, 39] or UCP3 [40, 41] levels does not induce any sign of mitochondrial uncoupling. The results of the present study demonstrate that this could also be the case for pancreatic beta cells. This lack of effect could be due to the absence or limited levels of an endogenous activator or suggest that UCP2 has no role in proton leak.
As an increase in UCP2 did not uncouple the beta cell or modify GSIS, the possible role of the protein in ROS production was examined after short or long exposure to high glucose concentrations. No increase in ROS generation was measured in either condition. These data are in agreement with recent investigations demonstrating that high glucose concentrations do not increase but rather decrease ROS generation in rat pancreatic beta cells [42]. Mitochondrial ROS generation has also been reported in beta cells after cytokine exposure [33]. Our results show that increasing UCP2 protein levels promoted a decrease in IL1-β-induced ROS production. These data can be related to observations in endothelial cells after high glucose or angiotensin II addition [43], or cardiomyocytes exposed to oxidative stress [23], in which an increase in UCP2 prevented ROS generation. The mechanism of action of UCP2 and the exact nature of the ROS induced by cytokines is presently being investigated. In addition to preventing ROS production, increasing UCP2 also attenuated cytokine-induced cell death [44].
In conclusion, the results of the present study demonstrate, in two independent models, that an increase in UCP2 protein per se does not induce any sign of mitochondrial uncoupling or alteration in glucose-induced mitochondrial hyperpolarisation, ATP/ADP ratio and insulin secretion. In contrast, increases in UCP2 protein decreased cytokine-induced ROS generation, an observation in agreement with the role described for UCP2 in other cell types [23, 43]. These data indicate that the protective effect of UCP2 on ROS production may overcome its potential deleterious effects on glucose-stimulated insulin secretion.
Abbreviations
- CCCP:
-
carbonylcyanide m-chlorophenylhydrazone
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- GSIS:
-
glucose-stimulated insulin secretion
- GTT:
-
glucose tolerance test
- PPAR:
-
peroxisome proliferator-activated receptor
- UCP2:
-
uncoupling protein-2
References
Henquin JC (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Argyropoulos G, Harper ME (2002) Uncoupling proteins and thermoregulation. J Appl Physiol 92:2187–2198
Stuart JA, Harper JA, Brindle KM, Jekabsons MB, Brand MD (2001) Physiological levels of mammalian uncoupling protein 2 do not uncouple yeast mitochondria. J Biol Chem 276:18633–18639
Negre-Salvayre A, Hirtz C, Carrera G et al (1997) A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11:809–815
Arsenijevic D, Onuma H, Pecqueur C et al (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26:435–439
Chan CB, MacDonald PE, Saleh MC, Johns DC, Marban E, Wheeler MB (1999) Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes 48:1482–1486
Chan CB, De Leo D, Joseph JW et al (2001) Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes 50:1302–1310
Hong Y, Fink BD, Dillon JS, Sivitz WI (2001) Effects of adenoviral overexpression of uncoupling protein-2 and -3 on mitochondrial respiration in insulinoma cells. Endocrinology 142:249–256
Lameloise N, Muzzin P, Prentki M, Assimacopoulos-Jeannet F (2001) Uncoupling protein 2: a possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes 50:803–809
Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V (2002) Induction of uncoupling protein 2 mRNA in beta-cells is stimulated by oxidation of fatty acids but not by nutrient oversupply. Endocrinology 143:1371–1377
Medvedev AV, Robidoux J, Bai X et al (2002) Regulation of the uncoupling protein-2 gene in INS-1 beta-cells by oleic acid. J Biol Chem 277:42639–42644
