
Abstract
Deregulation of innate immune signalling and cell death form the basis of most human disease pathogenesis. Inhibitor of APoptosis (IAP) protein-family members are frequently overexpressed in cancer and contribute to tumour cell survival, chemo-resistance, disease progression and poor prognosis. Although best known for their ability to regulate caspases, IAPs also influence ubiquitin-dependent pathways that modulate innate immune signalling by activation of NF-κB. Recent advances in our understanding of the molecular mechanisms through which IAPs influence cell death and innate immune responses have provided new insights into novel strategies for treatment of cancer. In this review we discuss our current understanding of IAP-mediated NF-κB signalling, as well as elaborate on unexpected insights into the involvement of IAPs in regulating the ‘Ripoptosome’, a novel intrinsic cell death-inducing platform. We propose an evolutionarily conserved concept whereby IAPs function as guardians of killer platforms such as the apoptosome in Drosophila and the Ripoptosome in mammals.
Similar content being viewed by others
Facts
-
Although best known for their ability to regulate caspases, IAPs also influence ubiquitin-dependent pathways that modulate innate immune signalling by activation of NF-κB.
-
IAPs contribute to cell survival by acting as key regulators of cell death-activating platforms, such as the apoptosome in Drosophila and the Ripoptosome in mammals.
-
The Ripoptosome is a novel death-inducing complex containing the core components RIPK1, caspase-8 and FADD that forms independently from death receptors/ligands.
-
Formation of the apoptosome in Drosophila and the Ripoptosome in mammals follows a common principle as they both spontaneously assembly after depletion of IAPs in response to cellular stress and/or developmental cues.
Open Questions
-
What are the signals that stimulate Ripoptosome assembly?
-
How do IAPs regulate the Ripoptosome, and what are the molecular mechanisms that lead to Ripoptosome formation?
-
What is the role of RIPK1 in health and disease?
-
How can we exploit Ripoptosome formation to improve treatment outcomes?
The realisation that members of the IAP protein family are frequently deregulated in cancer and contribute to chemo-resistance and treatment failure1 has sparked renewed interest in the development of small pharmacologic inhibitors of IAPs.2 Consistent with the notion that different types of cancer cells are addicted to IAPs for their survival, inactivation of IAPs, particularly when combined with other treatments, results in the death of many tumour cells.3 While it is clear that IAPs are frequently deregulated in cancer, less clear is how these versatile E3 ligases mediate their pro-tumourigenic effects. Although best known for their ability to block caspases and apoptosis, IAPs also modulate inflammatory signalling and immunity, copper homeostasis, mitogenic kinase signalling, proliferation and mitosis as well as cell invasion and metastasis. More recently, cIAP1, cIAP2 and XIAP were also found to protect cells from spontaneous formation of the Ripoptosome,4, 5 a large multi-protein complex that has the capability to kill cancer cells in a caspase-dependent and caspase-independent manner.
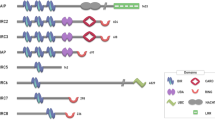
IAPs are defined by the presence of one or more BIR (baculoviral IAP repeat) domains, a protein interaction domain of ∼70 amino acids that mediates protein–protein interactions, and is essential for the anti-apoptotic potential of most IAPs6, 7, 8 (Figure 1). The mammalian IAPs that regulate cell survival, cIAP1, cIAP2 and XIAP, harbour three BIR domains. In addition, these IAPs contain a UBA (ubiquitin (Ub)-associated domain) domain that enables them to bind to poly-ubiquitin chains9, 10 as well as a RING (Really Interesting New Gene) domain that provides them with Ub E3 ligase activity.11 cIAP1 and cIAP2 also contain a CARD (caspase recruitment domain) that is required to shut-down cIAP1's E3 ligase activity under steady-state conditions.12

(a) Domain architecture of a selection of mammalian and Drosophila IAPs. cIAPs, XIAP and DIAP2 carry three BIR domains. BIR domains provide interaction with proteins such as TRAF2, caspases and IAP antagonists. The UBA domain binds to poly-Ub chains. The CARD of cIAP1 is required to shut down cIAP1's E3 ligase activity under steady-state conditions. The C-terminal RING domain provides these IAPs with E3 ligase activity. It also functions as a dimerisation interface and docking site for E2s. (b) Shown are structures of the BIR3 (bound to an SM), UBA, CARD and RING. The UBA domain is a predicted model
