
Abstract
Purpose
To report the association and prevalence of reticular pseudodrusen (RPD) in eyes with newly presenting adult onset foveomacular vitelliform dystrophy (AFVD). To compare the strength of association with other pathologies resulting from dysfunction of the choroid-Bruch’s membrane–retinal pigment epithelium (RPE) complex, including eyes with geographic atrophy (GA) and angioid streaks.
Methods
Retrospective single-centre review of all consecutive newly presenting AFVD. Multimodal imaging with spectral domain optical coherence tomography, fundus photographs, red-free/blue light images, and fundus fluorescein angiograms were graded for the presence of RPD. For comparison, all consecutive newly presenting cases of GA and eyes with angioid streaks were studied.
Results
Fifteen (15) patients were identified with AFVD (mean age of 77.3 years; 73.3% female). Mean age of patients with AFVD and RPD was 80.5 years (SD 3.7), whereas that of patients with AFVD without RPD was 75.1 years (SD 7.0). This age difference did not reach statistical significance, P=0.1. Six (40%) had identifiable RPD; being a bilateral finding in 100% of patients. No males with AFVD and RPD were identified. A total of 92 eyes presented with GA. Twenty-three (23) of these (25.0%) had RPD. Twelve (12) patients presented with identifiable angioid streaks, with 4 (36.4%) having RPD.
Conclusion
RPD are a frequent finding in eyes with newly presenting AFVD; not being restricted to AMD, but a finding common among diseases where pathophysiological mechanisms involve damage to Bruch’s membrane and the RPE, whether genetic or degenerative. Our study supports the concept that they occur with high but variable frequencies in eyes with various pathologies.
Similar content being viewed by others


Introduction
Reticular pseudodrusen (RPD) were first described by Mimoun et al in 1990 as a particular yellowish pattern in the fundus of patients with age-related macular degeneration (AMD) that were more clearly visible with red-free or blue light.1 Interest in them quickly increased as their importance was recognised when a strong association between RPD and AMD was observed in 1995, as Arnold et al2 reported that two-thirds of eyes with RPD had or would develop neovascular AMD (nAMD). Since then several groups have reported a high prevalence of RPD in eyes with both geographic atrophy (GA) and nAMD.3, 4, 5, 6, 7 It has been shown that 30–50% of eyes with RPD will progress to AMD within 5 years4, 8 and RPD have been shown to increase the risk for the development of AMD four- to sixfold when compared to eyes with other stages of age-related maculopathy only.8, 9 In the Beaver Dam Eye Study (BDES), RPD were demonstrated to confer a higher risk of visual impairment. New developments in retinal imaging technologies has allowed for their improved visualisation, revealing their location to be internal to the retinal pigment epithelium (RPE).10, 11, 12
Adult-onset foveomacular vitelliform dystrophy (AFVD) was first described by Gass in 1974 as a ‘peculiar foveomacular dystrophy’.13 Since this initial description, which used fundus fluorescein angiography (FFA), many groups have used a wide range of terminologies to describe this relatively uncommon macular disease. In some cases there has been lack of consensus with regard to the diagnostic criteria and pathogenesis of AFVD.14 Previous terms used include adult vitelliform macular dystrophy,15, 16 adult vitelliform macular degeneration,17, 18 and pseudovitelliform macular degeneration19 among others. AFVD has historically been included among a spectrum of pattern dystrophies, such as reticular dystrophy of the RPE and butterfly-shaped pigment dystrophy,14, 20 and there can be significant overlap between these heterogeneous groups. There appears to be a wide range of phenotypes for AFVD, and although there is significant variability in the age of onset, patients generally remain asymptomatic until at least the fifth decade. As the course of the disease may be benign for many, others can develop severe visual symptoms from the deposition of yellowish subretinal material within the macular area, which can lead to RPE alterations, atrophy, or the development of choroidal neovascularisation (CNV).21
The pathophysiological mechanisms underlying the formation of both RPD and AFVD have not been fully elucidated. Until now, only a minority of AFVD cases have known genetic defects, and most cases appear to be sporadic.14 Different genotypes can have their own typical phenotypic features and ages of presentation. The PRH2 gene has been linked to AFVD and has been shown to account for ~2–18% of patients in cohorts to date.22, 23 IMPG1 and IMPG2 are two new causal genes for AFVD, identified in 8% of families in a recently published cohort,24 and in these cases the vitelliform dystrophies were characterised by late-onset moderate visual impairment and the frequent (69% of patients) association with drusen-like lesions23 in the macular area. Limited genetic studies are currently available regarding RPD. Puche et al evaluated four