
Abstract
PAK3-related mental retardation represents a rare cause of X-linked mental retardation associated with behavioural symptoms. So far, four families carrying PAK3 mutations have been reported, and in most cases PAK3 dysfunction resulted from missense mutations thought to affect either the catalytic or the N-terminal regulatory domain activity. Here, we report on a Tunisian family of X-linked moderate mental retardation with behavioural symptoms, common dysmorphic features, oro-motor impairment and secondary microcephaly. Linkage analysis showed that affected male subjects and obligate carrier female subjects share a common haplotype in the Xp21.31 – Xq23 region that contains the PAK3 gene. Direct sequencing of PAK3 coding exons and flanking intronic sequences allowed us to identify the first splice mutation in PAK3 gene located at the 5′ end of intron 6 (c.276+4A>G), which results in a complete switch-off of the genuine donor splice site and an activation of a cryptic donor splice site (GTAAG) located four nucleotides downstream to the genuine one. RT-PCR experiments using the RNA from the patient's lymphoblasts showed that PAK3 transcripts contain four additional nucleotides that lead to a disruption of reading frame with a premature stop codon at position 128. Together with previously reported observations, our data further confirm that PAK3 mutations result in a specific form of X-linked mental retardation with fairly constant clinical features.
Similar content being viewed by others

Introduction
X-linked mental retardations are clinically and genetically heterogeneous entities, commonly divided into syndromic forms with a specific pattern of neurological, physical as well as imaging and/or biochemical abnormalities, and non-syndromic forms with no other consistent features than mental retardation (MR).1 To date, more than 60 causative genes are known.2, 3, 4 Among these, mutations in the PAK3 gene have been found in four different families of X-linked MR with neuropsychiatric and morphological features.5, 6, 7, 8 PAK3 gene transcripts encode p21-activated kinase (PAK) protein isoforms shown to be critical downstream ρGTPases effectors that link Rac1 and Cdc42 to actin cytoskeleton and MAP kinases cascades.5, 9 The most investigated PAK3 protein isoform contains 544 amino acids with two critical domains, consisting of an N-terminal regulatory region that includes a CRIB motif that binds to GTP bound forms of Cdc42 and Rac1, and a C-terminal catalytic domain with a serine/threonine (STK) protein kinase domain.10 To our knowledge, the described PAK3 mutations include one nonsense mutation (p.R419X) and three missense mutations located either in the regulatory or the catalytic domain without any evidence of correlation with the severity of the phenotype.5, 6, 7, 8
Here, we report the first PAK3 gene splice mutation that activates a cryptic donor splice site identified in a family with X-linked MR. Although PAK3-related MR is still up to now considered as a non syndromic form of MR, our data together with those reported in the literature suggest that some of the common features may allow the delineation of a distinguishable PAK3-related MR syndrome.
Case report
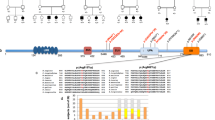
The pedigree of the family is shown in Figure 1a.

(a) Pedigree of the family. Filled squares denote affected male subjects and dotted circles denote carrier female subjects. Asterisks indicate individuals for whom DNA was available. Results of the X-inactivation are indicated under the circles corresponding to the tested two carrier female subjects. (b) Haplotypes of the Xp21.32-Xp23 region. Maximum Lod scores are indicated on the right. (c) Sequence chromatograms of a control individual and the affected family member III.2 showing a partial sequence of exon 6 and flanking intron sequences of PAK3 gene. Asterisks indicate the nucleotidic variation c.276+4A>G. D. RT-PCR analysis using the primers 5F and 7R located in exons 5 and 7 of the PAK3 cDNA. Sequences of the primers are as follow: 5F : CACTCAACCACAGCTCCAAA, primer 7R : TGGGTTCTTCTTCTGTTCCAA. (d) Agarose gel electrophoresis of RT-PCR products using RNA extracted from patient III.2 and a control individual lymphoblasts showing the amplified product of about 285 bp. (e) Sequence analysis of RT-PCR products from patient III.2 and control individual shows the presence in the patient's RT-PCR fragment an insertion of four nucleotides (GTAG) between exon 6 and exon 7. (f) Schematic representation of the genomic and protein structure of PAK3 gene and PAK3 protein with positions of the functional domains (CRIB domain and kinase domain), as well as the position of previously described mutations (c.199C>T/p.R67C6, 11 ; c.1049C>A/p.A365E,7 c.1255C>T/p.R419X,5 c.1337G>A/p.W446S8) and of the new mutation described in this report (c.276+4A>G/p.G92VfsX35). The truncated polypeptide predicted to result from the premature stop codon at position 128 is also illustrated.
