
Abstract
Mitotic catastrophe (MC) has long been considered as a mode of cell death that results from premature or inappropriate entry of cells into mitosis and can be caused by chemical or physical stresses. Whereas it initially was depicted as the main form of cell death induced by ionizing radiation, it is today known to be triggered also by treatment with agents influencing the stability of microtubule, various anticancer drugs and mitotic failure caused by defective cell cycle checkpoints. Although various descriptions explaining MC exist, there is still no general accepted definition of this phenomenon. Here, we present evidences indicating that death-associated MC is not a separate mode of cell death, rather a process (‘prestage’) preceding cell death, which can occur through necrosis or apoptosis. The final outcome of MC depends on the molecular profile of the cell.
Similar content being viewed by others


Historical Remarks
During mitosis, proliferating cells undergo several structural and molecular changes, characterized by chromatin condensation, spindle formation, nuclear envelope fragmentation and cytoskeleton reorganization.1 Chromosome segregation is carried out by a complex machinery – the mitotic spindle – that is based on a bipolar array of microtubules. Microtubules are highly dynamic polymers that continuously grow and shrink, and in the spindle, this behavior is regulated by proteins that bind to the sides or ends of microtubules.2 The rate of microtubule disassembly, from a state of polymerization to depolymerization, is designated as the catastrophe rate.3, 4 The process of microtubule disintegration was described in 1984 as a ‘microtubule catastrophe’.5 Thereby, the first link between two words – catastrophe and mitosis – became apparent. However, the expression ‘mitotic catastrophe’ (MC) was not utilized until 1986, when it was used in an attempt to illustrate the phenotype of a yeast strain raised from combining an activated cdc2 allele (cdc2-3w) with a recessive temperature-sensitive weel mutation (wee 1–50).6 These cells were not arrested in the cell cycle; instead, they entered mitosis prematurely without completing it effectively. The lethal phenotype of this yeast strain was not due to premature entry into mitosis, it rather seemed to be an aberrant execution of mitosis, particularly with respect to chromosome segregation and septum formation.7 Nowadays, the term ‘mitotic catastrophe’ is used to explain a mechanism of a delayed mitotic-linked cell death, a sequence of events that results from premature or inappropriate entry of cells into mitosis that can be caused by chemical or physical stresses. It can be triggered with agents influencing the stability of microtubule, various anticancer drugs and mitotic failure caused by defective cell cycle checkpoints.8 MC was also depicted as the main form of cell death induced by ionizing radiation.
The first observations of MC were made in the late 1930s and early 1940s when cells in exponential growth phase were exposed to radiation.9, 10 It was noticed that the fraction of cells in the mitotic stage instantly declined in response to radiation and did not reappear until several hours following treatment. Microscopy examinations revealed that cells began to die at or after the first post-irradiation mitotic peak (reappearance of mitotic cells)11 and were shown to display abnormal configurations and spatial rearrangements of chromosomes. Accordingly, this death was called mitotic or division death, which emphasized the association with mitosis and cell division, or delayed death, which highlighted the appearance of dead cells several hours after irradiation.
The Current Concept of MC
Mitotic catastrophe has been described as an aberrant form of mitosis associated with various morphological and biochemical changes (see below). The final step of MC is almost always characterized by the formation of nuclear envelopes around individual clusters of missegregated chromosomes. MC is also correlated with incomplete DNA synthesis and premature chromosome condensation (PCC).12, 13 Initially, PCC was not linked to mitotic events, as the appearance of multinucleated cells with ‘pulverized’ chromosomes was observed when interphase and mitotic cells were fused using Sendai virus. The interphase chromatin was in this case forced to condense into discrete units known as prematurely condensed chromosomes.14, 15 Later, another type of PCC that occurred in only a fraction of metaphase chromosomes was described. This metaphase-associated PCC, also called spontaneous PCC, appeared in cells possessing micronuclei as a result of radiation-induced incomplete DNA synthesis.16, 17 Interestingly, the well-defined mode of cell death – apoptosis – is also characterized by chromatin condensation; however, the morphology of apoptosis is distinguished from MC and illustrated by cytoplasm shrinkage and nuclear fragmentation. In heated HeLa cells, the appearance of spontaneous PCC is followed by asymmetric segregation of chromatid clusters and premature reformation of the nuclear membrane. Then, instead of nuclear fragmentation, cells are characterized by an accumulation of multiple micronuclei that, subsequently, exhibit features similar to necrotic cell death.18
Mitotic catastrophe has also been described as a delayed form of reproductive death based on observations that the multinucleated giant cells can be temporarily viable.19, 20 The term ‘reproductive death’ denotes the loss of the ability of a cell to generate viable progeny that reproduced continuously.21 As most of the cells undergoing MC eventually die, cellular processes that lead to irreversible growth arrest and those that are termed ‘reproductive death’ may better fit to conditions known as senescence.22 Senescent cells are generally characterized by a reduction in proliferative capacity, adoption of a flattened and enlarged cell shape and an increase of β-galactosidase (SA-β-gal) activity.23 Although senescence was associated with MC,24, 25 it has been shown that the polyploid giant cells expressing senescent marker (SA-β-gal activity) may overcome the state of growth arrest and even undergo de-polyploidization.26
Based on observations that abnormalities in the cell cycle and mitosis eventually lead to cell death, MC has been defined in morphological terms as a mechanism of cell death occurring during or after aberrant mitosis.8 Alternatively, MC has been classified not as a mode of cell death but as a special example of apoptosis. This classification is based on the observation that MC shares several biochemical hallmarks with apoptosis, namely mitochondrial membrane permeabilization and caspase activation.27, 28 However, it is still unclear whether MC results in death that requires caspase-dependent or caspase-independent mechanisms.29 In fact, arguments have raised the more general question of whether MC is a mode of cell death or whether this is a process that leads to apoptosis or necrosis.30, 31, 32, 33 In addition, there are several examples that permit to define MC as a cell survival mechanism of tumors.34 Moreover, in some cases, MC may represent a process through which cells switch from an abnormal to mitotic cell cycle.35 Despite, or maybe owing to, these various definitions at present, there is no generally accepted classification of MC. Here, we present evidence indicating that death-associated MC is a process (‘prestage’) preceding necrosis or apoptosis (Figure 1).

