
Abstract
The proper functioning of the endoplasmic reticulum (ER) is critical for numerous aspects of cell physiology. Accordingly, all eukaryotes react rapidly to ER dysfunction through a set of adaptive pathways known collectively as the ER stress response (ESR). Normally, this suite of responses succeeds in restoring ER homeostasis. However, in metazoans, persistent or intense ER stress can also trigger programmed cell death, or apoptosis. ER stress and the apoptotic program coupled to it have been implicated in many important pathologies but the regulation and execution of ER stress-induced apoptosis in mammals remain incompletely understood. Here, we review what is known about the ESR in both yeast and mammals, and highlight recent findings on the mechanism and pathophysiological importance of ER stress-induced apoptosis.
Similar content being viewed by others

Overview: The Endoplasmic Reticulum Stress Response
The endoplasmic reticulum (ER) encompasses about half the total membrane area and one-third the newly translated proteins in a typical eukaryotic cell.1, 2 Proper ER function is therefore critical for numerous aspects of cell physiology, including vesicle trafficking, lipid and membrane biogenesis and protein targeting and secretion. Accordingly, metazoan cells react rapidly to ER dysfunction through a set of adaptive pathways known collectively as the ER stress response (ESR).2, 3, 4, 5, 6
The ESR can be triggered by disparate perturbations in normal ER function, such as the accumulation of unfolded, misfolded or excessive protein, ER lipid or glycolipid imbalances, or changes in the redox or ionic conditions of the ER lumen.2, 3, 5, 6 In response to such dysfunction, the ESR acts both to increase the capacity of the ER to fold and process client proteins, and to alleviate the burden on the organelle by reducing the amount of protein inside the ER. These effects are achieved through three major pathways: (I) the unfolded protein response (UPR), a transcription-dependent induction of ER lumenal chaperone proteins and other components of the secretory apparatus to augment the polypeptide folding and processing capacity of the ER;5, 7 (II) the activation of proteasome-dependent ER-associated degradation (ERAD) to remove proteins from the ER;8, 9, 10, 11 and (III) the control of protein translation to modulate the polypeptide traffic into the ER.12, 13 Normally, this suite of responses succeeds in restoring ER homeostasis. However, in metazoans, persistent or intense ER stress can also trigger programmed cell death, or apoptosis.14, 15, 16 ER stress and the apoptotic program coupled to it have been implicated in many important pathologies, including diabetes, obesity, neurodegenerative disorders, viral infection and a variety of ER storage diseases.5, 17, 18, 19, 20, 21 However, the regulation and execution steps of ER stress-induced apoptosis in mammals remain poorly understood, making this area an important topic for future research.
The ESR in Yeast
The foundation of our knowledge of the ESR rests on the genetic analysis of the budding yeast Saccharomyces. cerevisiae.2, 3, 22 In yeast, ER stress is monitored by Ire1p, an ER transmembrane protein with an N-terminal lumenal domain and a C-terminal serine/threonine kinase and endonuclease domains in the cytoplasm or nucleus.23, 24 Normally, the ER lumenal domain of a single Ire1p molecule interacts with Kar2p, an ER lumenal chaperone of the Hsp70 family that assists in folding nascent polypeptides entering the lumen.25 Under ER stress conditions, such as when unfolded protein accumulates inside the ER, Kar2p is titrated away from Ire1p by unfolded protein.25 The removal of Kar2p permits the oligomerization of Ire1p through its lumenal domain and the subsequent activation of Ire1p and trans-autophosphorylation via the C-terminal kinase domain.23, 24, 25
Activated Ire1p signals to the nucleus to initiate the transcription-dependent UPR. The endonuclease domain of activated Ire1p splices the mRNA of the HAC1 gene, which encodes a bZIP transcription factor that drives expression of UPR-responsive genes.26, 27, 28, 29 HAC1 mRNA is constitutively transcribed under resting conditions but contains an intron with secondary structure that blocks translation.26, 28, 29 During ER stress, the translation attenuator intron is spliced from the HAC1 message by activated Ire1p in a novel reaction that is independent of the spliceosome but dependent on tRNA ligase.26, 30 The splicing of HAC1 mRNA both facilitates its translation and causes a frame-shift that replaces the C-terminal portion of the protein with a more potent transactivation domain, thereby promoting efficient UPR induction by mature Hac1p in two distinct ways.26, 29, 31
DNA microarray analyses have shown that genes involved in all stages of the secretory pathway are upregulated by Hac1p, including regulators of protein folding, translocation, glycosylation, vesicular transport, cell wall biosynthesis, vacuolar protein targeting and phospholipid metabolism.32, 33 These targets of Hac1p reduce the burden on the ER by increasing the capacity and efficiency of protein folding and export in the lumen. Remarkably, an estimated 5% of all yeast genes are induced in response to ER stress,32 suggesting that the accumulation of unfolded proteins in the ER may remodel global cell physiology.
In addition to upregulating components of the secretory pathway, ER stress also activates a novel proteolytic pathway known as ERAD.3, 8, 9, 10, 11 In ERAD, misfolded proteins in the ER membrane or lumen are actively retro-translocated, or ‘dislocated’, into the cytoplasm, where they are targeted for ubiquitin-mediated degradation by the proteasome. ERAD can be divided into four conceptual steps: the recognition and targeting of an unfolded substrate to the dislocation apparatus, its transport across the ER membrane, its release into the cytosol and its degradation.8 The components that mediate each step are not fully understood and, in fact, appear to vary by substrate, including differences between soluble lumenal and membrane proteins.34 However, recent research has provided important insight into the major features of the ERAD machinery.