Joseph JW, Koshkin V, Zhang CY et al (2002) Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes 51:3211–3219
Brown JE, Thomas S, Digby JE, Dunmore SJ (2002) Glucose induces and leptin decreases expression of uncoupling protein-2 mRNA in human islets. FEBS Lett 513:189–192
Tordjman K, Standley KN, Bernal-Mizrachi C et al (2002) PPARalpha suppresses insulin secretion and induces UCP2 in insulinoma cells. J Lipid Res 43:936–943
Armstrong MB, Towle HC (2001) Polyunsaturated fatty acids stimulate hepatic UCP-2 expression via a PPARalpha-mediated pathway. Am J Physiol Endocrinol Metab 281:E1197–E1204
Ravnskjaer K, Boergesen M, Rubi B et al (2005) Peroxisome proliferator-activated receptor alpha (PPARalpha) potentiates, whereas PPARgamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology 146:3266–3276
Ito E, Ozawa S, Takahashi K et al (2004) PPAR-gamma overexpression selectively suppresses insulin secretory capacity in isolated pancreatic islets through induction of UCP-2 protein. Biochem Biophys Res Commun 324:810–814
Patane G, Anello M, Piro S, Vigneri R, Purrello F, Rabuazzo AM (2002) Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-gamma inhibition. Diabetes 51:2749–2756
Zhang CY, Baffy G, Perret P et al (2001) Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105:745–755
Joseph JW, Koshkin V, Saleh MC et al (2004) Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem 279:51049–51056
Echtay KS, Roussel D, St-Pierre J et al (2002) Superoxide activates mitochondrial uncoupling proteins. Nature 415:96–99
Mattiasson G, Shamloo M, Gido G et al (2003) Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 9:1062–1068
Teshima Y, Akao M, Jones SP, Marban E (2003) Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res 93:192–200
Maechler P, Gjinovci A, Wollheim CB (2002) Implication of glutamate in the kinetics of insulin secretion in rat and mouse perfused pancreas. Diabetes 51(Suppl 1):S99–S102
Schultz V, Sussman I, Bokvist K, Tornheim K (1993) Bioluminometric assay of ADP and ATP at high ATP/ADP ratios: assay of ADP after enzymatic removal of ATP. Anal Biochem 215:302–304
Wang H, Iynedjian PB (1997) Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc Natl Acad Sci USA 94:4372–4377
Labarca C, Paigen K (1980) A simple, rapid, and sensitive DNA assay procedure. Anal Biochem 102:344–352
Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145:667–678
Itoh Y, Kawamata Y, Harada M et al (2003) Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422:173–176
Yaney GC, Korchak HM, Corkey BE (2000) Long-chain acyl CoA regulation of protein kinase C and fatty acid potentiation of glucose-stimulated insulin secretion in clonal beta-cells. Endocrinology 141:1989–1998
Robertson RP, Harmon J, Tran PO, Poitout V (2004) Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 53(Suppl 1):S119–S124
Kilpatrick ED, Robertson RP (1998) Differentiation between glucose-induced desensitization of insulin secretion and beta-cell exhaustion in the HIT-T15 cell line. Diabetes 47:606–611
Lortz S, Tiedge M, Nachtwey T, Karlsen AE, Nerup J, Lenzen S (2000) Protection of insulin-producing RINm5F cells against cytokine-mediated toxicity through overexpression of antioxidant enzymes. Diabetes 49:1123–1130
Nedergaard J, Ricquier D, Kozak LP (2005) Uncoupling proteins: current status and therapeutic prospects. EMBO Rep 6:917–921
Bai Y, Onuma H, Bai X et al (2005) Persistent nuclear factor-kappa B activation in Ucp2−/− mice leads to enhanced nitric oxide and inflammatory cytokine production. J Biol Chem 280:19062–19069
Hammar EB, Irminger JC, Rickenbach K et al (2005) Activation of NF-kappaB by extracellular matrix is involved in spreading and glucose-stimulated insulin secretion of pancreatic beta cells. J Biol Chem 280:30630–30637
Norlin S, Ahlgren U, Edlund H (2005) Nuclear factor-κB activity in β cells is required for glucose-stimulated insulin secretion. Diabetes 54:125–132