genes with known associations with AMD (ARMS2/HTRA1, apolipoprotein E, C3, and CFH), and found all of the genotypes studied were similarly observed in patients with and without RPD. The current evidence seems to suggest that RPD occur in the same genotype and epidemiological background as AMD.25 A recent publication by Gliem et al26 has also reported a high prevalence (52%) of RPD in eyes with pseudoxanthoma elasticum (PXE). This observation is an interesting one, in that PXE is normally an autosomal recessive genetic disease with an established pathophysiology. It is associated with a mutation in the ABCC6 gene with a corresponding phenotype that involves calcification and fragmentation of connective tissue rich in elastic fibres, such as the Bruch’s membrane (BM) of the eye.27 Interestingly, the mean age of the patients with PXE and RPD in that report was 48.6 years, which is considerably lower than that for isolated RPD. The authors surmised that this association revealed that RPD occurred not only in individuals with AMD and of advanced age but also may be the end result of dysfunction of the BM. More recently, a similar association between RPD and Sorsby fundus dystrophy has been reported.28
Both RPD29 and cuticular drusen30, 31 have previously been associated with vitelliform-type dystrophies. There is, however, very limited literature available on the association of RPD and AFVD. To the best of our knowledge, Zweifel et al29 were the only group that has reported the association other than in case reports. This was, however, a small study with seven eyes of six patients only, and did not report the prevalence of RPD within eyes presenting consecutively with AFVD. It is, therefore, difficult to assess the strength of the association. Knowledge of the prevalence of RPD in eyes with newly presenting AFVD will allow comparison to similar populations with other diseases such as nAMD. Any significant differences may give important information regarding possible genetic associations between the two conditions and may lead to a better understanding of any pathophysiological mechanisms involved with the aetiology of the conditions.
We report a consecutive case series of newly diagnosed AFVD and report the associated prevalence of RPD within this cohort. Within our defined population, we also evaluated the prevalence of RPD within eyes with newly presenting GA and eyes with angioid streaks to allow direct comparisons between prevalence rates.
Materials and methods
All the different multimodal imaging for consecutive patients that were referred to a rapid access Macular Clinic in Kings Mill Hospital, Sutton-in-Ashfield, over a 4-year period (February 2009–February 2013) with newly presenting AFVD were retrospectively reviewed.
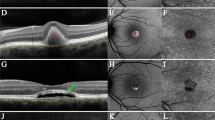
Inclusion criteria were all new patients with a clinical diagnosis of AFVD, confirmed by the presence of a sub-foveal, round, yellowish, and predominantly homogenous lesion with the corresponding presence of hyper-reflective subretinal material on spectral domain optical coherence tomography (SD-OCT) as illustrated in Figure 1. Cuticular drusen were specifically differentiated from RPD by their sub-RPE location on SD-OCT imaging and their intense starry-sky appearance on FFA, when available. All patients underwent a complete ophthalmological assessment at their clinic attendance, including slit lamp examination, SD-OCT, colour fundus photographs (FP), and FFA (as indicated) to rule out CNV. Exclusion criteria included all AFVD eyes with comorbid CNV, established GA, other macular pathologies or poor quality images that would confound grading. All available SD-OCT, FP, red-free and blue light images, and FFA were graded for the presence of RPD. As there is no recognised gold standard imaging method for RPD, their presence was considered definite if they were detected with either the SD-OCT or the FP. SD-OCT images were acquired using the 3D OCT-1000 instrument (Topcon, Tokyo, Japan). The field of view used was a 6mm by 6mm area centred on the fovea. A raster scan consisting of a total of 128 frames each made up of 512 axial scans was performed for each eye within this field. All 128 frames within each raster scan were reviewed for the presence of RPD. RPD were considered present if there were five or more discrete hyper-reflective collections in the subretinal space that were sufficient to alter the contour of the presumed inner segment-outer segment junction, as previously described by others,10 and illustrated in Figure 2. In addition, some individuals had further imaging with the Spectralis SD-OCT (Heidelberg Engineering, Heidelberg, Germany). With FP, RPD were considered present if there was a definite reticular pattern of round or oval yellow-white lesions that joined to form an ill-defined network of broad, interlacing ribbons. An example is shown in Figure 3. The FP was initially graded without digital alteration and subsequently graded with adjustment to the red-free and the blue channels of the fundus photograph using the IMAGEnet i-base programme.