The recognition and targeting of irreparably misfolded polypeptides to the dislocation machinery are mediated, at least in part, by protein glycosylation and chaperone proteins in the ER lumen. In particular, chaperones such as Kar2p and protein disulfide isomerase 1 (PDI1) play an important role in the case of soluble ERAD substrates and perhaps integral membrane proteins as well.35, 36, 37, 38 In some cases, glycosylation of misfolded proteins is also necessary for recognition by the ERAD machinery, possibly via a distinct set of chaperones.39, 40 However, the precise reason for this requirement is not yet clear.
Some experiments indicate that the dislocation step itself occurs, at least in some instances, through the Sec61 channel, which also mediates the entry of nascent polypeptides into the lumen from ER-associated ribosomes.41, 42 Both genetic and biochemical evidence suggests that several ERAD substrates require Sec61 function, implying that the channel may act bidirectionally.35, 43, 44 However, the impairment of ERAD in some Sec61 loss-of-function genetic systems may be an indirect effect of the disruption of ER homeostasis due to blocked import of ER-targeted proteins, rather than to a direct effect on an ERAD dislocation channel. In addition, it is unclear whether all ERAD substrates require Sec61 to exit the ER, nor is there any mechanistic model for how soluble misfolded proteins in the lumen could be threaded into the Sec61 channel.8 Therefore, other means of ER egress may exist.
The release and ubiquitination of ERAD substrates are likely coordinated.8, 9, 10 Cdc48p, a cytosolic member of the AAA family of ATPases, participates in pulling ERAD substrates out of the ER in an ATP-dependent manner in cooperation with Ufd1p and Npl4p.45, 46, 47, 48, 49 In concert with Cdc48p action, ERAD substrates are ubiquitinated and targeted for degradation via the proteasome. Ubiquitin conjugating enzymes participating in ERAD include Ubc1p, Ubc6p, Ubc7p and its membrane-anchored binding partner Cue1p.50, 51, 52 Other proteins physically and genetically interact with the ubiquitin-conjugating machinery during dislocation, including Der1p, Der3p/Hrd1p and Hrd3p,53, 54, 55, 56 although the exact biochemical functions of all these components in yeast are unknown. In addition to signaling for degradation, ubiquitination may also assist in removing some substrates from the ER via a ratchet mechanism, whereby polyubiquitinated chains are prevented from passively sliding backwards through the dislocation channel. Consistent with this model, inhibiting polyubiquitination causes the accumulation of ERAD substrates inside the ER.46, 50
ERAD and the UPR are reciprocally regulated. On the one hand, DNA microarray experiments showed that critical ERAD genes, such as DER1 and HRD3, are strongly induced by treatment with the N-linked glycosylation inhibitor tunicamycin (Tm) in wild-type but not ire1Δ yeast, indicating that the UPR can upregulate ERAD.32, 52 Indeed, the ire1Δ defect was overcome by forced expression of mature Hac1p, implicating the core signaling machinery of the UPR in ERAD induction.32, 33 On the other hand, in ERAD-defective strains, such as ubc1Δ, ubc7Δ, der1Δ, hrd1Δ or hrd3Δ, the UPR is constitutively activated, indicating a feedback control of ERAD over the UPR.32, 52 Therefore, in ERAD-deficient mutants, misfolded protein in the ER cannot be removed, causing the cells to activate the UPR to restore ER homeostasis. Importantly, single mutations impairing either the UPR or ERAD do not grossly affect yeast cell viability, but combining them results in synthetic toxicity, in part through the accumulation of reactive oxygen species (ROS) in the ER.32, 52, 57 Thus, ERAD and the UPR cooperate and the ESR is critical for cell viability, even in the absence of unusual, exogenous stress.
For years, it was thought that yeast lacked the third major ESR pathway outlined above, that of translational control. Recently, however, a bioinformatic approach revealed that the transcription factor Gcn4p is upregulated by ER stress via a translational mechanism and is required for the full activation of a variety of UPR genes.58 The best known role of Gcn4p is in the response to amino-acid starvation, which causes the translational upregulation of GCN4 mRNA through the activity of Gcn2p, a kinase that phosphorylates the eukaryotic translation initiation factor 2 subunit α (eIF2α).59 Upon its induction, Gcn4p upregulates the expression of amino-acid biosynthesis and transport genes.59 Interestingly, it was recently shown that the same Gcn2p/eIF2α/Gcn4p pathway is also activated by ER stress and participates in UPR induction, demonstrating that a form of translational control exists in the yeast ESR.58 Further work is needed to determine whether the phosphorylation of eIF2α in yeast has important consequences during ER stress beyond Gcn4p upregulation, as it does in mammals.