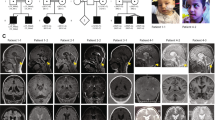
Individual III-1 is a 21-year-old male subject with mild MR and behaviour disturbance. He was born by spontaneous delivery after an uneventful pregnancy. The neonatal period was normal. Birth weight, length and head circumference were normal. From birth, his developmental milestones were delayed with a marked hypotonia and early oro-motor difficulties with permanent drooling. He was able to walk independently at the age of 3 and did not have distinct speech until the age of 6. At the last evaluation, height and weight were normal with microcephaly (third percentile). Dysmorphic features were noticed with a flat face, low forehead, up-slanting palpebral fissures, short nose with upturned nasal tips and large ears. His mouth was large, with thick upper lips, high vault palate and irregular and large permanent teeth (Figure 2). Testicular size was normal.

Photos of the affected male III-1 at age 20 years showing the dysmorphic features. Note the low forehead, large ears and thick upper lips.
Neurological examination disclosed brisk reflex without spasticity and oro-motor difficulties with a hasty and indistinct speech. Neuropsychological assessment gave a full scale IQ at 54 (WAIS III) with a mild impairment of visuo-spatial and constructional skills, as well as of attentional and executive functions. After attending special school, he goes to a sheltered job.
During infancy, he also presented two occasional epileptic seizures. However, repeated EEGs were normal. Behavioural disturbances started during infancy and mainly consisted of aggressive, clastic and screaming episodes. Neither autistic-like features, nor sleep disturbance were reported.
Individual III-2, the proband's brother, aged 27 years, exhibits the same phenotype. The neonatal period was normal, and growth parameters were within the normal range. From the first months of life, severe hypotonia was noticed with oral hypotonia with drooling and tendency to keep the mouth open. He learned to walk at 3 years and first words were developed at 6 years, but with single words and poorly articulated speech. Behavioural disturbances started at 3 years with high levels of hyperactivity, extreme agitation and aggression. No sleep disturbances were reported. At the age of evaluation, height and weight were within the normal range, and secondary microcephaly (third percentile) was also observed. Dysmorphic features were similar to those of his brother. Neurological examination showed motor clumsiness, brisk reflex without spasticity. No seizures were reported. He went to a special school and had an adapted occupational training. Although IQ testing was not performed, clinical history as well as schooling performances indicated the presence of a significant intellectual delay and impaired adaptative skills compatible with moderate MR. Overall, both individuals presented mild MR associated with common features that consisted of aggressive behaviour, agitation, dysmorphic face, microcephaly and hypotonia.
Individual II-2, the proband's mother as well as her three brothers II-3, II-4 and II-5 were considered normal. In particular, all these individuals had a regular job, and no psychiatric disorders. Though others individuals of this family were not thoroughly evaluated, the family reported that individual II-8, also presented mild MR aggravated at the age of puberty by an aggressive behaviour and psychotic features.
Molecular genetic studies
On the basis of the pedigree of this family (Figure 1a), we suspected X-linked MR (XLMR). Following informed consent, DNA was extracted from peripheral blood using standard methods for individuals II-2, III-1, III-2, III-3, I-4 and II-8. Segregation analysis using 30 polymorphic markers evenly distributed on the X chromosome allowed us to exclude implication in the MR phenotype of most regions of the X chromosome, except the region in Xq21.13-Xq23 between the markers DXS1196 and DXS8088. At this region, the affected male subjects were found to share a haplotype spanning about 25 Mb and the highest two-point Lod-score within the interval was 1.11 at DXS8020 (Figure 1b). This region contains two genes shown to be involved in non-syndromic forms of XLMR, namely ACSL4 (CoA synthetase long chain family member 4) and PAK3 genes. Both genes were screened for mutations by direct sequencing of coding exons and their flanking intronic sequences, and a unique variation in the intron 6 (c.276+4A>G) was identified that was reported neither in the literature, nor in online databases as polymorphism (Figure 1c). Available DNA from other members of the family was analysed and the variation was found to segregate with the phenotype and in obligate carriers. We also analysed DNA from 200 control individuals and did not find this variation.
Bioinformatic analysis of the variation G>A (gtaagtaagctc>gtaggtaagctc) consequences on the natural donor splice site (gtaagtaa) at the side of exon 6 using the online programme ‘splice prediction by neuronal network’ available at http://www.fruitfly.org/seq_tools/splice.html predicted an abolition of the efficiency of the natural splice site with a score shift from 0.92 to 0. Moreover, the programme also predicted that the variation is associated with the creation of a potentially strong (score of 1) novel donor splice site (gtaggtaagctc) located immediately after the changed nucleotide.