An example of AFVD associated with RPD using the Heidelberg SD-OCT. A sub-foveal, round, yellowish, and predominantly homogenous lesion corresponds to hyper-reflective subretinal material on OCT. There is incidental vitreomacular adhesion. The near-infrared reflectance image clearly identifies the RPD giving a target lesion appearance, with the centre being brighter than its surroundings.

Heidelberg SD-OCT demonstrating discrete hyper-reflective collections in the subretinal space (subretinal drusenoid deposits), causing undulation of the ellipsoid band. One appears to protrude towards the outer nuclear layer.

A colour fundus photograph demonstrating reticular pseudodrusen. There are multiple round and oval yellow-white lesions that join to form an ill-defined network of broad, interlacing ribbons that are predominantly located in the superotemporal location. There are both ‘dot’ and ‘ribbon’ types of RPD. This photo is only for illustration purposes. It has an incidental peripapillary choroidal neovascular membrane, rather than vitelliform lesion. RPD can be subtle on colour fundus photography.
For all eyes identified as having RPD and AFVD, the topographical distribution of RPD was evaluated. In brief, an Early Treatment Diabetic Retinopathy Study (EDTRS) Grid was placed on all available FP or SD-OCT images, and the presence of RPD was recorded for all nine subfields, along with outside the grid and nasal to the optic disc, as described elsewhere.9 The EDTRS grid was opened and initially aligned automatically within either the IMAGEnet i-base programme (for the review of FP) or within the Topcon 3D OCT-1000 viewer programme, for OCT image analysis. If required, manually repositioning was performed.
All consecutive newly presenting cases of pure GA and all eyes with identifiable angioid streaks were also reviewed and graded for the presence of RPD. GA was diagnosed if there was a sharply demarcated area of RPE loss that was at least 175 μm in diameter within the EDTRS grid on FP. The lesion should be roughly round or oval in shape, with at least two of the following features: scalloped edges, visible choroidal vessels that are more prominent than in the surrounding areas and well defined margins in-keeping with the clarity of the fundus photograph. If any part of the area of GA was contiguous with peripapillary atrophy, then it was not graded as GA. Any eye with combined GA and nAMD was graded as the latter. Angioid streaks were diagnosed of FP if there were irregular orange-red lines radiating from the optic nerve head and passing peripherally across the retina, corresponding to breaks in BM. They would correspond to a window defect with FFA if performed.
All retinal images were graded by at least two ophthalmologists with appropriate experience in AMD grading (CW, MP, AL or WA). Side by side grading with immediate open adjudication was used. If disagreement between the two grading ophthalmologists occurred, or a finding was graded as questionable, adjudication by a third ophthalmologist was performed.
Eyes with newly presenting AFVD were divided into those with and without RPD. The two groups were compared for gender using the Fisher’s exact test. The mean ages of the two groups were calculated. Subsequent to this analysis all eyes with newly presenting GA were also reviewed to allow comparison regarding the strength of association with RPD between the various pathologies within the defined population.
Results
A total of 15 consecutive patients were identified as having newly diagnosed AFVD. All these individuals were Caucasian with 11 (73.3%) of these patients being female. The mean age at presentation was 77.3 years, with a standard deviation (SD) of 6.3 years. One patient had a disciform scar in the contralateral eye. Excluding this individual, AFVD was a bilateral finding in 12 out of 14 patients (85.7%). In total, a cohort of 27 eyes was identified with AFVD and 10 (37%) of these eyes had RPD. Six patients (40%) with AFVD had RPD. Within this cohort RPD were a bilateral finding in 100% of patients. The mean age of patients presenting with AFVD and having comorbid RPD was 80.5 years (SD 3.7), although the mean age of patients presenting with AFVD without RPD was 75.1 years (SD 7.0). This age difference did not reach statistical significance (with an unpaired t-test) P=0.1. There were no males with AFVD that were identified as having RPD. However, Fisher’s exact test did not show this difference to be statistically significant (P value=0.10).