ESR in Mammals
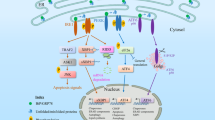
The mammalian ESR retains several key features of the yeast program but is significantly more complex (see Figure 1).2, 3, 4, 5, 6, 13 Mammals have two homologs of Ire1p (Ire1α and Ire1β) and a homolog of Kar2p (BiP/Grp78), which serve as sentinels of ER stress and are believed to function similarly to the corresponding yeast proteins. Ire1α is ubiquitously expressed, while Ire1β is detected only in the intestine.60, 61 Like yeast Ire1p, the overexpression of mammalian Ire1α activates the UPR in the absence of any ER stress signal.60 However, while Ire1p is absolutely required in yeast for initiation of the UPR, cells derived from Ire1α−/−/Ire1β−/− embryos show no obvious UPR defect, demonstrating that compensatory pathways exist.13

Mammalian ESR signaling. Unfolded protein in the ER lumen titrates BiP away from three sentinels of ER stress: PERK, IRE1 and ATF6. Activated PERK phosphorylates the translation initiation factor eIF2α to slow global protein synthesis temporarily and upregulate certain stress-inducible messages, such as ATF4. Activated IRE1 splices the mRNA for XBP-1 to allow the translation of mature XBP-1 protein, a transcription factor that mediates the transcriptional upregulation of numerous genes involved in mammalian ER function and the secretory pathway in general. Similarly, during ER stress ATF6 trafficks to the Golgi, where it is cleaved by S1P/S2P proteases and thereby released from the membrane to activate a distinct but overlapping set of genes in the nucleus. See text for details
The mammalian X-box-binding protein-1 (XBP-1), a bZIP member of the CREB/ATF family of transcription factors, serves as a functional homolog of yeast Hac1p.62, 63 XBP-1 mRNA is ubiquitous in adult tissues but preferentially expressed in the fetal exocrine glands, osteoblasts, chondroblasts and liver.64 Like HAC1, newly synthesized XBP-1 transcript must be spliced by activated Ire1 to a mature, efficiently translated mRNA.62, 63 When activated by ER stress, Ire1 excises an intron from the immature XBP-1 transcript, resulting in the replacement of the C-terminal domain of the protein and the translational activation of the mRNA, analogous to the splicing of HAC1 mRNA.62, 63, 65
Unlike HAC1 mRNA, mammalian XBP-1 mRNA is not expressed at high levels in unstressed cells.63 Instead, the expression of XBP-1 mRNA is regulated by the transcription factor ATF6, which is itself an important component of the ESR.63, 66, 67, 68 The overall structure of ATF6 does not resemble Hac1p and ATF6 protein levels are not regulated by mRNA processing.67 However, ATF6 does share significant sequence identity with Hac1p in its N-terminal basic region and ATF6 overexpression activates many targets of the mammalian UPR.66, 67, 68, 69
Like HAC1 and XBP-1, ATF6 is activated by ER stress via an unusual mechanism. ATF6 is a type II transmembrane protein in the ER of unstressed cells.67 As with Ire1, the ER lumenal domain of ATF6 interacts with BiP, which binds to a Golgi localization signal (GLS) sequence on ATF6.70 During ER stress, BiP is titrated away from ATF6, exposing the GLS and allowing ATF6 to traffic to the Golgi.70, 71 There, ATF6 is cleaved by the site-1 and site-2 proteases, the same enzymes that cleave membrane-bound sterol response element binding proteins during conditions of sterol starvation.67, 71 When ATF6 is cleaved, its N-terminal bZIP domain is liberated from the membrane and enters the nucleus to activate transcription of XBP-1 and other UPR target genes.67, 71 Interestingly, time course studies have shown that ATF6 action precedes that of XBP-1 during ER stress, suggesting a time-dependent shift in the UPR transcriptional response.72 ATF6 and XBP-1 therefore play distinct but complementary roles in adjusting UPR-mediated transcription during persistent stress.
Mammalian ERAD pathways are less well understood than those of yeast, but several parallels are clear.8, 9 As in yeast, glycosylation probably participates in the recognition of mammalian ERAD substrates,73 as do BiP and PDI.74, 75 Similarly, at least some mammalian ERAD substrates may be dislocated through the Sec61 channel.76, 77 Ubiquitination machinery similar to that of yeast is also employed, and a homolog of Cdc48p, termed p97, has been shown to cooperate with mammalian homologs of Ufd1p and Npl4p in substrate dislocation.78, 79, 80 Recently, two groups using independent functional criteria identified Derlin1 as a mammalian homolog of Der1p and demonstrated that it mediates the association of p97 with the ER membrane to facilitate ERAD.81, 82 Another protein in this p97 targeting complex, VIMP, was also identified in one of these studies.82
As might be expected, mammalian ERAD pathways are more complex than those of yeast and several novel components have been described. For example, several groups recently identified the U-box protein CHIP as a ubiquitin E3 ligase that binds to chaperone and E2 enzymes to mediate ERAD.83, 84, 85, 86 Interestingly, Parkin, a gene associated with a recessive form of juvenile Parkinson's disease (PD), encodes another ERAD ubiquitin E3 ligase.87, 88, 89 Parkin mediates the ERAD degradation of a novel form of α-synuclein, a protein also implicated in inherited forms of PD and the major constituent of Lewy inclusion bodies observed in the neurons of PD patients.87 Parkin mutants associated with familial PD are unable to act on α-synuclein, suggesting that Parkin-mediated ERAD processing of α-synuclein is critical for avoiding PD.87 It is likely that other substrate- and cell type-specific components of the mammalian ERAD pathway remain to be identified.
The third major pathway of the mammalian ESR depends on the control of protein translation.13 As in yeast, translational control in the mammalian ESR is mediated by the transient phosphorylation of the translation initiation factor eIF2α. EIF2 is a tripartite protein complex that binds and hydrolyzes GTP during its role in recruiting the initiator methionyl-tRNA to the 40S ribosome to begin mRNA translation.90 In order to initiate subsequent rounds of translation, eIF2 must exchange its bound GDP for GTP, a process facilitated by the guanine nucleotide exchange factor (GEF), eIF2B.90, 91, 92 However, when the α subunit of eIF2 is phosphorylated on Ser51, it binds to eIF2B with much greater affinity.90, 91, 92 This interaction physically sequesters eIF2B, which is stoichiometrically limiting in the cell, and prevents it from mediating the exchange of GDP for GTP by eIF2. Therefore, phosphorylated eIF2α downregulates global translation by inhibiting its own GEF.