The summary of current concepts of MC (for details see the text)
DNA Damage-Induced MC
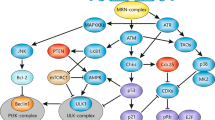
It is well known that tumors differ in their sensitivity to treatment. Sensitive cells, in response to various chemotherapeutic agents, are dying in interphase, before entering mitosis. Even low doses of irradiation can induce interphase death of some tumors, especially of hematopoietic origin. In this case, morphology and biochemistry of dying cells resemble apoptosis. Cells that do not undergo death in interphase may become arrested in G1 and/or G2. If the cellular machinery is not able to repair the injury during the arrest, DNA damage can lead to different death-associated consequences. Alternatively, when repair processes are accomplished, cells reenter the cell cycle. Checkpoint regulations have evolved to coordinate the response to different types of DNA damage36 and is mediated through activation of two protein kinase pathways: the ATM (ataxia-telangiectasia-mutated) and the ATR (ataxia-telangiectasia and Rad3-related). The alert signal is modulated through the action of the so-called checkpoint mediators (adaptors) and further transduced through phosphorylation of effector kinases Chk1 and Chk2.37 Delays in G1, S or G2 phases are controlled by the ATM/ATR-Chk2/Chk1 genotoxic response and each cell cycle segment requires different downstream key substrates to cope with DNA replication and mitosis. The p53/MDM2-p21 conduit is dominant in G1/S arrest where the DNA damage is repaired before chromosome replication takes place. DNA synthesis is thereby blocked by silencing of G1/S promoting cyclin E/Cdk2. Independent of p53, a late G1 arrest may also occur by downregulation of Cdc25A. A similar Cdc25A degradation cascade controlled by ATM/ATR operates during S-phase to hold back DNA replication. The G2/M checkpoint reversibly halts the cell cycle progression before mitotic entry, preventing the segregation of damaged chromosomes.38 Mitotic phase entry is directed by the cyclin B/Cdk1 kinase and Cdc25C, which also is a critical target of checkpoint control.39 Induced expression of cell cycle inhibitors, such as the Cdk inhibitor p21 and the 14-3-3σ protein, by p53 and checkpoint mediator BRCA1 probably affects duration of the G2/M arrest. Cells with impaired or lost checkpoint functions are unable to maintain this arrest and enter mitosis prematurely in the presence of unrepaired DNA40, 41, 42, 43 (Table 1). The fate of such cells depends on multiple parameters and attempts to define possible outcomes are ongoing (Figure 2). For example, some tumor cells can die by apoptosis after one or even repeated mitotic cycles. In other circumstances, the premature mitosis may lead to MC. However, depending on the severity of DNA damage, cells can also exit mitosis to form 4N G1 cells, and after second arrest at G1, they die or survive. Importantly, the extent of cell death and loss of clonogenicity are correlated with the duration of growth arrest.44 Dying cells are characterized by multiple mitotic abnormalities, including multipolar meta- or anaphase, lagging telophase, random distribution of condensed chromosomes throughout the cells, and so on. Biochemical mechanisms responsible for these differences still require additional investigation. Crucially, without prematurely entering mitosis, a cell cannot undergo MC. Thus, the abrogation of G1 and/or G2 checkpoints is essential for MC (Figure 2). In both scenarios, the damage in the interphase is expanded within mitosis and activates the mitotic checkpoints. It is important to note that cells that are sensitive to apoptosis and die in interphase have very little or no possibility to display features of MC. There are several experimental models that have been utilized to clarify biochemical changes associated with MC, but most observations were made using cell lines with induced disturbances in key elements of checkpoint control. The inability of HCT116 colon carcinoma cells to sequester cyclin B and Cdc2 in the cytoplasm following doxorubicin-induced DNA damage is already a well-established MC model.45 However, more recent data indicate that Chk2 and not 14-3-3σ may be the main negative regulator of MC in this model.46, 47 Abrogation of the ATR-initiated checkpoint cascade mediated through Chk1 also directs cells into MC and has been analyzed with respect to specific Chk1 phosphorylations.48, 49 Other cell-cycle-regulating proteins whose normal function seems to inhibit entering mitosis are the checkpoint mediators 53BP1,50 Brca1,51 p7352 and p53.32 In the absence of p53, cells have the capability to activate alternative routes to halt the cell cycle. One of the proteins that may be involved in this process in addition to Chk1 is p38MAPK. Depletion of p38MAPK/MK2 only in p53-deficient cells leads to checkpoint deregulations and MC irrespective of the type of DNA damage induced.53 This two-hit model suggests that MC is the outcome of DNA damage only when multiple central checkpoint proteins are suppressed and other options for cell death and survival are minimized. This may also be true for heterogeneous and genetically instable clinical tumor samples exposed to radiation.54, 55, 56 Most experimental facts concerning the biology of MC are based on observations obtained using cells growing in monolayer, and even if these may serve well as models, extrapolation to three-dimensional in vivo structures should be done with great consideration.57 In fact, specific cell–cell and cell–matrix interactions may be as important as the genomic background for the MC response, although this point of view is frequently neglected.