Although overexpression of most IAPs can protect cells against apoptotic stimuli,13 XIAP is the only inhibitor of caspases in a strict biochemical sense, blocking caspases through a ‘key–lock’ type of mechanism.14 Residues within a small segment N-terminal to XIAP's BIR2 domain directly bind to the active-site pocket of caspase-3 and caspase-7, thereby preventing substrate entry. Other IAPs, such as cIAP1 and cIAP2, can also bind to caspases, but are inefficient in inhibiting them through mere physical interactions under in vitro conditions. Under physiological conditions, however, IAPs are likely to make use of their E3 ligase activity to neutralise caspases.15 Although IAPs are best known for their ability to regulate caspases, they also fulfil survival-signalling functions independent of controlling caspases. In particular, cIAP1 and cIAP2 modulate Ub-dependent signalling events that regulate activation of NF-κB transcription factors. cIAPs are required for stimulus dependent activation of the canonical and constitutive suppression of the non-canonical NF-κB pathways.16, 17, 18, 19, 20, 21, 22, 23 Moreover, cIAPs, and to some extent XIAP, also regulate NF-κB activation downstream from other innate immunity platforms such as the ones assembled by Toll-like receptors (TLR2, TLR3 and TLR4), NOD (nucleotide-binding oligomerisation-domain protein)-like receptors (NOD1 and NOD2) and RIG-I.24, 25, 26, 27, 28
The recent development of pharmacological inhibitors of cIAPs, dubbed Smac-mimetics (SMs), has helped to shed new light on the role of IAPs in normal physiology and cancer.29 SM compounds are a class of small-pharmacological molecules that mimic the N-terminal IAP-binding motif (IBM) (AVPI) of mature Smac (also known as DIABLO), a member of the loosely defined family of IAP antagonists.30 These compounds selectively bind to the BIR2 and BIR3 domains of IAPs. Although they bind to multiple IAPs, SMs exert their most prominent effects on cIAP1 and cIAP2. Binding of SMs to the BIR3 causes a conformational change of cIAP1, allowing RING dimerisation and E2 activation.31 This results in auto-ubiquitylation and proteasomal degradation of cIAP1 and cIAP2 within minutes of SM treatment. SM-mediated co-depletion of cIAP1 and cIAP2 has been instrumental in dissecting the physiological and cancer-related functions of cIAPs.32
cIAPs in TNF-R1 Signalling
Signalling by tumour necrosis factor (TNF) receptor (TNF-R1) can stimulate a plethora of seemingly opposing biological processes, ranging from inflammatory cytokine production, cell survival, cell proliferation and cell death.33 Originally, TNF was discovered as a macrophage-derived factor that selectively causes necrosis of mouse fibrosarcoma L-929 cells.34 After initial promising prospects for this discovery to be exploited in anticancer therapies, it quickly became clear that TNF not generally promotes tumour necrosis, but instead acts as one of the key mediators of cancer-related inflammation that drives tumour development and/or progression.35, 36 Consistently, constitutive production of TNF from the tumour micro-environment is seen in many malignant tumours, and is frequently associated with poor prognosis.37 A large body of effort, including the seminal work by Tschopp and co-workers, has greatly contributed to our current understanding of how TNF can drive NF-κB activation and cell survival, and under what conditions TNF stimulates tumour necrosis.
TNF mediates most of its effect through activating a set of transcription factors that are collectively referred to as NF-κB.38 Key to TNF-mediated activation of NF-κB is the assembly of a Ub-dependent signalling complex39 (Figure 2a). Ub thereby functions as a binding surface for proteins with Ub-binding domains (UBDs), also referred to as Ub receptors. Ub moieties can be conjugated to one another by different lysine (K) residues within Ub, allowing the formation of differently linked Ub chains.40 Additionally, Ub can also be conjugated to the amino-terminus, resulting in M1-linked chains.41 The different types of Ub chain linkages exert distinct functional outcomes.42 This is predominantly due to the fact that the different chain types adopt distinct topologies, which in turn are recognised by specific Ub receptors. Recent studies indicate that TNF signalling involves conjugation of chains linked through M1, K11, K48 and/or K63 of Ub.39 However, the importance of individual Ub linkage sub-types for TNF signalling is currently subject to intense debate.