To further evaluate the consequences of this mutation on the maturation of PAK3 transcripts, we performed RT-PCR using total RNA prepared from the patient's lymphoblasts and primers located in exons 5 and 7 (Figure 1d), and analysed RT-PCR products by electrophoresis and direct sequencing. These analyses showed the amplification of one RT-PCR fragment that has an apparent size comparable to the control, but a sequence differing by the presence of an insertion of four nucleotides (GTAG) between the exons 6 and 7 that correspond to those located between the 3′ end of exon 6 and the newly created donor splice site (Figure 1e). These experimental results were in perfect agreement with the bioinformatic programme prediction, and confirmed that this insertion results in a frame shift and, a premature stop codon at position 128, not far from the N-terminal part of the protein (Figure 1f). It is worth mentioning that the only detected RT-PCR product is the one with the insertion. This suggests that the splice mutation had a drastic effect on the normal splice site function that resulted in an absence of detectable residual level of the normal transcript. Though a very low level of the normal transcript cannot be excluded, it is of interest to note that the only detected RT-PCR product is the one with the insertion.
In this family, the absence of cognitive deficit in obligate carriers prompted us to study X-inactivation profiles and search for a potential skewed X-inactivation (data not shown).
Discussion
Our report describes the fifth family with MR co-segregating with a mutation in the PAK3 gene. This c.276+4A>G intronic mutation was detected in all affected male subjects, as well as in obligate carrier female subjects, but was absent from normal male relatives and from 200 anonymous normal control individuals. Unlike the three missense and one nonsense mutations in the four previous families,5, 6, 7, 8 RT-PCR experiments showed that this mutation (at position +4 of intron 6) is a splice mutation that leads to a disruption of the normal donor splice site of exon 6, and creation of a functional novel splice site 4 nucleotides downstream to the normal one. Moreover, we showed that the mutation resulted in the production and an abnormal out-of-frame PAK3 transcript bearing an insertion of the four nucleotides corresponding to the sequence between the 3′ end of exon 6 and the novel donor splice site (Figure 1f) and a premature stop codon at position 128 within the N-terminal regulatory domain. Though residual expression of the normal transcript, and/or production of a truncated PAK3 protein (a peptide with 128 amino acids) lacking the kinase domain can not be excluded, on the basis of RT-PCR results, it is reasonable to postulate that the mutation described here results in a PAK3 loss-of-function. Indeed, the presence at the 5′ end of the abnormal transcript of a premature stop codon is likely to induce PAK3 degradation through the nonsense mediated RNA decay (NMD) process that detects and degrades RNA transcripts containing nonsense and frameshift mutations.12, 13
In the family described here, X-inactivation status was tested in two obligate female carriers and found that they had complete skewed inactivation profiles. Though fine neuropsychological tests were not performed, they were clinically evaluated as asymptomatic with no behavioural problems. Recent data suggested that in some cases, female carriers could show borderline to mild MR that is not necessarily correlated with a biased X-inactivation status. Indeed, in a recent study reported by Pieppo et al.,8 2/4 mildly delayed female subjects did not have skewed X-inactivation, whereas a skewed inactivation (>80%; 20%) was found in two normal female carriers and one borderline MR female subject. Though analysis of additional families with PAK3 mutations is required, it appears that obligate carriers of PAK3 mutations do not systematically exhibit a skewed X-inactivation pattern in peripheral cells.
In the present family, the nature and the location of PAK3 mutations are different from previously reported mutations. However, clinical examination of affected patients suggests the presence of specific features similar to findings recently highlighted by Peippo et al.8 Including the present study, 31 patients from five families carrying PAK3 mutations have been reported. The most frequent features associated with the PAK3 mutations are: MR, secondary small head circumference or microcephaly, oral motor dysfunction and behavioural disturbance, each at variable degrees, from mild to moderate5, 6, 7, 8, 11 (Table 1). The relatively specific behavioural profile associated with PAK3 mutations that consist of aggressive behaviour, disinhibition with poor control of responses and behaviour or psychotic features could be related to abnormal functioning of the frontal lobe. It is noticeable that other neuropsychiatric signs usually observed in MR, such as autistic features, stereotypes or sleep disorder, have not been reported in PAK3-related disorders. In addition, generalised hypotonia is reported in most patients with PAK3 mutations, combined with oral motor dysfunction that leads to permanent drooling in early childhood. Indistinct speech in later infancy is also frequently observed. Conversely, among the 31 male subjects carrying PAK3 mutations, only 2 (and one described here) had epilepsy that mainly consisted of occasional generalised seizures without any specific EEG pattern. This suggests that epilepsy is not a specific feature of the PAK3-related MR. All together, these observations contribute to the phenotypic knowledge of patients with PAK3 mutations and allow the clinician to distinguish from other XLMR syndromes, such as conditions associated with PQBP1 (polyglutamine binding protein 1 gene) or ARX (Aristaless related homeobox gene) mutations. Indeed, in PQBP1 associated MR microcephaly is also a common feature, often associated with additional signs such as short stature, spastic paraplegia and striking facial appearance with long narrow faces, upward slant of the eyes, malar hypoplasia, prognathism, high-arched palate and nasal speech. In addition, small testes and midline defects as are seen in several patients (cardiac defects and cleft palate).14, 15 Oral motor dysfunction in PAK3 mutations is also different from orobuccal dyspraxia described in ARX associated MR. In both conditions, it leads to drooling and dysarthria, but in PAK3, it appears to be related with facial hypotonia, whereas in ARX mutations, it is combined with facial dystonia. In the most severe form, ARX related oral dystonia makes part of Partington syndrome (MIM 309510), that combines hand dystonia and ataxia.16, 17
In conclusion, our data further confirm that mentally retarded male subjects with PAK3 mutations share some common clinical and behavioural symptoms. The emerging PAK3-related phenotypes include small head size or microcephaly, oral motor dysfunction with persistent drooling and inarticulate speech, and behavioural and psychiatric symptoms. Though thorough description of additional patients is required to support this emerging phenotype, it is worth considering the analysis of PAK3 gene in patients who present with the emerging combination of symptoms highlighted in this study.