Over the same time period, 92 eyes presented with newly documented GA. Twenty-three (23) of these eyes (25.0%) had RPD present. The mean age of eyes presenting with GA was 82.1 years (SD 8.5), and within this group, eyes with GA and comorbid RPD had a higher mean age than those without RPD, (83.1 vs 81.5 years). This difference was not statistically significant (unpaired t-test, P=0.45). Eyes presenting with GA and RPD were more likely to be female (87 vs 13%); this gender difference reached statistical significance (P=0.01).
Twelve (12) patients also presented with identifiable angioid streaks with four (36.4%) having RPD in at least one eye. The mean age of these patients was 79.5 years. Within this group, 9 (75%) patients also had a newly presenting CNV, disciform scar and/or GA.
The topographical distribution or RPD in eyes with newly presenting AFVD is shown in Figure 4.

Topographical representation of the fundus distribution of RPD in eyes with AFVD. Eleven eyes were included as one eye was excluded given the presence of a disciform scar.
Discussion
There is very limited data available on the prevalence of RPD from large population based epidemiological studies.8, 9 It is known that RPD are an infrequent finding, particularly in younger individuals. In The Blue Mountains Eye Study (BMES) their prevalence was reported to be 1.95% in Australian subjects aged 49 years and older, with a 15-year cumulative incidence of 4.0% (n=95).8 In the younger American population (43 years and over) of BDES, the reported prevalence of RPD was lower at 0.7%. The overall 15-year incidence was, however, similar at 3%.9 It is well documented that their prevalence increases with age and was reported to be as high as 6.6% in the 75–86 year age group in the BDES.9 The highest 15-year cumulative incidence in the BMES (4.9%) was reported in the 75 years and over age range. Finger et al32 demonstrated persons with definite RPD were older than those with large drusen (over 125 μm) and no RPD (76±4 vs 68±9). The 40% prevalence of RPD in consecutive patients with newly presenting AFVD in this series is much higher than reported in otherwise healthy individuals from any of the reported large epidemiological studies. To the best of our knowledge, this is the first time the prevalence of RPD has been described within an AFVD population.
Also notable in the present study was the gender specific prevalence of RPD in females with newly diagnosed AFVD. This gender disparity did not reach a level of statistical significance, probably because of the small sample size. This finding is consistent with previous reports of RPD having a female preponderance. When adjusted for age, their 15-year cumulative incidence has been shown within the BMES to be twice more likely in females compared to males (5.6 vs 2.2%).2, 3, 7, 8, 33 Arnold et al2 reported 87% of patients with RPD were female, and Klein et al9 demonstrated that the prevalence of RPD to be 2.5 times higher in woman within normal subjects. Similarly, in the BMES, after adjusting for age, Joachim et al8 demonstrated the 15-year cumulative incidence of RPD was twice as likely in females. In the cohort of six patients reported by Zweifel et al,29 four individuals with AFVD and RPD were female but their series was not consecutive. Gliem et al8 reported no difference in sex distribution in their study on the association between RPD and PXE.
The reasons for this gender disparity remain unknown. In the BDES, Klein et al9 reported a 54% decreased survival rate in subjects with RPD at baseline. There is an increasing evidence to suggest an association between RPD and cardiovascular problems. Boddu et al34 reports patients with RPD are more likely to be hypertensive than those with large soft drusen. In the Melbourne Collaborative Cohort Study, Finger et al32 found that RPD were associated with a moderately elevated systolic blood pressure, current smoking and a trend for an association with a history of myocardial infarction or stroke. The association was, however, reported to disappear in their multivariate analysis, indicating a modest effect size. In the BDES, while controlling for age, RPD were associated not only with being female (OR 2.67, 95% CI: 1.16–6.17, P=0.02) but also having a lower income (OR per lower income group 1.75, 95% CI: 1.16–2.62, P=0.007), higher body mass index (OR per 1 kg/m2 1.08, 95% CI: 1.02–1.15, P=0.006), and more pack years smoked (OR 35 or more pack years smoked vs none 2.61, 95% CI: 1.17–5.85, P=0.02). In the BMES, Tan et al35 found that high-density lipopreoteins were inversely related to the incidence of late AMD. A history of any cardiovascular disease, including stroke, myocardial infarction or angina was also associated with incident early AMD and incident soft or reticular drusen.35 These associations with cardiovascular disease could explain the preponderance of RPD in older females, if young males with RPD were at increased risk of mortality from cardiac disease. This question clearly warrants further study.