cIAP-mediated regulation of canonical and non-canonical NF-κB. (a) Binding of TNF to TNF-R1 stimulates the formation of complex-I consisting of TNF-R1, TRADD, Sam68, RIPK1, TRAF2, TRAF5 and cIAP1/2. Of note, TRAF5 can also be part of this complex; however, as it is currently not clear how TRAF5 contributes to the formation of complex-I (TRAF5 does not bind to cIAPs) it was omitted from this figure for clarity reason. TRAF2 recruits the E3 ligases cIAP1 and cIAP2 to complex-I. Subsequently, cIAPs ubiquitylate several components of this complex, which is required to attract LUBAC. LUBAC subsequently consolidates this complex by facilitating/promoting further ubiquitylation, which results in stabilisation of complex-I, and also recruits IKK. This leads to activation of TAK1 and IKK, which in turn phosphorylates IκB leading to its destruction and liberation of NF-κB (p65/p50). NF-κB subsequently drives the expression of a plethora of target genes. NF-κB-mediated upregulation of FLIP suppresses cell death induced by complex-IIA. In the absence of NF-κB-mediated gene expression, or in the presence of cycloheximide, formation of complex-IIA can trigger caspase-8 activation and cell death. (b) Regulation of non-canonical NF-κB activation. Under resting conditions (left), NIK is constitutively targeted for degradation by the E3 complex TRAF3:TRAF2:cIAP1/2, keeping NIK levels low. TRAF3 and TRAF2 thereby function as adaptors that bring NIK into position for cIAP-mediated ubiquitylation. After ligation of CD40 (right), TRAF3:TRAF2:cIAP1/2 is recruited to the receptor where cIAPs now target TRAF3 for ubiquitylation and proteasomal destruction. Depletion of TRAF3 causes stabilisation and activation of NIK because NIK can no longer be ubiquitylated by cIAPs in the absence of TRAF3. Active NIK phosphorylates IKKα, which in turn phosphorylates p100. Phosphorylated p100 undergoes limited proteasome-mediated proteolysis of p100 to p52. RelB:p52 heterodimers then translocate to the nucleus and activate the expression of NF-κB target genes
According to the current dogma,43 binding of trimeric TNF to TNF receptor-1 (TNF-R1) triggers the initial recruitment of the adaptor proteins TRADD, Sam68, TRAF2 and TRAF5; the E3 ligases; cIAP1 and cIAP2; and the protein kinase RIPK1 (Figure 2a). This complex is frequently referred to as complex-I.44 cIAP-mediated conjugation of Ub to components of complex-I, such as RIPK1, allows subsequent recruitment of the Linear UB chain Assembly Complex (LUBAC, composed of HOIL/HOIP/Sharpin), and the kinase complexes TAK1/TAB2/TAB3, and IKK (composed of NEMO/IKKα/IKKβ.39 Ub-dependent recruitment of LUBAC, TAK1/TAB2/TAB3 and IKK is mediated by UBDs present in TAB2, NEMO and HOIP. Once recruited, LUBAC then modifies NEMO and RIPK1 with M1-linked Ub chains, resulting in increased stability of the complex. Additionally, binding of NEMO to M1-linked Ub chains causes a conformational change of IKK that is thought to facilitate its activation.45 After activation, IKKβ phosphorylates IκB, which targets it for ubiquitylation and proteasomal degradation. Depletion of IκB liberates NF-κB dimers, which subsequently translocate to the nucleus and drive the expression of target genes. As TNF-mediated activation of NF-κB critically relies on ubiquitylation, signalling is attenuated by de-ubiquitylating enzymes, such as CYLD and A20-like proteases, that remove the Ub adduct and disassemble complex-I.46
While activation of TNF-R1 results in the NF-κB-mediated expression of genes important for inflammation and cell survival, under certain circumstances TNF can also induce cell death. Micheau and Tschopp were the first to identify a secondary death-promoting complex that is formed after TNF stimulation. This cytoplasmic complex, which derives from complex-I, is frequently referred to as complex-II (also known as complex-IIA) (Figure 2a). This secondary complex consists of TRADD, FADD, caspase-8 and FLIP, and forms 2 h after ligation of TNF to TNF-R1. Importantly, the killing version of complex-II, which lacks FLIP, occurs only under conditions where expression of NF-κB target genes is blocked, such as upon genetic deletion of NF-κB, expression of the IκB-super repressor (IκBSR) or in the presence of cycloheximide.44, 47 However, if TNF successfully stimulates NF-κB-mediated expression of target genes, formation of complex-II does not lead to apoptosis. This is because NF-κB activation results in upregulation of anti-apoptotic proteins, such as FLIP, that ‘restrains’ caspase-8 in complex-II.44, 48 From the 11 known FLIP isoforms, the long (FLIPL) and the short (FLIPS) isoforms are prominently expressed in cultured tumour cells.49 FLIPL resembles caspase-8 in its domain architecture, but, unlike caspase-8, FLIPL lacks a catalytic cysteine necessary for enzymatic activity. Hence, FLIPL represents an inactive pseudo-caspase. FLIPS only encodes the prodomain, which contains the death effector domains (DED) necessary for FADD binding. While FLIPS completely inhibits caspase-8 activation, FLIPL actually promotes localised activation of caspase-8.50, 51, 52, 53 FLIPL-mediated activation of caspase-8 is achieved through dimerisation-induced conformational changes of the FLIPL–caspase-8 heterodimer. Heterodimerisation rotates the catalytic pocket of caspase-8 into position for catalysis, allowing FLIPL–caspase-8 heterodimers to be functionally active.51, 53 Although FLIPL imparts catalytic activity to caspase-8, this activity is not sufficient to trigger cell death, because FLIPL prevents cleavage of the prodomain of caspase-8 and subsequent release of active caspase-8 from FADD. Moreover, FLIPL–caspase-8 heterodimers show less proteolytic activity than caspase-8 homodimers on apoptotic substrates such as Bid and caspase-3.53, 54
cIAPs: Double-Edged Swords
cIAPs are not only positive regulators of canonical NF-κB signalling downstream from the TNF-R1 receptor, but also function as negative regulators of the non-canonical NF-κB pathway (Figure 2b).16, 17, 18, 19, 20, 21, 22, 55, 56 Activation of the non-canonical NF-κB pathway occurs in response to ligands of a subset of the TNF receptor superfamily that includes BAFF, CD40L and TWEAK. Under unstimulated conditions, non-canonical NF-κB signalling is normally suppressed because of constitutive proteasomal degradation of the kinase NIK by a Ub E3 ligase complex consisting of TRAF2, TRAF3 and cIAPs.16, 57 TRAF3 binds directly to NIK and recruits it to TRAF2 through its ability to heterodimerise with TRAF2. TRAF2 in turn recruits cIAP1 or cIAP2, which are responsible for conjugation of K48-linked Ub chains to NIK. While cIAPs constitutively shut down non-canonical NF-κB signalling, activation of this pathway is triggered upon ligation of BAFF, CD40L and TWEAK. Receptor ligation results in the recruitment of TRAF3–TRAF2–cIAP. This causes ubiquitylation and degradation of TRAF3, or coordinated depletion of TRAF2 and cIAP1, depending on the cellular context and receptor involved. In any case, depletion of the components of the TRAF3–TRAF2–cIAP E3 complex results in stabilisation of NIK, which subsequently phosphorylates IKKα and the NF-κB precursor p100. Activated IKKα homodimers phosphorylate additional residues in p100, which leads to its partial degradation to generate the p52 form. Like after receptor stimulation, genetic loss of TRAF2, TRAF3 or cIAPs also results in accumulation of NIK and constitutive activation of non-canonical NF-κB signalling.