Mammals have four eIF2α kinases that respond to diverse stress stimuli: GCN2, PKR, HRI and PERK. Like its counterpart in yeast, mammalian GCN2 is activated during amino-acid starvation by binding to uncharged tRNAs.93, 94 PKR is activated by double-stranded RNAs and is an important component of the interferon-mediated antiviral response.95, 96 HRI is restricted to erythroid cells and is inhibited by heme, such that it blocks the translation of new globin chains when insufficient heme exists to assemble holo-hemoglobin.97 Finally, PERK phosphorylates eIF2α in response to ER stress.98
Like Ire1, PERK is a type I ER transmembrane protein.99 The large ER lumenal domain of PERK is homologous to that of Ire1α and Ireβ, implying that the PERK activation mechanism may resemble that of mammalian and yeast Ire1 proteins.25, 100 Indeed, the lumenal domains of Ire1 and PERK interact with BiP under resting conditions but dimerize in a ligand-independent manner during ER stress.25, 100, 101 As in yeast, it is believed that excess unfolded protein accumulates during ER stress and competes with mammalian Ire1 and PERK for BiP binding, allowing Ire1 or PERK to homodimerize and self-activate by trans-autophosphorylation. Consistent with this model, the displacement of BiP from Ire1 or PERK correlates with the appearance of activated PERK and Ire1, and overexpression of BiP attenuates their activation.25
The transient phosphorylation of mammalian eIF2α (e.g., by PERK) downregulates the translation of most mRNAs. It has been suggested that this relatively brief window of translation attenuation alleviates the burden on the stressed ER by reducing the traffic of newly synthesized polypeptides into the organelle.99 Indeed, PERK−/− cells are hypersensitive to ER stress.102 Paradoxically, however, some components of the ESR are induced at the translational level when eIF2α is phosphorylated, including the transcription factor ATF4 and its downstream target CHOP, comprising a translation-activated response that promotes cell survival during ER and other stresses.93, 103
Although ATF4 mRNA is constitutively expressed, it is normally associated with monoribosomes and low molecular weight polyribosomes, indicating that it is inefficiently translated under resting conditions.93, 103 However, ATF4 mRNA quickly shifts to heavier ribosomal fractions and is selectively translated in a PERK-dependent fashion upon ER stress induction.93 Ribosomal bypass scanning of the upstream open reading frames (uORFs) in the 5′ leader is thought to allow the preferential translation of ATF4 mRNA even when most protein translation is halted by eIF2α phosphorylation.93 Indeed, this mechanism is reminiscent of the upregulation of GCN4 mRNA in yeast by Gcn2p-mediated eIF2α phosphorylation.58, 59, 90 Therefore, while eIF2α phosphorylation inhibits the efficient translation of most mRNAs, it activates the translation of a small subpopulation of transcripts that contain small uORFs, such as ATF4.59, 90, 93, 104, 105 Accumulating evidence suggests that this gene expression program is the major cytoprotective effect of eIF2α phosphorylation.
Genetic experiments indicate that ATF4 is not the only important gene subject to translational upregulation by the PERK/eIF2α phosphorylation pathway. Indeed, overexpression of ATF4 is insufficient to activate the full complement of ESR genes93 and the gross phenotype of ATF4−/− mice106 is different from that of PERK−/− mice99 and eIF2αA/A knockin mice, which harbor a homozygous Ser51 → Ala51 mutation at the critical phosphorylation site in the endogenous eIF2α locus.107 EIF2αA/A cells fail to upregulate about one-third of the genes normally induced by ER stress and are hypersensitive to ER stress-induced apoptosis,107 indicating that translational control impacts on a wide range of ER stress-responsive genes. However, eIF2αA/A cells also show transcription profile differences from wild-type cells under unstressed conditions,107 so the immediate consequences of the loss of eIF2α phosphorylation during ER stress remain to be dissected in detail. On the other hand, ATF4 is critical for the full response to stresses that induce eIF2α phosphorylation,108 probably through the direct transcriptional upregulation of UPR targets such as BiP and CHOP.108, 109, 110
CHOP is a bZIP transcription factor that contains an ESR element in its promoter and is transcriptionally upregulated by ATF6.109, 111, 112 Interestingly, CHOP protein induction also stringently depends on eIF2α phosphorylation, probably both because CHOP transcription is also activated by ATF4,109 and because the 5′ leader of the CHOP mRNA contains small uORFs.93, 113 CHOP can form heterodimers with members of the C/EBP and fos-jun families of transcription factors114, 115 and likely contributes to the regulation of many genes in orchestrating the transcriptional component of the ESR.116
EIF2α signaling is also controlled by protein dephosphorylation mediated by the general cellular serine/threonine phosphatase protein phosphatase 1 (PP1).117, 118 The catalytic subunit of PP1 shows little, if any, intrinsic substrate specificity and relies on the binding of non-enzymatic cofactors to direct its subcellular localization and substrate choice.119 In the case of eIF2α dephosphorylation, GADD34 functions as a cofactor for PP1 under ER stress conditions.118 Like CHOP and ATF4, the expression of GADD34 itself is activated by eIF2α phosphorylation, and GADD34 induction requires GCN2 or PERK under conditions of amino-acid starvation or ER stress, respectively.118 At least in some contexts, this may be mediated by upregulation of GADD34 mRNA by CHOP116 and/or the transcription factor ATF3, which is also upregulated by ATF4.120 Therefore, the regulation of eIF2α by GADD34/PP1 completes a feedback loop to control translation: during ER stress, PERK phosphorylates eIF2α, which induces the expression of ATF4, ATF3 and CHOP. These transcription factors then upregulate GADD34, which mediates the dephosphorylation of eIF2α by PP1. Thus, eIF2α phosphorylation is normally transient and protein translation returns to its baseline state after ER stress has abated. Consistent with this model, GADD34−/− cells are impaired in their ability to resume normal translation following ER stress.116, 121, 122
Other proteins can mediate eIF2α dephosphorylation as well. A constitutively expressed homolog of GADD34, termed CReP, binds to PP1 and keeps eIF2α unphosphorylated in the absence of stress.123 Indeed, RNAi against CReP induces eIF2α phosphorylation in unstressed cells.123 More recently, the SH2/SH3 domain-containing protein Nck-1 has been shown to antagonize PERK signaling in a phosphatase-dependent manner, probably through the direct dephosphorylation of activated PERK itself.124 Nck-1 activity also results in the dephosphorylation of eIF2α though whether it does so indirectly, by inactivating PERK, or directly, by mediating eIF2α dephosphorylation (or both), remains to be determined.124
The bZIP transcription factor Nrf2 was recently identified as a second substrate of PERK.125 Upon ESR induction, PERK phosphorylation causes Nrf2 to relocalize from the cytoplasm to the nucleus, where it upregulates a range of antioxidant response genes.126, 127, 128 Therefore, a loss of Nrf2 function may partly explain why PERK−/− cells experience increased oxidative stress in response to perturbations in ER function.108 Consistent with this model, Nrf2−/− cells are sensitized to ER stress,125 providing another way in which PERK signaling promotes cell survival.