The existing prevalence studies may have underestimated the frequency of RPD to some degree because of the sole use of FP in their grading. It is known that newer imaging modalities, such as SD-OCT, fundus autofluorescence, near-infrared reflectance, and scanning laser ophthalmoscopy have led to improvements in the diagnostic accuracy of RPD,33, 36, 37 and multimodal imaging is now becoming the norm for RPD identification. This discrepancy in imaging sensitivities would obviously not explain this significant disparity in prevalence. The association between AFVD and RPD appears to be stronger than that for GA, as in our series, RPD were present in only 25% of eyes presenting with GA. However, it is well documented that RPD can fade with the development of GA and CNV. It is, therefore, possible that some patients with GA may have had RPD which subsequently regressed.
We acknowledge, as pointed out by others,38 that although the subretinal drusenoid deposits identified resemble closely, and share the phenotypic and demographic features of RPD, there remains a lack of histopathological and biochemical correlation similar to that published for RPD occurring in AMD.2, 39 For this reason, some have recommended using the term ‘reticular pseudodrusen-like’ lesions until such a histopathological correlation become available for pathologies other than AMD. Within our study however, the topographical distribution of the reticular pseudodrusen-like lesions within eyes with AFVD had a distribution that was almost identical to that published by Klein et al,9 from the BDES population, with a significant preponderance of RPD within the inner and outer superior subfields along with outside the grid, particularly superiorly. We believe that this is supportive of the suggestion that they are indeed the same as RPD seen within AMD populations.
The prevalence of RPD (36.5%) in our patients with identifiable angioid streaks is very similar to that reported by Gliem et al26 in their prospective series of biopsy confirmed PXE patients. The mean age of patients is, however, considerably higher in our series (79.5 years) compared to that of 48.6 years reported by Gliem et al.26 It would be expected that given our much older population, the prevalence of RPD would be higher. The populations in the two studies obviously differ, however, in that the Gliem series comprised of patients with confirmed diagnosis of PXE either through genetic testing or through skin biopsy. Our patients had angioid streaks identified with no specific diagnosis regarding their aetiology being made. However, it is likely some of these patients may have had PXE.
RPD have been reported by others to have a prevalence of around 40% in newly presenting AMD.33 This present study confirms that RPD are not a finding restricted to eyes with AMD, but rather a finding common among diseases where the pathophysiological mechanisms primarily involve damage to the BM and the RPE, whether genetic in origin or degenerative. Our study supports the concept that RPD appear to occur with high but variable frequencies in eyes with various retinal and macular pathologies, ranging from monogenetic diseases such as PXE (52% as published by Gliem et al), angioid streaks (36.5% in our series), to other more poorly defined conditions with a strong genetic component such as AFVD (40%). Their prevalence in eyes with GA is likely underestimated in our retrospective series at 25%, most likely because of the known tendency for RPD to regress with the expansion of GA.40

References
Mimoun G, Soubrane G, Coscas G . Macular Drusen. J Fr Ophtalmol 1990; 13 (10): 511–530.
Arnold JJ, Sarks SH, Killingsworth MC, Sarks JP . Reticular pseudodrusen. A risk factor in age-related maculopathy. Retina 1995; 15 (3): 183–191.
Cohen SY, Dubois L, Tadayoni R, Delahaye-Mazza C, Debibie C, Quentel G . Prevalence of reticular pseudodrusen in age-related macular degeneration with newly diagnosed choroidal neovascularisation. Br J Ophthalmol 2007; 91 (3): 354–359.
Pumariega NM, Smith RT, Sohrab MA, Letien V, Souied EH . A prospective study of reticular macular disease. Ophthalmology 2011; 118 (8): 1619–1625.
Finger RP, Wu Z, Luu CD, Kearney F, Ayton LN, Lucci LM et al. Reticular pseudodrusen: a risk factor for geographic atrophy in fellow eyes of individuals with unilateral choroidal neovascularization. Ophthalmology 2014; 121 (6): 1252–1256.
Hogg RE, Silva R, Staurenghi G, Murphy G, Santos AR, Rosina C et al. Clinical characteristics of reticular pseudodrusen in the fellow eye of patients with unilateral neovascular age-related macular degeneration. Ophthalmology 2014; 121 (9): 1748–1755.