Depletion of cIAPs by SM treatment also results in stabilisation of NIK and spontaneous activation of non-canonical NF-κB signalling (Figure 3a).19, 20 In some cells (referred to SM-sensitive cells), SM-mediated activation of NF-κB leads to induction of TNF. Autocrine produced TNF subsequently stimulates TNF-R1 and, in the absence of cIAP-mediated ubiquitylation of components of complex-I, this leads to the formation of complex-IIB (Figure 3b).47, 58 This complex is distinct from complex-IIA as it requires RIPK1, instead of TRADD, to recruit FADD and caspase-8. Complex-IIB forms in response to TNF-R1 stimulation under conditions when cIAPs are missing or when cIAPs’ E3 activity is defective. Assembly of complex-IIB results in rapid activation of caspase-8 and induction of apoptosis. In most cases, this form of death is completely blocked by caspase inhibitors indicating that it is caspase-derived.19, 20, 58 However, in some cells, exposure to caspase inhibitors, or genetic defects that prevent caspase-8 activation, switches the apoptotic response to programmed necrosis, also referred to as necroptosis.59, 60, 61, 62, 63 The switch to necroptotic cell death depends on the levels of RIPK3,62, 64 a kinase similar to RIPK1. In cells that express high levels of RIPK3, RIPK3 is recruited to complex-IIB. Activation of the kinase activities of RIPK1 and RIPK3 then initiates a signalling cascade that results in necroptotic cell death.61, 62, 64

Non-canonical NF-κB signalling and SM-induced cell death. (a) Under resting conditions (left), NIK is constitutively targeted for degradation by the E3 complex TRAF3:TRAF2:cIAP1/2, keeping NIK levels low. TRAF3 and TRAF2 thereby function as adaptors that bring NIK into position for cIAP-mediated ubiquitylation. After exposure to SM, cIAPs rapidly undergo auto-ubiquitylation and proteasomal degradation. This causes stabilisation and activation of NIK. Active NIK phosphorylates IKKα, which in turn phosphorylates p100. Phosphorylated p100 undergoes limited proteasome-mediated proteolysis of p100 to p52. RelB:p52 heterodimers then translocate to the nucleus and activate the expression of NF-κB target genes. NF-κB-mediated induction of TNF results in activation of TNF-R1 (b). In the absence of cIAPs, RIPK1 recruits FADD and caspase-8 to form complex-IIB. Depending on the amount of RIPK3, RIPK3 is also recruited to complex-IIB contributing to cell death by necroptosis
Interestingly, activation of the non-canonical pathway also provides a negative feedback mechanism for SM-induced killing as it results in upregulation of the NF-κB target gene cIAP2. NF-κB-mediated cIAP2 gene expression can provide protection against the lethal effects of TNF, presumably by targeting RIPK1 and NIK for ubiquitylation.65, 66 Surprisingly, cIAP2 that is upregulated in response to SMs is resistant to SM-mediated degradation. This is because SM-mediated degradation of cIAP2 depends on the presence of cIAP1. Thus, only in the presence of cIAP1, SM treatment stimulates degradation of cIAP2. While treatment with SM leads to efficient degradation of both cIAP1 and cIAP2 at early time points, later on cIAP2 levels dramatically increase (as a result of NF-κB activation), whereas the ones of cIAP1 remain low. This has significant implications as it predicts that SM treatment will be less effective in cancers that lack cIAP1 and express high levels of cIAP2. Consistently, inhibition of cIAP2 upregulation renders cancer cells more susceptible to treatment with TNF and SMs.65, 66
IAPs as guardians of killer platforms
Although cIAPs are unlikely to inhibit caspases directly, they are essential to suppress activation of the extrinsic cell death pathway after ligation of TNF to TNF-R1. The role of cIAPs in regulating the intrinsic (or mitochondrial) cell death pathway is less clear. In mammals, the intrinsic pathway proceeds after mitochondrial outer membrane permeabilisation (MOMP), which leads to release of pro-apoptotic proteins into the cytosol.67 One of these proteins, cytochrome c, binds to the adaptor protein Apaf-1 and, together with dATP, induces a conformational change that leads to Apaf-1 oligomerisation and recruitment of caspase-9 to form the apoptosome. Upon its recruitment to the apoptosome, caspase-9 is activated, which in turn leads to activation of downstream effector caspases and apoptosis. The key decision point of the intrinsic cell death pathway appears to reside with members of the Bcl-2 family of proteins that control MOMP and release of cytochrome c.68 Accordingly, mice that lack the anti-apoptotic Bcl-2 member Bcl-x or Mcl1 die during embryogenesis owing to ectopic apoptosis.69, 70 While anti-apoptotic members of the Bcl-2 family function as the last line of defence, presence of IAPs appears to influence only the rate of cell death, but do not seem to provide clonogenic survival after MOMP.71 Further, although XIAP is able to inhibit caspase-3, caspase-7 and caspase-9,14 XIAP-deficient mice are viable and do not show any gross apoptotic phenotypes.72 This indicates that the ‘decision’ to undergo apoptosis (point of no return) is taken at the level of the mitochondria.