ESR and Apoptosis
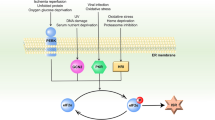
The ESR acts to restore normal ER homeostasis and is therefore cytoprotective. However, when a stress is so strong or persistent that ER dysfunction cannot be corrected, metazoan cells can initiate apoptosis, allowing the regulated destruction of cells that are irreparably damaged or a risk to the organism as a whole. A unified model for ER stress-induced apoptosis is only beginning to emerge, but recent interest in the field has generated an increasing amount of information (see Figure 2).

Simplified depiction of selected apoptotic pathways induced by ER stress. Physiological or experimentally induced ER stress leads to the activation of PERK and, eventually, the GADD34/PP1 phosphatase complex, which dephosphorylates eIF2α, promoting apoptosis. Genetic strategies or chemicals (e.g., salubrinal) that enforce eIF2α phosphorylation protect cells from ER stress-induced apoptosis. Caspase-12 (mice) or -4 (humans) is associated with the cytoplasmic face of the ER membrane and can be activated by ER stress in several ways, including via IRE1 and TRAF2, or by cleavage by calpain, itself activated by the release of calcium from ER stores. Bcl-2 family members also reside in the ER membrane and influence apoptosis induced by ER stress, both through the regulation of calcium flux and amplification of the apoptotic signal via the mitochondrial pathway (not shown). See text for details
Some core components of the protective ESR participate in ER stress-induced apoptosis as well. For example, mammalian Ire1 can activate JNK and downstream proapoptotic kinases such as ASK1, which may contribute to ER stress-induced apoptosis, as it does in response to other stimuli.129, 130 ER stress inducers such as Tm, thapsigargin (Tg, an inhibitor of the ER calcium-dependent ATPase) or reducing agents, as well as the overexpression of Ire1α, induce the activation of JNK.130 Ire1 also interacts with TRAF2, an adaptor protein involved in the signaling pathways of proinflammatory cytokines.130 This interaction recruits ASK1 to form an Ire1/TRAF2/ASK1 ternary complex, which in turn could activate JNK.130, 131 The functional importance of JNK activation in the ER stress pathway has not been fully explored, but ASK1−/− cells are partially resistant to ER stress-induced apoptosis,132 suggesting that JNK may promote apoptosis in this context. A recent report also indicates that ATF6 might mediate a proapoptotic signal during normal myoblast development,133 implicating this upstream UPR sentinel in apoptotic pathways as well.
CHOP has also been shown to promote apoptosis in several contexts,93, 112, 134 and this effect can be blocked by BiP overexpression,111 indicating that CHOP-activated apoptotic pathways are downstream from the ER. CHOP can transcriptionally downregulate the antiapoptotic protein Bcl-2135 and upregulate DR5, a member of the death receptor protein family.136, 137 Interestingly, CHOP also leads to a depletion of cellular glutathione and an increase in ROS in the ER, due in part to its induction of ERO1α, an ER oxidase.116, 135 Interfering with ERO1α function reduces the accumulation of ROS in the stressed ER, leading to cytoprotection.108 Therefore, ERO1α may be an important apoptotic effector downstream of CHOP. CHOP−/− MEFs are partially resistant to ER stress-induced cell death, although CHOP−/− mice are not resistant to lethal doses of Tm,112 suggesting that other proapoptotic pathways are also at work.
The eIF2α phosphatase cofactors GADD34 and CReP also mediate apoptotic signaling downstream from the ER. During ER stress, GADD34−/− cells display persistent eIF2α phosphorylation121, 122 and fewer misfolded protein aggregates in the ER lumen,116 suggesting that GADD34 function and the removal of eIF2α phosphorylation are proapoptotic in this context. Indeed, GADD34−/− mice are resistant to the toxic effects of Tm.116 Similarly, RNAi-mediated knockdown of CReP protects cells from a variety of stimuli, including ER stress.123 It seems likely that the cytoprotection provided by genetic loss of GADD34 or CReP is due primarily to increased eIF2α phosphorylation because enforcing eIF2α phosphorylation with constitutively active PERK had a similar effect.138 Consistent with this hypothesis, we have shown that a selective pharmacological inhibitor of eIF2α phosphatases, termed salubrinal, can protect cells from ER stress-induced apoptosis.139
ER stress can also activate well-known general regulators of mammalian apoptosis, including the Bcl-2 and caspase families of proteins. It has long been known that a pool of endogenous Bcl-2 resides in the ER membrane,140, 141 and while Bcl-2 family members are thought to function principally at the mitochondrial outer membrane,142 there is ample evidence that they influence homeostasis and apoptosis from the ER as well.143, 144 For example, variants of the antiapoptotic family members Bcl-2 or Bcl-XL targeted specifically to the ER membrane can block apoptosis induced by pharmacological kinase inhibition or by proapoptotic Bcl-2 family members.145, 146 Conversely, ER stress itself can upregulate or otherwise activate several ‘BH3-only’ proapoptotic members of the Bcl-2 family, including Bim,147 BIK,148, 149, 150, 151 and PUMA.152, 153 Therefore, efferent signaling from the stressed ER can engage the Bcl-2 death machinery directly.