Schematic illustration of the pathways leading from MC to cell death. Premature entry into mitosis as a consequence of abrogated G2/M arrest or adaptation in the presence of DNA damage or direct mitotic damage leads to arrest at the metaphase–anaphase transition due to spindle checkpoint and to catastrophic mitosis. During mitotic arrest, cells can die through caspase-dependent or caspase-independent apoptosis. MC cells can undergo endocycle and become polyploid. These cells can die by either necrosis or apoptosis. Cells being arrested at the metaphase–anaphase transition can escape mitosis through mitotic slippage and become tetraploid. Cells that cannot be arrested at the metaphase–anaphase transition due to defects in the spindle checkpoint also become tetraploid. These tetraploid cells either can arrest at G1 and die through p53-dependent apoptosis or do not arrest at G1 and enter S-phase (endoreplication) and die through necrosis
Defects in Mitosis Leading to MC
Mitotic catastrophe might be induced by drugs that directly affect progression through mitosis by disturbing the mitotic spindle formation and cell division. During mitosis, formation of the mitotic spindle is an essential step for accurate segregation of chromosomes. Before cytokinesis, chromosomes attain bipolar attachments from two opposing centrosomes in the metaphase and are pulled toward opposite poles at anaphase.58 Spindle poisons are mitotic inhibitors that influence microtubule dynamics by binding to tubulin dimers and prevent either their assembly (polymerization) or their disassembly (depolymerization). Microtubule-hyperpolymerizing agents include taxanes (paclitaxel, docetaxel), whereas vinca alkaloids (vinblastine, vincristin) act as depolymerizing agents.59 Changes in the microtubule dynamics induced by these agents lead to disturbances of the spindle formation. An aberrant mitotic spindle activates the mitotic checkpoint (spindle assembly checkpoint), where a ‘wait anaphase’ signal in the metaphase–anaphase transition is generated. Numerous proteins are involved in the signaling cascade that regulates mitotic cycle progression, and central in this process is suppression of the ubiquitin ligase activity mediated by the anaphase-promoting complex (APC).60 Defects in checkpoint signaling in the presence of mitotic failure may cause abortive centrosome duplication,61 and eventually multipolar mitosis and/or premature segregation of unaligned chromosomes, leading to an uneven delivery of genetic material into the cell offspring (aneuploidy).62, 63 Aneuploidy is a common characteristic of tumors and likely an explanation for tumor cell heterogeneity in vivo. However, MC, manifested in micronucleation, may occur in cells where mitosis completely fails, and may therefore precede formation of aneuploid cells (Figure 2). There are several examples indicating that deregulations in mitosis promote development of MC. Loss of the transcription factor FoxM1 affects genes that are essential for chromosome segregation and generates mitotic spindle defects and delays in mitosis, and induces MC.64 Survivin is a member of the chromosomal passenger complex implicated in kinetochore attachment, bipolar spindle formation and cytokinesis.65 Inhibition of survivin in glioma cells was followed by increased spontaneous apoptosis, MC and cell cycle arrest.66 In addition, it has been shown that, being the target for oxaliplatin, survivin downregulation leads to MC or apoptosis.67 Microtubule-associated proteins (MAPs), such as Op18, XKCM1 and MCAK, are certainly essential for the regulation of microtubule turnover, and alterations in the expression levels of these proteins generate defects in the mitotic spindle formation, including formation of monopolar, monoastral and small prometaphase-like spindles with poorly organized poles, as well as improperly attached chromosomes.68 MAP deregulations associate with mitotic arrest, and examples of MC-like conditions in selected cell types exist, both in vitro and in vivo.69 Yet, a general link between MAP function and MC is still not established (Table 1). Notably, MC in response to mitotic spindle perturbations or during experimental conditions, where mitotic deregulations have been introduced, depends on the genetic background of the cell (Figure 2). It is, therefore, tempting to speculate that cells that are prone to acquire an MC-like phenotype in response to spindle poisons or MAP defects also are defective with respect to mitotic checkpoint control.
Polo-like kinase 1 (PLK1) has been shown to play a critical role in the establishment of a bipolar spindle.70 A dominant-negative PLK1 selectively induced MC in HeLa cells. However, this was contrasted by an observed proapoptotic function of dominant-negative PLK1 in other tumor cells, although in non-tumor cell lines deregulated PLK1 induced mitotic arrest.71 Interestingly, depletion of hNuf2, a kinetochore protein involved in microtubule attachment, also arrests HeLa cells followed by mitotic cell death.72 Thus, nonproper assembly of mitotic spindle along with defects in the mitotic arrest is associated with initiation of MC. Although many biochemical details leading to MC remain to be clarified, the so-called centrosome inactivation checkpoint provides an interesting connection between DNA damage, MC and mitotic cell cycle progression.73
Checkpoint Adaptation and Mitotic Slippage
Checkpoint adaptation, first described in budding yeast, is the ability of cells to enter mitosis following a sustained checkpoint-imposed cell cycle arrest despite the presence of persistent DNA damage.74, 75 A checkpoint-regulated protein, claspin, mediates Chk1 activation when DNA replication is inhibited. After a prolonged G2/M arrest, claspin can dissociate from the chromatin and thereby inactivate Chk1, permitting the entry into mitosis.76 Cells that enter mitosis following adaptation are arrested in metaphase, which may represent another mechanism for the development of MC (Figure 2). Thus, adaptation might be served as an alternative elimination process of cells with unrepairable damage.77
Similar to adaptation, checkpoint activity can also be lost as a result of prolonged mitotic arrest. Through this mechanism, also known as mitotic slippage, cells with dearranged chromatin will exit mitosis and become tetraploid.78 Although many molecular details remain to be clarified, a central event in mitotic slippage seems to be proteosomal degradation of cyclin B.79 This protein drives cells into mitosis through its binding to Cdk1, where phosphorylation of lamins leads to disassembly of the nuclear envelope.80 During normal conditions, cells exit mitosis when cyclin B is degraded by the APC/C.81 This degradation is prevented in an aberrant mitosis and cyclin B remains stable. However, when the arrest is prolonged, cyclin B cannot evade from a slow but continuous proteosomal degradation and cells exit mitosis in the presence of chromosome abnormalities.79 Once a cell exits mitosis without cell division, nuclear envelope is then due to dephosphorylation of lamins reformed around random groups of chromosomes and cells become multinucleated.
Taking together, although pathways resulting in abnormal mitosis differ depending on the nature of the inducers and the status of cell cycle checkpoints, MC seems to be the main response of some mammalian cells, deficient in regulators of cell cycle control, to a variety of mitotic damages (Table 1).
Formation of Giant Cells During MC
One of the most prominent morphological characteristics of MC is the formation of giant cells with abnormal nuclei (Figure 3).20 The appearance of giant cells was first observed in colonies of high-dose-irradiated cells.82 Following irradiation, the volume of some cells continuously increased and finally reached a size that was 200 times bigger than the size of normal cells.83 The formation of giant cells was initially explained by several mechanisms: either by fusion of daughter cells after cell division or by abnormal cell division, or by a combination of both. The former assumption was based on time-lap studies demonstrating that the daughters of irradiated HeLa cells could form a single binucleated cell.84 This mechanism would require repeated divisions and fusions of the daughter cells that eventually become giant cells. As no metaphase or telophase transitions were observed in the giant cell population,82 it is unlikely that the appearance of these cells was a result of successive fusion events. Recently, it was shown that formation of giant cells is not a consequence of daughter cell fusion, but rather a result of the fusion between mitotic cells and cells in S- or G2 phase. Formation of these cells was dependent on asymmetric distribution of chromosomes during the metaphase, leading to the generation of three or more daughter cells, each with a different size of the nucleus.85 The second assumption is based on the suggestion that irradiated cells can go through a sequence of S-phase repetitions (endoreplication) before they enter mitosis in a polyploid state.86 If division then occurs, octaploid giant daughter cells are formed. However, most cells that were irradiated in these experiments became polyploid without endoreplication. Although endopolyploidy may be the main mechanism for the formation of giant cells, it seems that uncoupling of DNA synthesis from cell division (endocycling) is the primary cause,87 and not endoreplication, as previously proposed.