Regulation of the intrinsic apoptotic machinery in mammals seems to be in stark contrast to the situation in Drosophila where the Drosophila IAP 1 (DIAP1) acts as the last line of defence against caspase-mediated cell death (Figure 4).73 Physical association between DIAP1 and caspases is essential for cell survival. Embryos homozygous for diap1 loss-of-function mutations that abrogate binding to effector or initiator caspases die during embryogenesis owing to ectopic cell death. On the other hand, mutations that enhance DIAP1's ability to associate with caspases result in a gain-of-function phenotype.74, 75 Therefore, the DIAP1:caspase association represents the key decision point in the regulation of the apoptotic caspase cascade in flies. In the absence of DIAP1, the initiator caspase Dronc, which is the Drosophila orthologue of caspase-9, spontaneously binds to Dark/dApaf-1/Hac1 to from the apoptosome and induce cell death, independently from mitochondrial factors. Clearly, in Drosophila, IAPs act as the last line of defence before apoptotic destruction.

IAP-mediated regulation of caspase-activating platforms. In Drosophila (left), DIAP1 represents the last line of defence against caspase-mediated cell death. Under healthy conditions, DIAP1 targets Dronc for ubiquitin-mediated inactivation. DIAP1 also suppresses spontaneous cell death by targeting the apoptosome for ubiquitylation and inactivation.85 In response to DNA damage, expression/activation of IAP antagonists or after developmental signals, however, DIAP1 levels are rapidly depleted, which liberates Dronc from DIAP1 inhibition and results in accumulation of the apoptosome and cell death. Similarly in mammals (right), cIAP1, cIAP2 and XIAP constitutively prevent the formation of a RIPK1-dependent caspase-activating platform. All three IAPs are required to target RIPK1 and the components of the Ripoptosome (caspase-8 and FLIP) for ubiquitin-mediated inactivation. After genotoxic stress, release of IAP antagonists or SM treatment, cIAP1, cIAP2 and XIAP levels are rapidly depleted and/or inactivated. This allows the formation and accumulation of the Ripoptosome, and activation of caspase-8. In the presence of high levels of RIP3 this can lead to necroptosis. FLIP also regulates Ripoptosome-mediated cell death. FLIPL thereby prevents apoptosis and necrosis, whereas FLIPS inhibits apoptosis but promotes necroptosis
Recent evidence now indicates that mammalian IAPs might have a similar key role in controlling the intrinsic (death receptor-independent) pathway after all; albeit not through regulating the formation of the apoptosome, but instead by controlling the assembly of an upstream cell death-inducing platform dubbed Ripoptosome (Figure 4).4, 5 The Ripoptosome assembles in response to genotoxic stress-induced depletion of XIAP, cIAP1 and cIAP2, as well as after SM treatment. This large ∼2-MDa macromolecular complex contains the core components RIPK1, FADD and caspase-8, and can stimulate caspase-8-mediated apoptosis as well as caspase-independent necrosis. Importantly, formation of the Ripoptosome complex occurs independently of TNF, CD95L/FASL, TRAIL, death receptors and mitochondrial pathways. While the core components of this complex (RIPK1, FADD and caspase-8) are identical to the ones of complex-IIB, the fact that this complex forms independently of death ligands indicates that it cannot constitute complex-IIB, which, per definition, originates from complex-I.44, 47 To uncouple it from the TNF-dependent complex-II, it is therefore better referred to as ‘Ripoptosome’. The Ripoptosome can also include additional proteins such as caspase-10, FLIPL, RIPK3 and TRIF, depending on cell type and stimulus. Assembly of the Ripoptosome depends on the kinase activity of RIPK1. It is negatively regulated by cIAP1, cIAP2 and XIAP, as well as FLIP. Although cIAPs and XIAP are functionally distinct IAPs, it is clear that cIAP1, cIAP2 and XIAP jointly suppress Ripoptosome formation.4 Accordingly, in the absence of XIAP, SM more readily triggers Ripoptosome formation and cell death. IAP-mediated inactivation of RIPK1 and/or Ripoptosome occurs in a Ub-dependent manner, most likely by targeting RIPK1, and other components of the Ripoptosome, for proteasomal degradation. Of particular interest is the observation that cIAPs and XIAP can target cleaved FLIP and caspase-8 for ubiquitylation.4 Previous work by Tschopp and co-workers indicated that caspase-8-mediated cleavage of FLIP leads to the generation of FLIP(p43), which allows its binding to TRAF2 and the formation of the FLIP(p43)-caspase-8–TRAF2 tertiary complex.50, 76 TRAF2 then recruits cIAPs, which target cleaved FLIP and caspase-8 for ubiquitylation.4 Intriguingly, cIAP1-mediated ubiquitylation of FLIP is completely blocked by z-VAD-fmk, which blocks caspase-8-mediated cleavage of FLIP. This indicates that cIAP1 targets the ‘active’ FLIP–caspase-8 complexes for ubiquitylation and inactivation.