Several studies have also demonstrated that the proapoptotic multidomain Bcl-2 family proteins, Bax and Bak, regulate ER stress-induced apoptosis. Bax−/−/Bak−/− MEFs are remarkably resistant to many forms of apoptosis, including ER stress,154, 155 implying that many apoptotic signaling pathways converge on Bax and Bak. Interestingly, endogenous Bax and Bak regulate ER stress-induced apoptosis from both the mitochondrial and ER membranes. In the ER membrane, Bax and Bak are crucial for maintaining the resting level of lumenal calcium, probably through an interaction with the type 1 inositol trisphosphate receptor.156, 157, 158, 159, 160 As a result, Bax−/−/Bak−/− cells display reduced calcium release from the ER in response to such stimuli as arachidonic acid and oxidative stress, thereby attenuating apoptosis.159 However, in response to other ER stress stimuli, such as the ER-to-Golgi vesicle transport inhibitor brefeldin A (BFA), Bax and Bak must be present at both the ER and mitochondrial membranes for apoptotic execution to proceed normally.159 Therefore, Bax and Bak participate in signal integration between the ER and the mitochondria to influence cell survival choices from multiple locations within the cell. Interestingly, a recent study also suggested that Bak but not Bax can mediate structural changes in the ER itself in a Bcl-XL-dependent manner, suggesting a unique role for Bak in regulating ER homeostasis.161
The caspase family of proapoptotic cysteine proteases also plays a critical role in ER stress-induced apoptosis. Caspase-12, a murine protein associated with the ER membrane, is activated by ER stress-induced apoptosis but not by other non-ER stimuli162 and is required for cell death in response to both pharmacological ER stress162 and ER-targeted Bim.147 However, caspase-12 can be activated by ER stress in several ways. For example, the cytoplasmic calcium-activated protease calpain can cleave and activate caspase-12 in response to calcium flux from the ER, which is often triggered by ER stress.163 Interestingly, caspase-12 may also auto-activate through a direct association with Ire1α and the adaptor protein TRAF2,164 although how ER stress regulates the formation of this complex is not yet clear. Caspase-12 has also been detected in high molecular weight complex with apoptosis-linked gene-2 protein and p97 (also referred to as valosin-containing protein), the ERAD mediator.165 Interference with the formation of this complex protects cells from ER stress-induced apoptosis, presumably by blocking the activation of caspase-12 or caspase-9.165 Because p97 is also involved in ERAD, it may also mediate crosstalk between the prosurvival and proapoptotic pathways induced by ER stress.
Once activated, caspase-12 can initiate downstream apoptotic pathways. For example, ER stress can induce the activation of caspase-9 independent of Apaf-1, the usual mediator of caspase-9 activation.166 This probably occurs via the direct cleavage of caspase-9 by caspase-12.167 In addition, caspase-7 translocates to the ER in response to some apoptotic stimuli,168 and it has been proposed that caspase-7 can activate caspase-12 directly.169 However, other experiments suggest that caspase-12 cleavage precedes caspase-7 cleavage during ER stress (our unpublished observations), implying that the order of activation may be the opposite in some contexts. In addition, glycogen synthase kinase 3β may influence caspase-3 activation specifically during ER stress,170 although whether this is a direct effect or a far-upstream event remains unclear. A systematic search for caspase-12 substrates should provide additional insight into how ER stress ultimately leads to caspase-dependent cell death.
It is worth noting that the role of human homologs of caspase-12 in ER stress-induced apoptosis has been controversial.171, 172 However, it was recently shown that human caspase-4, which is 48% homologous to murine caspase-12, is localized to the ER membrane and is specifically activated by and required for ER stress-induced apoptosis.173 These data suggest that caspase-4 is the human functional counterpart of murine caspase-12.
As noted above, the apoptotic program initiated by ER stress has been implicated in a wide variety of human diseases.5, 17, 18, 19, 20, 21 Other reviews in this issue of Cell Death and Differentiation cover the role of ER stress and apoptosis in development, metabolic disorders, neurodegeneration and viral infection in more detail.
Future Challenges
Our understanding of the mammalian ESR and the apoptotic program coupled to it has increased greatly in recent years but there remains much to learn. In addition to continuing to identify and characterize the molecular components of the ESR, investigators face several major challenges. First, we know little or nothing about how the numerous signals from the stressed ER are integrated such that the cell can ‘decide’ whether to initiate some or all ESR pathways, activate apoptosis or pursue a different fate altogether. In particular, it is unclear how an individual cell judges when a given level of ER stress is too great or persistent to be corrected by the ESR, making cell death the appropriate course of action. Second, many of the current cell-based models of ER stress rely on chemical agents, such as inhibitors of protein glycosylation and folding, vesicle trafficking or ER calcium flux. In contrast to these acute, synchronous, severe forms of ER stress, the physiological intensity, duration and periodicity of ER stress experienced by cells in vivo are quite different. Therefore, new, physiologically faithful cell-based and biochemical systems are needed to understand ESR signaling in its native context. Third, ER stress has been implicated in myriad human diseases, but the functional role of the ESR in these contexts remains largely unexplored, including questions of which ESR pathways slow disease progression, and which exacerbate it. Finally, because of the importance of the ESR to both cell biology and human health, pharmacologically useful reagents and strategies for the rational manipulation of the ESR are needed. In the future, such research tools and drug candidates may prove invaluable to basic researchers and clinicians alike.