Confocal microscopy image and FACS analysis of control and cisplatin-treated SKOV-3 cells. Cells were stained with mAbs against cytochrome c to illustrate mitochondrial integrity and co-stained with Hoechst to visualize nuclei (b – untreated cells; d and f – 72 and 96 h following treatment, respectively). Changes in nuclear morphology visualized only by Hoechst staining (a – untreated cells; c and e – 72 and 96 h following treatment, respectively). After 72 h, many multinucleated giant cells with chromosome vesicles were observed, and at 96 h, most of the cells had started to collapse and detach from the plate, indicative of necrosis-like lysis. Accumulation of necrotic cells was documented using FACS analysis (g – untreated cells; h – 96 h after treatment)
As mentioned above, one of the early steps in MC is premature mitosis following genotoxic insult. Such cells arrest in mitosis by the spindle checkpoint with subsequent initiation of endocycling, or alternatively, cells escape the checkpoint after a prolonged arrest without completion of cell division and become tetraploid (Figure 2).79 The consequences of the former situation are failure of proper chromatin compaction as well as errors in chromosome alignment and segregation. Depending on the starting point of endocycling, either mono- or multi-nucleated giant cells can be formed (Figure 2).34 Chromosomes during endocycling are highly varied, which lead to the generation of endoploid cells.88, 89 Endocycling has been observed in tumor cells, and specifically in those lacking p53.90, 91
Formation of tetraploid cells is also a prerequisite for initiation of endocycling that in turn can lead to the appearance of polyploid giant cells with nuclear envelopes around clusters of chromosomes or chromosome fragments.88, 89 Although some polyploid giant cells might escape death through depolyploidization through efforts to repair their DNA by means of homologous recombination,34 the ultimate fate of most of them is death.
Death of Giant Cells
Several possible scenarios for the death of giant cells have been suggested. According to one of them, death is a direct cause of MC and it is distinct from apoptosis.8 This conclusion was based on dissimilarities in morphology between MC cells, illustrated by the formation of nuclear envelopes around individual clusters of missegregated, uncondensed chromosomes, as compared with apoptotic cells, which are recognized by a reduced volume, condensed chromatin, nuclear fragmentation, membrane blebbing and formation of apoptotic bodies.92 Despite their distinct morphology, some authors have suggested that both processes share several biochemical hallmarks. For example, by inhibition of Chk2 in syncytia generated by the fusion of asynchronous HeLa cells, metaphase-associated MC was accompanied by sequential caspase-2 activation, mitochondrial release of pro-apoptotic proteins, activation of caspase-3, DNA fragmentation and chromatin condensation.85 However, it has also been reported that dying multinucleated giant cells are characterized by uncondensed chromatin and the absence of DNA ladder formation.8, 93 It seems that the presence or absence of chromatin condensation during MC depends on different events, including the stage when mitotic arrest takes place. There is no consensus concerning the requirement of caspases for MC,27, 29, 32 but it is likely that progression of MC is caspase independent, as neither inhibition nor downregulation of caspases influence the appearance of giant cells (Figure 3). However, caspases are essential for the termination of MC, suggesting that MC-related morphological changes are followed by activation of the apoptotic machinery.32, 33 Thus, MC is not an ultimate manifestation of cell death but rather a process leading to apoptosis. Indeed, already in 1994, it was shown that irradiation of various tumors of lymphoid origin induced mitotic arrest and accumulation of giant cells, which seemed to die due to apoptosis.40, 94 It should also be noted that, apart from giant cells, other cell populations might result from MC and, finally, die due to apoptosis. Moreover, apoptosis may be the outcome of an abnormal mitotic exit (mitotic slippage).95
Mitotic arrest is strongly associated with apoptosis, but the exact molecular mechanism of this association remains to be elucidated.72, 96 Premature mitosis leading to an arrest during the metaphase–anaphase transition is a p53-independent event. Apoptosis in metaphase has also been suggested to occur independently from p53 but as a consequence of caspase-2 activation.85 However, as caspase-2 activation itself is dependent on p53,97, 98 it is unlikely that apoptosis induced by mitotic arrest would be p53 independent.
In response to irradiation, additional DNA damage in metaphase-arrested cells was shown to be a result of over-accumulation of cyclin B1 during delay in late interphase-S and interphase-G2.99 This additional DNA damage may also trigger p53 activity. However, during mitotic arrest, activation of p53 occurs in an ATM/ATR-independent fashion, suggesting that several mechanisms for p53 activation exist.100, 101 Interestingly, p53 can localize to centrosomes during mitosis but is displaced upon spindle damage, indicating that p53 can sense mitotic failure and initiate apoptosis.102 Thus, activation of p53 during mitotic arrest is required for induction of cell death. Notably, p73 have been demonstrated to induce mitotic-linked cell death, which is caspase independent and characterized by the activation of endonuclease G with subsequent DNA fragmentation.103 However, the molecular mechanism that regulates p73 activity in this situation is unknown.
Decreased level of anti-apoptotic proteins, such as Bcl-2, through inhibition of transcription and/or hyperphosphorylation upon prolonged mitotic arrest, represents another possibility to trigger apoptosis through activation of mitochondria-mediated pathway.104, 105 As a response to the microtubule-hyperpolymerizing agent paclitaxel, another Bcl-2 family protein, Bim, was shown to dissociate from microtubules, an event that was linked to the inhibition of Bcl-2.106
Metaphase arrest-associated apoptosis has been reported to involve survivin,65 an anti-apoptotic protein that is important for regulation of cell division.107 Survivin modulates several mitotic events, including spindle and interphase microtubule organization, the spindle assemble checkpoint and cytokinesis.108, 109, 110 Reduction or loss of survivin is associated with several mitotic defects, including hyperduplication of centrosomes108 and aberrant spindle assembly.93 The anti-apoptotic function of survivin is inhibited when the interaction between survivin and microtubules is disrupted by mitotic inhibitors leading to caspase-9 and caspase-3 activation.108 Cells might have a mitotic self-elimination mechanism in which some spindle checkpoint proteins could act as pro-apoptotic. For example, downregulation of Mad2 inhibits induction of apoptosis.111 Importantly, the two different functionalities of this protein seem to be independent from each other.