Wilde C, Patel M, Lakshmanan A, Morales MA, Dhar-Munshi S, Amoaku WM . Prevalence of reticular pseudodrusen in eyes with newly presenting neovascular age-related macular degeneration. Eur J Ophthalmol 2015; 26 (2): 128–134.
Joachim N, Mitchell P, Rochtchina E, Tan AG, Wang JJ . Incidence and progression of reticular drusen in age-related macular degeneration: findings from an older Australian cohort. Ophthalmology 2014; 121 (4): 917–925.
Klein R, Meuer SM, Knudtson MD, Iyengar SK, Klein BE . The epidemiology of retinal reticular drusen. Am J Ophthalmol 2008; 145 (2): 317–326.
Zweifel SA, Spaide RF, Curcio CA, Malek G, Imamura Y . Reticular pseudodrusen are subretinal drusenoid deposits. Ophthalmology 2010; 117 (2): 303–312 e301.
Schmitz-Valckenberg S, Steinberg JS, Fleckenstein M, Visvalingam S, Brinkmann CK, Holz FG . Combined confocal scanning laser ophthalmoscopy and spectral-domain optical coherence tomography imaging of reticular drusen associated with age-related macular degeneration. Ophthalmology 2010; 117 (6): 1169–1176.
Spaide RF, Curcio CA . Drusen characterization with multimodal imaging. Retina 2010; 30 (9): 1441–1454.
Gass JD . A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc 1974; 72: 139–156.
Chowers I, Tiosano L, Audo I, Grunin M, Boon CJ . Adult-onset foveomacular vitelliform dystrophy: a fresh perspective. Prog Retin Eye Res 2015; 47: 64–85.
Brecher R, Bird AC . Adult vitelliform macular dystrophy. Eye 1990; 4 (Pt 1): 210–215.
Renner AB, Tillack H, Kraus H, Kohl S, Wissinger B, Mohr N et al. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina 2004; 24 (6): 929–939.
Epstein GA, Rabb MF . Adult vitelliform macular degeneration: diagnosis and natural history. Br J Ophthalmol 1980; 64 (10): 733–740.
Greaves AH, Sarks JP, Sarks SH . Adult vitelliform macular degeneration: a clinical spectrum. Aust N Z J Ophthalmol 1990; 18 (2): 171–178.
Sabates R, Pruett RC, Hirose T . Pseudovitelliform macular degeneration. Retina 1982; 2 (4): 197–205.
Marmor MF, Byers B . Pattern dystrophy of the pigment epithelium. Am J Ophthalmol 1977; 84 (1): 32–44.
Francis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J et al. Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol 2005; 89 (9): 1115–1119.
Boon CJ, den Hollander AI, Hoyng CB, Cremers FP, Klevering BJ, Keunen JE . The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res 2008; 27 (2): 213–235.
Meunier I, Senechal A, Dhaenens CM, Arndt C, Puech B, Defoort-Dhellemmes S et al. Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: a rationale for molecular analysis. Ophthalmology 2011; 118 (6): 1130–1136.
Meunier I, Manes G, Bocquet B, Marquette V, Baudoin C, Puech B et al. Frequency and clinical pattern of vitelliform macular dystrophy caused by mutations of interphotoreceptor matrix IMPG1 and IMPG2 genes. Ophthalmology 2014; 121 (12): 2406–2414.
Puche N, Blanco-Garavito R, Richard F, Leveziel N, Zerbib J, Tilleul J et al. Genetic and environmental factors associated with reticular pseudodrusen in age-related macular degeneration. Retina 2013; 33 (5): 998–1004.
Gliem M, Hendig D, Finger RP, Holz FG, Charbel Issa P . Reticular pseudodrusen associated with a diseased bruch membrane in pseudoxanthoma elasticum. JAMA Ophthalmol 2015; 133 (5): 581–588.
Le Saux O, Martin L, Aherrahrou Z, Leftheriotis G, Varadi A, Brampton CN . The molecular and physiological roles of ABCC6: more than meets the eye. Front Genet 2012; 3: 289.
Gliem M, Muller PL, Mangold E, Bolz HJ, Stohr H, Weber BH et al. Reticular pseudodrusen in sorsby fundus dystrophy. Ophthalmology 2015; 122 (8): 1555–1562.
Zweifel SA, Spaide RF, Yannuzzi LA . Acquired vitelliform detachment in patients with subretinal drusenoid deposits (reticular pseudodrusen). Retina 2011; 31 (2): 229–234.