Although treatment with genotoxic stress (etoposide and teniposide) and SM results in cIAP depletion and the formation of the Ripoptosome, etoposide is clearly more effective in stimulating Ripoptosome-mediated cell death than SMs. This may be due, at least in part, to the ability of etoposide to deplete XIAP77 and FLIP78, 79, 80, 81 in addition to cIAP1 and cIAP2.77 Moreover, SM treatment causes NIK-mediated upregulation of cIAP2.65 Therefore, the different potency in inducing cell death may result from their different capabilities to deplete IAPs and FLIP.
Even though SM treatment does not necessarily lead to immediate cell death, SM-induced Ripoptosome formation primes cells to death as it converts pro-inflammatory signals into pro-death stimuli.4, 5 For example, while activation of TLR3 normally signals for inflammatory responses through NF-κB and type-I interferon induction, in the presence of SM (or absence of cIAPs) TLR3 signalling leads to Ripoptosome-mediated cell death. Likewise, the pro-inflammatory cytokines TNF, TWEAK and LIGHT trigger Ripoptosome-mediated cell death in the presence of etoposide- or SM-mediated depletion of IAPs. This suggests that mere formation of the Ripoptosome is not sufficient to induce cell death. For death to occur, an additional ‘activating’ signal is required. This can be provided in the form of DNA damage or cytokine signalling.
The Ripoptosome can also mediate caspase-independent necroptosis. This form of death depends on the presence of RIPK3 and generation of reactive oxygen species (ROS).82 Interestingly, different isoforms of FLIP determine whether the Ripoptosome induces RIPK3-dependent necroptosis or caspase-mediated apoptosis.4, 5 Whereas FLIPL can protect cells against both forms of cell death, FLIPS actively promotes RIPK3-dependent necroptosis, while blocking apoptosis. This paradoxical role of the different isoforms of FLIP is likely due to the fact that FLIPL allows localised activation of caspase-8, which results in cleavage and inactivation of RIPK1,53 and suppression of necroptotic cell death. This observation also provides a mechanistic explanation why genetic deletion of FADD, FLIPL or caspase-8 causes embryonic lethality. While caspase-8- and FADD-deficient mice die at embryonic stage 10.5, they are rescued, at least in part, by co-deletion of RIPK1 and RIPK3.52, 83, 84 Therefore, caspase-8 appears to be required to suppress caspase-independent necroptosis mediated by RIPK3.
The complex that protects cells from RIPK-dependent necrosis is likely to contain RIPK1/3, FADD, caspase-8 and FLIP; however, the precise mechanism by which this complex assembles to prevent RIPK-mediated cell death is not presently known. It is intriguing to speculate on a role of the Ripoptosome. Clearly, more work is needed to clarify whether the Ripoptosome is required for caspase-8-mediated inactivation of RIPK, and how IAPs might regulate this. Much anticipated is the analysis of cIAP1/cIAP2 double-knockout animals and whether their putative phenotype can be rescued by simultaneous co-deletion of RIPK1 or RIPK3. Further, it will be essential to determine the contribution of TNF signalling.
Concluding Remarks
All in all, a common theme emerges whereby IAPs contribute to cell survival by acting as key regulators of cell death-activating platforms, such as the apoptosome in Drosophila and the Ripoptosome in mammals. Formation of these macromolecular platforms follows a common principle as they both spontaneously assembly after depletion of IAPs in response to cellular stress or developmental cues. While DIAP1 regulates the apoptosome by targeting Dark and Dronc, cIAPs and XIAP suppress Ripoptosome formation by ubiquitylating RIPK1 and caspase-8. Ubiquitylation of RIPK1 further contributes to pro-survival signalling as it enables RIPK1-mediated activation of NF-κB and induction of pro-survival transcriptional programmes.