In addition to metaphase arrest-induced apoptosis, tetraploid cells that are generated through catastrophic mitosis followed by mitotic slippage also undergo apoptosis. An immediate induction of p21 after mitotic slippage is an indicator of a p53-dependent checkpoint response in G1, which would act as a second ‘fail-system’ after an aberrant mitosis.101, 112 However, as there are some examples when cells do not halt in response to G1 checkpoint even in the presence of a functional p53, it is not clear whether activation of this arrest has any role in cell death initiation.113, 114 Despite this knowledge, it is obvious that tetraploid cells require activation of p53 and its target genes for induction of the mitochondrial apoptotic pathway.115 In addition, p53 is also responsible for initiating apoptosis after endomitosis and endoreplication (Figure 2).116, 117 Thus, besides its central role in apoptosis induced by DNA damage, p53 appears to be an important regulator of MC.25, 118
Apoptosis, however, is not always required for MC lethality, as some giant cells can undergo slow death in a necrosis-like manner. This conclusion is based on some similarities between morphological changes during MC and necrosis (the loss of nuclear and plasma membrane integrities).25, 119 Indeed, mitotic arrest in docetaxel-treated tumor cells was followed by massive cell destruction by means of cell lysis.120 Appearance of multinucleated giant cells that were terminated through a necrosis-like lysis was also observed in cisplatin-treated ovarian carcinoma cell line, SKOV-3 (Figure 3).32 Necrosis following MC could be an effect of genetic instability caused by aneuploidy and/or polyploidy.121 Taken together, it is important to note that cells that are facing a mitotic-linked cell death can in fact die by two separate mechanisms, by either apoptosis or necrosis.
Concluding Remarks
The concept of programmed cell death was introduced in the mid-1960s122 but attracted researchers only after apoptosis was verified as ‘a basic biological phenomenon with wide-ranging implication in tissue kinetics’.92 Since that time, many different cell death pathways has been described. Recently, the Nomenclature Committee on Cell Death suggested definitions for eight mechanism-based types of cell death.123 Yet, some researchers depict up to 11 pathways of cell death in mammals.124 However, an agreement whether all of these pathways illustrate examples of programmed cell death is far yet to be achieved. According to the seminal publications, programmed cell death refers specifically to a cell autonomous genetic developmental death program. If we try to fully pursue this definition, then many of the cell death pathways described do not fulfill this criterion. This is particularly true for the process of MC. Addressing the question of the actual numbers of different modes of cell death, some authors did not even consider MC as a separate mode.124 Is MC an example of programmed cell death?
Firstly, as discussed above, there are examples when cells that are morphologically related to MC can survive and produce progeny that is potentially dangerous. On the other hand, in most cases, MC leads to death, suggesting that both survival and cell death pathways might be a result of MC. A similar discussion relates to autophagy, another destiny of cells. Let us try to separate survival from cell death function and concentrate on the latter one.
Secondly, it is clear, to some extent, that various morphological and biochemical hallmarks of each described modes of cell death are overlapping. This makes the distinction between different cell death modes more difficult. All cells undergoing accidental death are using a part of a machinery that is genetically regulated and encoded. If so, then all mechanisms leading to cell death, including the very poorly defined MC, are programmed. Notably, features reminiscent of MC have been observed during normal development. Similar sequence of polyploidization events have been described during the development of trophoblast,125 and of heart myocytes during the first postnatal week,126 and during reproduction of gland cells and neurons.127 These observations suggest that MC cannot be considered a simple dysregulation process activated in response to DNA damage but rather represents an example of a programmed event. Importantly, by activation of any cell-elimination mechanisms and depending on trigger and/or cell specificity, an individual cell engages various pathways to die, but it is still unclear whether all pathways can be classified as a distinct mode of cell death. There is a consensus that MC is the major pathway of tumor cell death activated after treatment with ionizing radiation or certain chemotherapeutic agents. There is also a consensus that MC is an outcome of aberrant mitosis that results in the formation of cells with abnormal nuclei. Nevertheless, then the opinion concerning description of the MC phenomenon is differing. As mentioned above, some groups are arguing that MC constitutes a special case of apoptosis;27 others believe that MC is fundamentally different from apoptosis8 (Figure 1). However, accumulating evidences are indicating that cells in MC can die in various ways (Figure 2). Indeed, recently, a new type of mitotic cell death, termed ‘chromosome fragmentation’, has been described, suggesting that the outcome of chromosomal fragmentation can be apoptosis.128 Importantly, chromosome fragmentation could also represent a form of autophagy, as it has been shown that mitotic chromosomes can be enveloped by autophagic vesicles upon cellular insult.129 In DNA-damaged cells, the absence of p53 and downregulation of Chk-2-related proteins following abrogation of caspase activation drives cells from MC to necrosis. On the other hand, downregulation of Chk-2-related proteins in the presence of a functional p53 is not sufficient to result in necrosis and cells die by apoptosis that can be initiated either in the metaphase-arrested cells or after exit from the aberrant mitosis. The increased p53 expression, followed by caspase activation, occurs subsequent to the morphological changes that characterize MC, indicating that drug-induced MC precedes apoptosis, which is delayed owing to a primary mitotic arrest.
Therefore, it is most likely that MC is a process resulting from aberrant mitosis, characterized by the formation of multinucleated cells and leading to cell death. Cell death might occur either during or after dysregulated mitosis. Thus, MC represents a prestage of apoptosis or necrosis (Figure 1). This suggestion is based on studies indicating that apoptosis might follow, rather than precede, MC.20, 95, 130 The outcome of MC depends on the molecular profile of the cells, which might have a potential relevance for tumor-type treatment. Further studies are required for the identification of additional proteins involved in the MC regulation and the detailed understanding of the mechanisms of this process. The obtained information will assist in improvement of the efficacy of therapy, providing opportunities for the development of new drugs.
Abbreviations
- MC:
-
mitotic catastrophe
- PCC:
-
premature chromosome condensation
- Chk1-2:
-
checkpoint kinase
- MAP:
-
microtubule-associated protein
- PLK1:
-
Polo-like kinase 1
- ATM:
-
ataxia-telangiectasia-mutated
- ATR:
-
ataxia-telangiectasia and Rad3-related
References
Murray AW . Cell-cycle control: turning on mitosis. Curr Biol 1993; 3: 291–293.
Karsenti E, Newport J, Kirschner M . Respective roles of centrosomes and chromatin in the conversion of microtubule arrays from interphase to metaphase. J Cell Biol 1984; 99: 47s–454s.
Kirschner M, Mitchison T . Beyond self-assembly: from microtubules to morphogenesis. Cell 1986; 45: 329–342.
Walker RA, O’Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP et al. Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J Cell Biol 1988; 107: 1437–1448.
McIntosh JR . Cell biology. Microtubule catastrophe. Nature 1984; 312: 196–197.
Russell P, Nurse P . cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell 1986; 45: 145–153.
Molz L, Booher R, Young P, Beach D . cdc2 and the regulation of mitosis: six interacting mcs genes. Genetics 1989; 122: 773–782.
Roninson IB, Broude EV, Chang BD . If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4: 303–313.
Glücksmann A, Spear FG . The effect of gamma radiation on cells in vivo. Part II. Br J Radiol 1939; 12: 486–499.