Abbreviations
- BFA:
-
brefeldin A
- eIF2α:
-
eukaryotic translation initiation factor 2 subunit α
- ER:
-
endoplasmic reticulum
- ERAD:
-
ER-associated degradation
- ESR:
-
ER stress response
- GEF:
-
guanine nucleotide exchange factor
- GLS:
-
Golgi localization sequence
- PD:
-
Parkinson's disease
- PDI1:
-
protein disulfide isomerase 1
- PP1:
-
protein phosphatase 1
- ROS:
-
reactive oxygen species
- Tg:
-
thapsigargin
- Tm:
-
tunicamycin
- uORF:
-
upstream open reading frame
- UPR:
-
unfolded protein response
- XBP-1:
-
X-box binding protein 1
References
Alberts B, Johnson A, Lewis J, Raff M, Roberts K and Walter P (2002) Molecular Biology of the Cell (New York: Garland Publishing)
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13: 1211–1233
Mori K (2000) Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101: 451–454
Ma Y and Hendershot LM (2001) The unfolding tale of the unfolded protein response. Cell 107: 827–830
Schroder M and Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat. Res. 569: 29–63
Schroder M and Kaufman RJ (2005) The Mammalian unfolded protein response. Annu. Rev. Biochem. 74: 739–789
Rutkowski DT and Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol. 14: 20–28
Tsai B, Ye Y and Rapoport TA (2002) Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell. Biol. 3: 246–255
Jarosch E, Lenk U and Sommer T (2003) Endoplasmic reticulum-associated protein degradation. Int. Rev. Cytol. 223: 39–81
Hampton RY (2002) ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14: 476–482
Plemper RK and Wolf DH (1999) Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem. Sci. 24: 266–270
Brostrom CO and Brostrom MA (1998) Regulation of translational initiation during cellular responses to stress. Prog. Nucleic Acid. Res. Mol. Biol. 58: 79–125
Harding HP, Calfon M, Urano F, Novoa I and Ron D (2002) Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18: 575–599
Ferri KF and Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3: E255–E263
Rao RV, Ellerby HM and Bredesen DE (2004) Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ 11: 372–380
Breckenridge DG, Germain M, Mathai JP, Nguyen M and Shore GC (2003) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608–8618
Aridor M and Balch WE (1999) Integration of endoplasmic reticulum signaling in health and disease. Nat. Med. 5: 745–751
Paschen W (2003) Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium 34: 365–383
Rutishauser J and Spiess M (2002) Endoplasmic reticulum storage diseases. Swiss Med. Wkly. 132: 211–222
Aridor M and Hannan LA (2000) Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic 1: 836–851
Aridor M and Hannan LA (2002) Traffic jams II: an update of diseases of intracellular transport. Traffic 3: 781–790
Patil C and Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 13: 349–355
Shamu CE and Walter P (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15: 3028–3039
Welihinda AA and Kaufman RJ (1996) The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 271: 18181–18187
Bertolotti A, Zhang Y, Hendershot LM, Harding HP and Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2: 326–332
Cox JS and Walter P (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87: 391–404
Mori K, Kawahara T, Yoshida H, Yanagi H and Yura T (1996) Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1: 803–817
Chapman RE and Walter P (1997) Translational attenuation mediated by an mRNA intron. Curr. Biol. 7: 850–859
Kawahara T, Yanagi H, Yura T and Mori K (1997) Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell 8: 1845–1862
Sidrauski C, Cox JS and Walter P (1996) tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87: 405–413
Mori K, Ogawa N, Kawahara T, Yanagi H and Yura T (2000) mRNA splicing-mediated C-terminal replacement of transcription factor Hac1p is required for efficient activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 97: 4660–4665
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS and Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101: 249–258
Casagrande R, Stern P, Diehn M, Shamu C, Osario M, Zuniga M, Brown PO and Ploegh H (2000) Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell 5: 729–735
Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL and Michaelis S (2004) Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 279: 38369–38378
Plemper RK, Bohmler S, Bordallo J, Sommer T and Wolf DH (1997) Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388: 891–895
Plemper RK, Egner R, Kuchler K and Wolf DH (1998) Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J. Biol. Chem. 273: 32848–32856
Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB and McCracken AA (1999) The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274: 3453–3460
Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ and Romisch K (1999) Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol. 147: 1443–1456
Knop M, Hauser N and Wolf DH (1996) N-glycosylation affects endoplasmic reticulum degradation of a mutated derivative of carboxypeptidase yscY in yeast. Yeast 12: 1229–1238
Spear ED and Ng DT (2005) Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J. Cell Biol. 169: 73–82
Rapoport TA, Matlack KE, Plath K, Misselwitz B and Staeck O (1999) Posttranslational protein translocation across the membrane of the endoplasmic reticulum. Biol. Chem. 380: 1143–1150
Clemons Jr WM, Menetret JF, Akey CW and Rapoport TA (2004) Structural insight into the protein translocation channel. Curr. Opin. Struct. Biol. 14: 390–396
Pilon M, Schekman R and Romisch K (1997) Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 16: 4540–4548
Zhou M and Schekman R (1999) The engagement of Sec61p in the ER dislocation process. Mol. Cell 4: 925–934
Richly H, Rape M, Braun S, Rumpf S, Hoege C and Jentsch S (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120: 73–84
Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH and Sommer T (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 4: 134–139
Braun S, Matuschewski K, Rape M, Thoms S and Jentsch S (2002) Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 21: 615–621
Hitchcock AL, Krebber H, Frietze S, Lin A, Latterich M and Silver PA (2001) The conserved npl4 protein complex mediates proteasome-dependent membrane-bound transcription factor activation. Mol. Biol. Cell 12: 3226–3241
Rape M, Hoppe T, Gorr I, Kalocay M, Richly H and Jentsch S (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107: 667–677
Biederer T, Volkwein C and Sommer T (1997) Role of Cue1p in ubiquitination and degradation at the ER surface. Science 278: 1806–1809
Sommer T and Jentsch S (1993) A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 365: 176–179