Finger RP, Charbel Issa P, Kellner U, Schmitz-Valckenberg S, Fleckenstein M, Scholl HP et al. Spectral domain optical coherence tomography in adult-onset vitelliform macular dystrophy with cuticular drusen. Retina 2010; 30 (9): 1455–1464.
Gass JD, Jallow S, Davis B . Adult vitelliform macular detachment occurring in patients with basal laminar drusen. Am J Ophthalmol 1985; 99 (4): 445–459.
Finger RP, Chong E, McGuinness MB, Robman LD, Aung KZ, Giles G et al. Reticular pseudodrusen and their association with age-related macular degeneration: the Melbourne Collaborative Cohort Study. Ophthalmology 2015; 123 (3): 599–608.
Zweifel SA, Imamura Y, Spaide TC, Fujiwara T, Spaide RF . Prevalence and significance of subretinal drusenoid deposits (reticular pseudodrusen) in age-related macular degeneration. Ophthalmology 2010; 117 (9): 1775–1781.
Boddu S, Lee MD, Marsiglia M, Marmor M, Freund KB, Smith RT . Risk factors associated with reticular pseudodrusen versus large soft drusen. Am J Ophthalmol 2014; 157 (5): 985–993 e982.
Tan JS, Mitchell P, Kifley A, Flood V, Smith W, Wang JJ . Smoking and the long-term incidence of age-related macular degeneration: the Blue Mountains Eye Study. Arch Ophthalmol 2007; 125 (8): 1089–1095.
Ueda-Arakawa N, Ooto S, Tsujikawa A, Yamashiro K, Oishi A, Yoshimura N . Sensitivity and specificity of detecting reticular pseudodrusen in multimodal imaging in Japanese patients. Retina 2013; 33 (3): 490–497.
Lois N, Owens SL, Coco R, Hopkins J, Fitzke FW, Bird AC . Fundus autofluorescence in patients with age-related macular degeneration and high risk of visual loss. Am J Ophthalmol 2002; 133 (3): 341–349.
Querques G . Reticular pseudodrusen: a common pathogenic mechanism affecting the choroid-bruch's membrane complex and retinal pigment epithelium for different retinal and macular diseases. Invest Ophthalmol Vis Sci 2015; 56 (10): 5914–5915.
Curcio CA, Messinger JD, Sloan KR, McGwin G, Medeiros NE, Spaide RF . Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: morphology, prevalence, topography, and biogenesis model. Retina 2013; 33 (2): 265–276.
Marsiglia M, Boddu S, Bearelly S, Xu L, Breaux BE Jr, Freund KB et al. Association between geographic atrophy progression and reticular pseudodrusen in eyes with dry age-related macular degeneration. Invest Ophthalmol Vis Sci 2013; 54 (12): 7362–7369.
Acknowledgements
This research was funded in part by a Research Grant from the Macular Society, UK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Professor Winfried Amoaku, MBChB, has disclosed the following relevant financial relationships: Served as an advisor or consultant for: Alcon Laboratories, Inc.; Alimera Sciences; Allergan, Inc.; Bayer HealthCare Pharmaceuticals; Novartis Pharmaceuticals Corporation; Pfizer Inc; Roche; ThromboGenics, Inc. Served as a speaker or a member of the speaker's bureau for: Alimera Sciences; Allergan, Inc.; Novartis Pharmaceuticals Corporation; Pfizer. Received grants for clinical research from: Allergan, Inc.; CenterVue; Novartis Pharmaceuticals Corporation; Pfizer. Other (Educational Travel Grants): Allergan, Inc.; Bayer HealthCare Pharmaceuticals; Novartis Pharmaceuticals Corporation. The remaining authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Wilde, C., Lakshmanan, A., Patel, M. et al. Prevalence of reticular pseudodrusen in newly presenting adult onset foveomacular vitelliform dystrophy. Eye 30, 817–824 (2016). https://doi.org/10.1038/eye.2016.46
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2016.46
This article is cited by
-
Natural course of adult-onset vitelliform lesions in eyes with and without comorbid subretinal drusenoid deposits
International Ophthalmology (2020)
-
Haller’s vessels patterns in non-neovascular age-related macular degeneration
Graefe's Archive for Clinical and Experimental Ophthalmology (2020)