Spear FG, Glücksmann A . The effect of gamma-radiation on cells in vivo. Part III. Br J Radiol 1941; 14: 65–77.
Gray LH . Actions of radiations on living cell, 1946 and after the second Douglas Lae memorial lecture. Br J Radiol 1952; 25: 235–244.
Mackey MA, Morgan WF, Dewey WC . Nuclear fragmentation and premature chromosome condensation induced by heat shock in S-phase Chinese hamster ovary cells. Cancer Res 1988; 48: 6478–6483.
Ianzini F, Mackey MA . Spontaneous premature chromosome condensation and mitotic catastrophe following irradiation of HeLa S3 cells. Int J Radiat Biol 1997; 72: 409–421.
Kato H, Sandberg AA . Chromosome pulverization in human cells with micronuclei. J Natl Cancer Inst 1968; 40: 165–179.
Johnson RT, Rao PN, Hughes HD . Mammalian cell fusion. 3. A HeLa cell inducer of premature chromosome condensation active in cells from a variety of animal species. J Cell Physiol 1970; 76: 151–157.
Obe G, Beek B . The human leulocyte test system. VII. Further investigations concerning micronucleus-derived premature chromosome condensation. Humangenetik 1975; 30: 143–154.
Obe G, Beek B, Vaidya VG . The human leukocyte test system. III. Premature chromosome condensation from chemically and x-ray induced micronuclei. Mutat Res 1975; 27: 89–101.
Swanson PE, Carroll SB, Zhang XF, Mackey MA . Spontaneous premature chromosome condensation, micronucleus formation, and non-apoptotic cell death in heated HeLa S3 cells. Ultrastructural observations. Am J Pathol 1995; 146: 963–971.
Hall LL, Th’ng JP, Guo XW, Teplitz RL, Bradbury EM . A brief staurosporine treatment of mitotic cells triggers premature exit from mitosis and polyploid cell formation. Cancer Res 1996; 56: 3551–3559.
Waldman T, Lengauer C, Kinzler KW, Vogelstein B . Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 1996; 381: 713–716.
Schneider DO, Whitmore GF . Comparative effects of neutrons and x-rays on mammalian cells. Radiat Res 1963; 18: 286–306.
Hayflick L, Moorhead PS . The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92: 9363–9367.
Shay JW, Roninson IB . Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004; 23: 2919–2933.
Eom YW, Kim MA, Park SS, Goo MJ, Kwon HJ, Sohn S et al. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005; 24: 4765–4777.
Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR . Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int 2006; 6: 25.
Castedo M, Kroemer G . [Mitotic catastrophe: a special case of apoptosis]. J Soc Biol 2004; 198: 97–103.
Bataller M, Portugal J . Apoptosis and cell recovery in response to oxidative stress in p53-deficient prostate carcinoma cells. Arch Biochem Biophys 2005; 437: 151–158.
Mansilla S, Priebe W, Portugal J . Mitotic catastrophe results in cell death by caspase-dependent and caspase-independent mechanisms. Cell Cycle 2006; 5: 53–60.
Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T et al. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene 2004; 23: 6548–6558.
Chu K, Teele N, Dewey MW, Albright N, Dewey WC . Computerized video time lapse study of cell cycle delay and arrest, mitotic catastrophe, apoptosis and clonogenic survival in irradiated 14-3-3sigma and CDKN1A (p21) knockout cell lines. Radiat Res 2004; 162: 270–286.
Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B . DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ 2008; 15: 555–566.
Skwarska A, Augustin E, Konopa J . Sequential induction of mitotic catastrophe followed by apoptosis in human leukemia MOLT4 cells by imidazoacridinone C-1311. Apoptosis 2007; 12: 2245–2257.
Erenpreisa J, Kalejs M, Ianzini F, Kosmacek EA, Mackey MA, Emzinsh D et al. Segregation of genomes in polyploid tumour cells following mitotic catastrophe. Cell Biol Int 2005; 29: 1005–1011.
Erenpreisa J, Cragg MS . Cancer: a matter of life cycle? Cell Biol Int 2007; 31: 1507–1510.
Kastan MB, Bartek J . Cell-cycle checkpoints and cancer. Nature 2004; 432: 316–323.
Bartek J, Lukas J . Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3: 421–429.
Nyberg KA, Michelson RJ, Putnam CW, Weinert TA . Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 2002; 36: 617–656.
Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P . Chk1 instability is coupled to mitotic cell death of p53-deficient cells in response to virus-induced DNA damage signaling. J Mol Biol 2007; 372: 397–406.
Radford IR, Murphy TK . Radiation response of mouse lymphoid and myeloid cell lines. Part III. Different signals can lead to apoptosis and may influence sensitivity to killing by DNA double-strand breakage. Int J Radiat Biol 1994; 65: 229–239.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–1501.
Kil WJ, Cerna D, Burgan WE, Beam K, Carter D, Steeg PS et al. In vitro and in vivo radiosensitization induced by the DNA methylating agent temozolomide. Clin Cancer Res 2008; 14: 931–938.
Kodym E, Kodym R, Choy H, Saha D . Sustained metaphase arrest in response to ionizing radiation in a non-small cell lung cancer cell line. Radiat Res 2008; 169: 46–58.
Chang BD, Watanabe K, Broude EV, Fang J, Poole JC, Kalinichenko TV et al. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis, senescence, and age-related diseases. Proc Natl Acad Sci USA 2000; 97: 4291–4296.
Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B . 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 1999; 401: 616–620.
Castedo M, Perfettini JL, Roumier T, Yakushijin K, Horne D, Medema R et al. The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene 2004; 23: 4353–4361.
Andreassen PR, Lacroix FB, Lohez OD, Margolis RL . Neither p21WAF1 nor 14-3-3sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res 2001; 61: 7660–7668.
Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M . Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol 2007; 27: 2572–2581.
Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, Nakanishi M . Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem 2005; 280: 39246–39252.
Xia Z, Morales JC, Dunphy WG, Carpenter PB . Negative cell cycle regulation and DNA damage-inducible phosphorylation of the BRCT protein 53BP1. J Biol Chem 2001; 276: 2708–2718.
Tominaga Y, Wang A, Wang RH, Wang X, Cao L, Deng CX . Genistein inhibits Brca1 mutant tumor growth through activation of DNA damage checkpoints, cell cycle arrest, and mitotic catastrophe. Cell Death Differ 2007; 14: 472–479.
Ichimiya S, Nakagawara A, Sakuma Y, Kimura S, Ikeda T, Satoh M et al. p73: structure and function. Pathol Int 2000; 50: 589–593.
Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB . p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11: 175–189.