Disclosure
The authors confirm that they have read the journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
Kerr B, Turner G, Mulley J, Gedeon A, Partington M : Non-specific X linked mental retardation. J Med Genet 1991; 28: 378–382.
Ropers HH, Hamel BC : X-linked mental retardation. Nat Rev Genet 2005; 6: 46–57.
Chelly J, Khelfaoui M, Francis F, Cherif B, Bienvenu T : Genetics and pathophysiology of mental retardation. Eur J Hum Genet 2006; 14: 701–713.
de Brouwer AP, Yntema HG, Kleefstra T et al: Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum Mutat 2007; 28: 207–208.
Allen KM, Gleeson JG, Bagrodia S et al: PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet 1998; 20: 25–30.
Bienvenu T, des Portes V, McDonell N et al: Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet 2000; 93: 294–298.
Gedeon AK, Nelson J, Gecz J, Mulley JC : X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am J Med Genet A 2003; 120: 509–517.
Peippo M, Koivisto AM, Sarkamo T et al: PAK3 related mental disability: further characterization of the phenotype. Am J Med Genet A 2007; 143: 2406–2416.
Meng J, Meng Y, Hanna A, Janus C, Jia Z : Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci 2005; 25: 6641–6650.
Obermeier A, Ahmed S, Manser E, Yen SC, Hall C, Lim L : PAK promotes morphological changes by acting upstream of Rac. EMBO J 1998; 17: 4328–4339.
des Portes V, Soufir N, Carrie A et al: Gene for nonspecific X-linked mental retardation (MRX 47) is located in Xq22.3-q24. Am J Med Genet 1997; 72: 324–328.
Hentze MW, Kulozik AE : A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999; 96: 307–310.
Wagner E, Lykke-Andersen J : mRNA surveillance: the perfect persist. J Cell Sci 2002; 115: 3033–3038.
Kalscheuer VM, Freude K, Musante L et al: Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat Genet 2003; 35: 313–315.
Kleefstra T, Franken CE, Arens YH et al: Genotype-phenotype studies in three families with mutations in the polyglutamine-binding protein 1 gene (PQBP1). Clin Genet 2004; 66: 318–326.
Partington MW, Mulley JC, Sutherland GR, Hockey A, Thode A, Turner G : X-linked mental retardation with dystonic movements of the hands. Am J Med Genet 1988; 30: 251–262.
Frints SG, Froyen G, Marynen P, Willekens D, Legius E, Fryns JP : Re-evaluation of MRX36 family after discovery of an ARX gene mutation reveals mild neurological features of Partington syndrome. Am J Med Genet 2002; 112: 427–428.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rejeb, I., Saillour, Y., Castelnau, L. et al. A novel splice mutation in PAK3 gene underlying mental retardation with neuropsychiatric features. Eur J Hum Genet 16, 1358–1363 (2008). https://doi.org/10.1038/ejhg.2008.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.103
Keywords
This article is cited by
-
PAK3 activation promotes the tangential to radial migration switch of cortical interneurons by increasing leading process dynamics and disrupting cell polarity
Molecular Psychiatry (2024)
-
Key role of Rho GTPases in motor disorders associated with neurodevelopmental pathologies
Molecular Psychiatry (2023)
-
The p21-activated kinases in neural cytoskeletal remodeling and related neurological disorders
Protein & Cell (2022)
-
Clinical and Molecular Aspects of the Neurodevelopmental Disorder Associated with PAK3 Perturbation
Journal of Molecular Neuroscience (2021)
-
Novel PAK3 gene missense variant associated with two Chinese siblings with intellectual disability: a case report
BMC Medical Genetics (2020)