Freeman H, Shimomura K, Horner E, Cox RD, Ashcroft FM (2006) Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metab 3:35–45
Pecqueur C, Alves-Guerra MC, Gelly C et al (2001) Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 276:8705–8712
MacLellan JD, Gerrits MF, Gowing A, Smith PJ, Wheeler MB, Harper ME (2005) Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes 54:2343–2350
Mozo J, Ferry G, Studeny A et al (2006) Expression of UCP3 in CHO cells does not cause uncoupling, but controls mitochondrial activity in the presence of glucose. Biochem J 393:431–439
Martens GA, Cai Y, Hinke S, Stange G, Van de Casteele M, Pipeleers D (2005) Glucose suppresses superoxide generation in metabolically responsive pancreatic beta cells. J Biol Chem 280:20389–20396
Park JY, Park KG, Kim HJ et al (2005) The effects of the overexpression of recombinant uncoupling protein 2 on proliferation, migration and plasminogen activator inhibitor 1 expression in human vascular smooth muscle cells. Diabetologia 48:1022–1028
Zengaffinen N, Perreten H, Lameloise N, Keller P, Muzzin P, Assimacopoulos-Jeannet F (2005) Uncoupling protein 2 overexpression prevents cytokine-induced reactive oxygen species production and apoptosis in pancreatic beta cells. Diabetologia 48(Suppl 1):A38
Acknowledgements
The excellent technical assistance of F. Califano is greatly acknowledged. We thank P. Iynedjian and H. Wang for the gift of INS-1 r9 cells and helpful advice, P. Maechler for the gift of the AdRIP-Luc viral construct and very helpful discussions, and J. Pakay for the gift of recombinant UCP2. This study was supported by grant 32.63624.00 from the Swiss National Science Foundation and by a grant from the De Reuter Foundation to F.A-J. P.Herrera is supported by grants from the Swiss National Science Foundation, the JDRF1 and the NIH/NIDDK. The project was part of the Geneva Program for Metabolic Disorders.
Author information
Authors and Affiliations
Corresponding author
Additional information
N. Produit-Zengaffinen and N. Davis-Lameloise contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table 1
Plasma and pancreatic parameters of control and RIP-UCP2 transgenic mice (L233)(doc 33 kb)
Figure 1
Fig 1
Increased levels of UCP2 in pancreatic beta cells do not affect glucose tolerance and insulin secretion in the second mouse line generated (L233). a Level of UCP2 in pancreatic islets of control and transgenic mice. UCP2 and GAPDH levels were determined by Western blotting in islets from wild-type (WT) mice and from the two lines of transgenic mice (L233 and L261) as described in the Materials and methods section. b Glucose tolerance test in control and transgenic mice. Glucose (1.0 mg/g body weight) was injected i.p. in overnight fasted 12- to 14-week-old control (open squares) and RIP-UCP2 (L233) mice (black squares) and plasma glucose measured. Basal insulin levels were 58.5 ± 16.9 and 68.8 ± 15.5 pmol/l for control and transgenic mice, respectively, and 110.1 ± 25.8 and 120.4 ± 15.5 pmol/l 15 min after glucose administration. Results are mean ± SE for four animals per group. c Insulin secretion by isolated pancreatic islets of control (open bars) and transgenic mice (black bars). Insulin release in response to 4, 8 and 16 mmol/l glucose or glucose + oleate (ol) 0.4 mmol/l was measured in isolated islets from control and RIP-UCP2 (L233) mice. Results are mean ± SE of four to five experiments (doc 98 kb)
Rights and permissions
About this article
Cite this article
Produit-Zengaffinen, N., Davis-Lameloise, N., Perreten, H. et al. Increasing uncoupling protein-2 in pancreatic beta cells does not alter glucose-induced insulin secretion but decreases production of reactive oxygen species. Diabetologia 50, 84–93 (2007). https://doi.org/10.1007/s00125-006-0499-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0499-6