Ianzini F, Domann FE, Kosmacek EA, Phillips SL, Mackey MA . Human glioblastoma U87MG cells transduced with a dominant negative p53 (TP53) adenovirus construct undergo radiation-induced mitotic catastrophe. Radiat Res 2007; 168: 183–192.
Seidl C, Port M, Gilbertz KP, Morgenstern A, Bruchertseifer F, Schwaiger M et al. 213Bi-induced death of HSC45-M2 gastric cancer cells is characterized by G2 arrest and up-regulation of genes known to prevent apoptosis but induce necrosis and mitotic catastrophe. Mol Cancer Ther 2007; 6: 2346–2359.
Qutob SS, Multani AS, Pathak S, Feng Y, Kendal WS, Ng CE . Comparison of the X-radiation, drug and ultraviolet-radiation responses of clones isolated from a human colorectal tumor cell line. Radiat Res 2004; 161: 326–334.
Indovina P, Rainaldi G, Santini MT . Three-dimensional cell organization leads to a different type of ionizing radiation-induced cell death: MG-63 monolayer cells undergo mitotic catastrophe while spheroids die of apoptosis. Int J Oncol 2007; 31: 1473–1483.
Rieder CL, Salmon ED . Motile kinetochores and polar ejection forces dictate chromosome position on the vertebrate mitotic spindle. J Cell Biol 1994; 124: 223–233.
Schmidt M, Bastians H . Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat 2007; 10: 162–181.
May KM, Hardwick KG . The spindle checkpoint. J Cell Sci 2006; 119: 4139–4142.
Gallant P, Nigg EA . Cyclin B2 undergoes cell cycle-dependent nuclear translocation and, when expressed as a non-destructible mutant, causes mitotic arrest in HeLa cells. J Cell Biol 1992; 117: 213–224.
Rieder CL, Maiato H . Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell 2004; 7: 637–651.
Weaver BA, Cleveland DW . Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res 2007; 67: 10103–10105.
Wonsey DR, Follettie MT . Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res 2005; 65: 5181–5189.
Rosa J, Canovas P, Islam A, Altieri DC, Doxsey SJ . Survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol Biol Cell 2006; 17: 1483–1493.
Zhen HN, Li LW, Zhang W, Fei Z, Shi CH, Yang TT et al. Short hairpin RNA targeting survivin inhibits growth and angiogenesis of glioma U251 cells. Int J Oncol 2007; 31: 1111–1117.
Ngan CY, Yamamoto H, Takagi A, Fujie Y, Takemasa I, Ikeda M et al. Oxaliplatin induces mitotic catastrophe and apoptosis in esophageal cancer cells. Cancer Sci 2008; 99: 129–139.
Kline-Smith SL, Walczak CE . Mitotic spindle assembly and chromosome segregation: refocusing on microtubule dynamics. Mol Cell 2004; 15: 317–327.
Soltani MH, Pichardo R, Song Z, Sangha N, Camacho F, Satyamoorthy K et al. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am J Pathol 2005; 166: 1841–1850.
Lane HA, Nigg EA . Cell-cycle control: POLO-like kinases join the outer circle. Trends Cell Biol 1997; 7: 63–68.
Cogswell JP, Brown CE, Bisi JE, Neill SD . Dominant-negative polo-like kinase 1 induces mitotic catastrophe independent of cdc25C function. Cell Growth Differ 2000; 11: 615–623.
DeLuca JG, Moree B, Hickey JM, Kilmartin JV, Salmon ED . hNuf2 inhibition blocks stable kinetochore-microtubule attachment and induces mitotic cell death in HeLa cells. J Cell Biol 2002; 159: 549–555.
Loffler H, Lukas J, Bartek J, Kramer A . Structure meets function – centrosomes, genome maintenance and the DNA damage response. Exp Cell Res 2006; 312: 2633–2640.
Sandell LL, Zakian VA . Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell 1993; 75: 729–739.
Toczyski DP, Galgoczy DJ, Hartwell LH . CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90: 1097–1106.
Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG . Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004; 117: 575–588.
Bartek J, Lukas J . DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19: 238–245.
Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE et al. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell 2005; 8: 49–59.
Brito DA, Rieder CL . Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol 2006; 16: 1194–1200.
Sebastian B, Kakizuka A, Hunter T . Cdc25M2 activation of cyclin-dependent kinases by dephosphorylation of threonine-14 and tyrosine-15. Proc Natl Acad Sci USA 1993; 90: 3521–3524.
Kotani S, Tanaka H, Yasuda H, Todokoro K . Regulation of APC activity by phosphorylation and regulatory factors. J Cell Biol 1999; 146: 791–800.
Puck TT, Marcus PI . Action of x-rays on mammalian cells. J Exp Med 1956; 103: 653–666.
Tolmach LJ, Marcus PI . Development of x-ray induced giant HeLa cells. Exp Cell Res 1960; 20: 350–360.
Pomerat CM, Kent SP, Logie LC . Irradiation of cells in tissue culture. I. Giant cell induction in strain cultures versus elements from primary explants. Z Zellforsch Mikrosk Anat 1957; 47: 158–174.
Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004; 23: 4362–4370.
Yu CK, Sinclair WK . Polyploidy induced by X-rays in Chinese hamster cells in vitro. Science 1964; 145: 508–510.
Erenpreisa J, Kalejs M, Cragg MS . Mitotic catastrophe and endomitosis in tumour cells: an evolutionary key to a molecular solution. Cell Biol Int 2005; 29: 1012–1018.
Nagl W . Polyploidy in differentiation and evolution. Int J Cell Cloning 1990; 8: 216–223.
Edgar BA, Orr-Weaver TL . Endoreplication cell cycles: more for less. Cell 2001; 105: 297–306.
Illidge TM, Cragg MS, Fringes B, Olive P, Erenpreisa JA . Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int 2000; 24: 621–633.
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G . Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23: 2825–2837.
Kerr JF, Wyllie AH, Currie AR . Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26: 239–257.
Okada H, Mak TW . Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 2004; 4: 592–603.
Tauchi H, Sawada S . Analysis of mitotic cell death caused by radiation in mouse leukaemia L5178Y cells: apoptosis is the ultimate form of cell death following mitotic failure. Int J Radiat Biol 1994; 65: 449–455.
Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L . Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res 1996; 56: 816–825.
Woods CM, Zhu J, McQueney PA, Bollag D, Lazarides E . Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway. Mol Med 1995; 1: 506–526.
Vakifahmetoglu H, Olsson M, Orrenius S, Zhivotovsky B . Functional connection between p53 and caspase-2 is essential for apoptosis induced by DNA damage. Oncogene 2006; 25: 5683–5692.