Given that resistance to apoptosis is one of the hallmarks in cancer, the finding that the Ripoptosome also can trigger cell death independent of caspases provides new opportunities on fundamental mechanisms by which chemotherapeutics may kill cancer cells. Further, the observation that Ripoptosome formation converts pro-inflammatory cytokines into pro-death signals has important implications for the design of anticancer therapies aimed at exploiting cancer-related inflammation.
Abbreviations
- IAP:
-
Inhibitor of APoptosis
- BIR:
-
baculoviral IAP repeat
- RING:
-
Really Interesting New Gene
- CARD:
-
caspase recruitment domain
- NOD:
-
(nucleotide-binding oligomerisation-domain protein)-like receptors
- UBA:
-
ubiquitin (Ub)-associated domain
- SM:
-
Smac mimetic
- IBM:
-
IAP-binding motif
References
Hunter AM, LaCasse EC, Korneluk RG . The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007; 12: 1543–1568.
Chen DJ, Huerta S . Smac mimetics as new cancer therapeutics. Anticancer Drugs 2009; 20: 646–658.
LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG . IAP-targeted therapies for cancer. Oncogene 2008; 27: 6252–6275.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43: 432–448.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M et al. cIAPs block Ripoptosome formation, a RIP1/faspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449–463.
Birnbaum MJ, Clem RJ, Miller LK . An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol 1994; 68: 2521–2528.
Hinds MG, Norton RS, Vaux DL, Day CL . Solution structure of a baculoviral inhibitor of apoptosis (IAP) repeat. Nat Struct Biol 1999; 6: 648–651.
Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 1999; 401: 818–822.
Gyrd-Hansen M, Darding M, Miasari M, Santoro MM, Zender L, Xue W et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kappaB as well as cell survival and oncogenesis. Nat Cell Biol 2008; 10: 1309–1317.
Blankenship JW, Varfolomeev E, Goncharov T, Fedorova AV, Kirkpatrick DS, Izrael-Tomasevic A et al. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2(1). Biochem J 2009; 417: 149–160.
Vaux DL, Silke J . IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol 2005; 6: 287–297.
Lopez J, Wicky Joh S, Tenev T, Tautureau GJP, Hinds MG, Francalanci F et al. CARD-mediated autoinhibition of cIAP1's E3 ligase activity suppresses cell proliferation and migration. Mol Cell 2011; 42: 569–583.
Salvesen GS, Duckett CS . IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 2002; 3: 401–410.
Eckelman BP, Salvesen GS . The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem 2006; 281: 3254–3260.
Bader M, Steller H . Regulation of cell death by the ubiquitin–proteasome system. Curr Opin Cell Biol 2009; 21: 878–884.
Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol 2008; 9: 1364–1370.
Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689–700.
Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 2008; 283: 24295–24299.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007; 131: 682–693.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA 2008; 105: 11778–11783.
Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res 2007; 67: 11493–11498.
Bonizzi G, Karin M . The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25: 280–288.
Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 2009; 106: 14524–14529.
Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M . Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009; 30: 789–801.
Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J 2008; 27: 373–383.
Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA . NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem 2007; 282: 36223–36229.
Damgaard RB, Gyrd-Hansen M . Inhibitor of apoptosis (IAP) proteins in regulation of inflammation and innate immunity. Discov Med 2011; 11: 221–231.
Dynek JN, Vucic D . Antagonists of IAP proteins as cancer therapeutics. Cancer Lett 2010 (in press).
Vaux DL, Silke J . Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun 2003; 304: 499–504.
Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM et al. SMAC-mimetics activate the E3 ligase activity of cIAP1 by promoting RING dimerisation. J Biol Chem 2011; 286: 17015–17028.
Gyrd-Hansen M, Meier P . IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 2010; 10: 561–574.
Lobito AA, Gabriel TL, Medema JP, Kimberley FC . Disease causing mutations in the TNF and TNFR superfamilies: focus on molecular mechanisms driving disease. Trends Mol Med 2011; 17: 494–505.
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B . An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 1975; 72: 3666–3670.
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457: 102–106.
Wu Y, Zhou BP . TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer 2010; 102: 639–644.
Lin WW, Karin M . A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 2007; 117: 1175–1183.
Campbell KJ, Perkins ND . Regulation of NF-kappaB function. Biochem Soc Symp 2006; 73: 165–180.
Behrends C, Harper JW . Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol 2011; 18: 520–528.
Komander D . The emerging complexity of protein ubiquitination. Biochem Soc Trans 2009; 37 (Part 5): 937–953.
Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 2006; 25: 4877–4887.
Hoeller D, Dikic I . Targeting the ubiquitin system in cancer therapy. Nature 2009; 458: 438–444.