Tinel A, Tschopp J . The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004; 304: 843–846.
Ianzini F, Mackey MA . Delayed DNA damage associated with mitotic catastrophe following X-irradiation of HeLa S3 cells. Mutagenesis 1998; 13: 337–344.
Stewart GS, Last JI, Stankovic T, Haites N, Kidd AM, Byrd PJ et al. Residual ataxia telangiectasia mutated protein function in cells from ataxia telangiectasia patients, with 5762ins137 and 7271T → G mutations, showing a less severe phenotype. J Biol Chem 2001; 276: 30133–30141.
Vogel C, Kienitz A, Hofmann I, Muller R, Bastians H . Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004; 23: 6845–6853.
Morris VB, Brammall J, Noble J, Reddel R . p53 localizes to the centrosomes and spindles of mitotic cells in the embryonic chick epiblast, human cell lines, and a human primary culture: an immunofluorescence study. Exp Cell Res 2000; 256: 122–130.
Niikura Y, Dixit A, Scott R, Perkins G, Kitagawa K . BUB1 mediation of caspase-independent mitotic death determines cell fate. J Cell Biol 2007; 178: 283–296.
Scatena CD, Stewart ZA, Mays D, Tang LJ, Keefer CJ, Leach SD et al. Mitotic phosphorylation of Bcl-2 during normal cell cycle progression and Taxol-induced growth arrest. J Biol Chem 1998; 273: 30777–30784.
Blagosklonny MV . Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007; 6: 70–74.
Puthalakath H, Huang DC, O’Reilly LA, King SM, Strasser A . The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 1999; 3: 287–296.
Altieri DC . Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer 2008; 8: 61–70.
Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998; 396: 580–584.
Carvalho A, Carmena M, Sambade C, Earnshaw WC, Wheatley SP . Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J Cell Sci 2003; 116: 2987–2998.
Skoufias DA, Mollinari C, Lacroix FB, Margolis RL . Human survivin is a kinetochore-associated passenger protein. J Cell Biol 2000; 151: 1575–1582.
Vogel C, Hager C, Bastians H . Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res 2007; 67: 339–345.
Margolis RL, Lohez OD, Andreassen PR . G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem 2003; 88: 673–683.
Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409: 355–359.
Taylor SS, McKeon F . Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell 1997; 89: 727–735.
Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS ONE 2007; 2: e1337.
Casenghi M, Mangiacasale R, Tuynder M, Caillet-Fauquet P, Elhajouji A, Lavia P et al. p53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp Cell Res 1999; 250: 339–350.
Di Leonardo A, Khan SH, Linke SP, Greco V, Seidita G, Wahl GM . DNA rereplication in the presence of mitotic spindle inhibitors in human and mouse fibroblasts lacking either p53 or pRb function. Cancer Res 1997; 57: 1013–1019.
Huang X, Tran T, Zhang L, Hatcher R, Zhang P . DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci USA 2005; 102: 1065–1070.
Dewey WC, Ling CC, Meyn RE . Radiation-induced apoptosis: relevance to radiotherapy. Int J Radiat Oncol Biol Phys 1995; 33: 781–796.
Schimming R, Mason KA, Hunter N, Weil M, Kishi K, Milas L . Lack of correlation between mitotic arrest or apoptosis and antitumor effect of docetaxel. Cancer Chemother Pharmacol 1999; 43: 165–172.
Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C . The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer 2003; 3: 695–701.
Lockshin RA, Williams CM . Programmed cell death – I. cytology of degeneration in the intersegmental muscles of the pernyi silkmoth. J Insect Physiol 1965; 11: 123–133.
Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 2005; 12 (Suppl 2): 1463–1467.
Melino G, Knight RA, Nicotera P . How many ways to die? How many different models of cell death? Cell Death Differ 2005; 12 (Suppl 2): 1457–1462.
Zybina EV, Zybina TG . Polytene chromosomes in mammalian cells. Int Rev Cytol 1996; 165: 53–119.
Brodsky WY, Arefyeva AM, Uryvaeva IV . Mitotic polyploidization of mouse heart myocytes during the first postnatal week. Cell Tissue Res 1980; 210: 133–144.
Anisimov AP, Kirsanova IA . [Somatic polyploidy in neurons of the gastropod mollusca. III. Mitosis and endomitosis in the postnatal development of neurons in the Succinea snail central nervous system]. Tsitologiia 2002; 44: 981–987.
Stevens JB, Liu G, Bremer SW, Ye KJ, Xu W, Xu J et al. Mitotic cell death by chromosome fragmentation. Cancer Res 2007; 67: 7686–7694.
Sit KH, Paramanantham R, Bay BH, Chan HL, Wong KP, Thong P et al. Sequestration of mitotic (M-phase) chromosomes in autophagosomes: mitotic programmed cell death in human Chang liver cells induced by an OH* burst from vanadyl(4). Anat Rec 1996; 245: 1–8.
Demarcq C, Bunch RT, Creswell D, Eastman A . The role of cell cycle progression in cisplatin-induced apoptosis in Chinese hamster ovary cells. Cell Growth Differ 1994; 5: 983–993.
Acknowledgements
We express our gratitude to Professor Sten Orrenius for permanent support. BZ would like to thank Professor Lockshin and Dr. Zakeri for always inspiring discussions and for long-standing friendship. Work in the authors’ laboratory was supported by grants from The Swedish and Stockholm Cancer Societies, The Swedish Childhood Cancer Foundation, The Swedish Research Council, the EC-FP-6 (Oncodeath and Chemores) and EC-FP-7 (APO-SYS). We apologize to authors whose primary references could not be cited due to space limitations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Vakifahmetoglu, H., Olsson, M. & Zhivotovsky, B. Death through a tragedy: mitotic catastrophe. Cell Death Differ 15, 1153–1162 (2008). https://doi.org/10.1038/cdd.2008.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.47
Keywords
This article is cited by
-
Role of microRNAs in regulation of doxorubicin and paclitaxel responses in lung tumor cells
Cell Division (2023)
-
Necrocide 1 mediates necrotic cell death and immunogenic response in human cancer cells
Cell Death & Disease (2023)
-
A guide to cell death pathways
Nature Reviews Molecular Cell Biology (2023)
-
CD30 stimulation induces multinucleation and chromosomal instability in HTLV-1-infected cell lines
International Journal of Hematology (2023)
-
The relationship of the efficiency of energy conversion into growth as an indicator for the determination of the optimal dose for mutation breeding with the appearance of chromosomal abnormalities and incomplete mitosis after gamma irradiation of kernels of Triticum turgidum ssp. durum L.
Radiation and Environmental Biophysics (2023)