Bianchi K, Meier P . A tangled web of ubiquitin chains: breaking news in TNF-R1 signaling. Mol Cell 2009; 36: 736–742.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009; 136: 1098–1109.
Harhaj EW, Dixit VM . Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res 2011; 21: 22–39.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J . NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 2001; 21: 5299.
Budd RC, Yeh WC, Tschopp J . cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol 2006; 6: 196–204.
Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem 2002; 277: 45162–45171.
Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS . Activation of caspases-8 and -10 by FLIP(L). Biochem J 2004; 382 (Part 2): 651–657.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C et al. Catalytic activity of the caspase-8–FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471: 363–367.
Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J 2011; 433: 447–457.
Hughes MA, Harper N, Butterworth M, Cain K, Cohen GM, MacFarlane M . Reconstitution of the death-inducing signaling complex reveals a substrate switch that determines CD95-mediated death or survival. Mol Cell 2009; 35: 265–279.
Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY . Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet 2007; 39: 1397–1402.
Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P et al. TWEAK–FN14 signaling induces lysosomal degradation of a cIAP1–TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 2008; 182: 171–184.
Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9: 1371–1378.
Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007; 12: 445–456.
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998; 187: 1477–1485.
Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P . Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 2003; 278: 5622–5629.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Zheng L, Bidere N, Staudt D, Cubre A, Orenstein J, Chan FK et al. Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol Cell Biol 2006; 26: 3505–3513.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J et al. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ 2011; 18: 1376–1386.
Petersen SL, Peyton M, Minna JD, Wang X . Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proc Natl Acad Sci USA 2010; 107: 11936–11941.
Green DR, Kroemer G . The pathophysiology of mitochondrial cell death. Science 2004; 305: 626–629.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR . The BCL-2 family reunion. Mol Cell 2010; 37: 299–310.
Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ . Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 2000; 14: 23–27.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995; 267: 1506–1510.
Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ et al. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol 2004; 165: 835–842.
Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB . Characterization of XIAP-deficient mice. Mol Cell Biol 2001; 21: 3604–3608.
Orme M, Meier P . Inhibitor of apoptosis proteins in Drosophila: gatekeepers of death. Apoptosis 2009; 14: 950–960.
Goyal L, McCall K, Agapite J, Hartwieg E, Steller H . Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J 2000; 19: 589–597.
Zachariou A, Tenev T, Goyal L, Agapite J, Steller H, Meier P . IAP-antagonists exhibit non-redundant modes of action through differential DIAP1 binding. EMBO J 2003; 22: 6642–6652.
Kataoka T, Tschopp J . N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol 2004; 24: 2627–2636.
Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD . Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000; 288: 874–877.
Ganten TM, Haas TL, Sykora J, Stahl H, Sprick MR, Fas SC et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ 2004; 11 (Suppl 1): S86–S96.
Song JH, Song DK, Herlyn M, Petruk KC, Hao C . Cisplatin downregulation of cellular Fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory proteins to restore tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human melanoma cells. Clin Cancer Res 2003; 9: 4255–4266.
Verbrugge I, Maas C, Heijkoop M, Verheij M, Borst J . Radiation and anticancer drugs can facilitate mitochondrial bypass by CD95/Fas via c-FLIP downregulation. Cell Death Differ 2010; 17: 551–561.
Stagni V, Mingardi M, Santini S, Giaccari D, Barila D . ATM kinase activity modulates cFLIP protein levels: potential interplay between DNA damage signalling and TRAIL-induced apoptosis. Carcinogenesis 2010; 31: 1956–1963.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Gentle IE, Wong WW, Evans JM, Bankovacki A, Cook WD, Khan NR et al. In TNF-stimulated cells, RIPK1 promotes cell survival by stabilizing TRAF2 and cIAP1, which limits induction of non-canonical NF-{kappa}B and activation of caspase-8. J Biol Chem 2011; 286: 13282–13291.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011; 471: 368–372.
Shapiro PJ, Hsu HH, Jung H, Robbins ES, Ryoo HD . Regulation of the Drosophila apoptosome through feedback inhibition. Nat Cell Biol 2008; 10: 1440–1446.
Acknowledgements
We thank the members of the Apoptosis Lab for support and critical reading of the manuscript. We acknowledge the NHS funding to the NIHR Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
This article is cited by
-
Drice restrains Diap2-mediated inflammatory signalling and intestinal inflammation
Cell Death & Differentiation (2022)
-
Increased sensitivity to SMAC mimetic LCL161 identified by longitudinal ex vivo pharmacogenomics of recurrent, KRAS mutated rectal cancer liver metastases
Journal of Translational Medicine (2021)
-
MIND bomb 2 prevents RIPK1 kinase activity-dependent and -independent apoptosis through ubiquitylation of cFLIPL
Communications Biology (2021)
-
Gene expression of PLAT and ATS3 proteins increases plant resistance to insects
Planta (2021)
-
RIP1 promotes proliferation through G2/M checkpoint progression and mediates cisplatin-induced apoptosis and necroptosis in human ovarian cancer cells
Acta Pharmacologica Sinica (2020)