Friedlander R, Jarosch E, Urban J, Volkwein C and Sommer T (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2: 379–384
Hampton RY, Gardner RG and Rine J (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3- methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7: 2029–2044
Knop M, Finger A, Braun T, Hellmuth K and Wolf DH (1996) Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 15: 753–763
Bordallo J, Plemper RK, Finger A and Wolf DH (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9: 209–222
Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R and Wolf DH (1999) Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 112: 4123–4134
Haynes CM, Titus EA and Cooper AA (2004) Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15: 767–776
Patil CK, Li H and Walter P (2004) Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2: E246
Hinnebusch AG (1997) Translational regulation of yeast GCN4. A window on factors that control initiator-TRNA binding to the ribosome. J. Biol. Chem. 272: 21661–21664
Tirasophon W, Welihinda AA and Kaufman RJ (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12: 1812–1824
Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB and Ron D (2001) Increased sensitivity to dextran sodium sulfate colitis in IRE1beta- deficient mice. J. Clin. Invest. 107: 585–593
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark S and Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96
Yoshida H, Matsui T, Yamamoto A, Okada T and Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891
Clauss IM, Gravallese EM, Darling JM, Shapiro F, Glimcher MJ and Glimcher LH (1993) In situ hybridization studies suggest a role for the basic region- leucine zipper protein hXBP-1 in exocrine gland and skeletal development during mouse embryogenesis. Dev. Dyn. 197: 146–156
Lee AH, Iwakoshi NN and Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23: 7448–7459
Yoshida H, Haze K, Yanagi H, Yura T and Mori K (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose- regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273: 33741–33749
Haze K, Yoshida H, Yanagi H, Yura T and Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10: 3787–3799
Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M and Mori K (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20: 6755–6767
Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ and Prywes R (2000) Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 275: 27013–27020
Shen J, Chen X, Hendershot L and Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3: 99–111
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS and Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6: 1355–1364
Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K and Mori K (2003) A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4: 265–271
Tokunaga F, Brostrom C, Koide T and Arvan P (2000) Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J. Biol. Chem. 275: 40757–40764
Tsai B, Rodighiero C, Lencer WI and Rapoport TA (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104: 937–948
Knittler MR, Dirks S and Haas IG (1995) Molecular chaperones involved in protein degradation in the endoplasmic reticulum: quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 92: 1764–1768
Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA and Ploegh HL (1996) Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384: 432–438
Bebok Z, Mazzochi C, King SA, Hong JS and Sorscher EJ (1998) The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61beta and a cytosolic, deglycosylated intermediary. J. Biol. Chem. 273: 29873–29878
Ye Y, Meyer HH and Rapoport TA (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414: 652–656
Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K and Hampton RY (2001) HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol. Biol. Cell 12: 4114–4128
Meyer HH, Shorter JG, Seemann J, Pappin D and Warren G (2000) A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 19: 2181–2192
Lilley BN and Ploegh HL (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429: 834–840
Ye Y, Shibata Y, Yun C, Ron D and Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429: 841–847
Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J and Patterson C (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3: 93–96
Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J and Patterson C (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276: 42938–42944
Meacham GC, Patterson C, Zhang W, Younger JM and Cyr DM (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3: 100–105
Murata S, Minami Y, Minami M, Chiba T and Tanaka K (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2: 1133–1138
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS and Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293: 263–269
Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL and Dawson TM (2001) Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat. Med. 7: 1144–1150
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y and Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105: 891–902
Hinnebusch A (2000) Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB, eds (Cold Spring Harbor: Cold Spring Harbor Laboratory Press) pp. 185–244
Clemens MJ (2001) Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog. Mol. Subcell Biol. 27: 57–89
Proud CG (2005) eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 16: 3–12
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M and Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6: 1099–1108
Sood R, Porter AC, Olsen DA, Cavener DR and Wek RC (2000) A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha. Genetics 154: 787–801
Kaufman RJ (2000) Double-stranded RNA-activated protein kinase, PKR. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB, eds (Cold Spring Harbor: Cold Spring Harbor Laboratory Press) pp. 503–528
Williams BR (1999) PKR; a sentinel kinase for cellular stress. Oncogene 18: 6112–6120
Chen JJ (2000) Heme-regulated eIF2α kinase. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB, eds (Cold Spring Harbor: Cold Spring Harbor Laboratory Press) pp. 529–546
Ron D and Harding HP (2000) PERK and translational control by stress in the endoplasmic reticulum. In Translational Control of Gene Expression Sonenberg N, Hershey JWB, Mathews MB, eds (Cold Spring Harbor: Cold Spring Harbor Laboratory Press) pp. 547–560
Harding HP, Zhang Y and Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum- resident kinase. Nature 397: 271–274
Liu CY, Schroder M and Kaufman RJ (2000) Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 275: 24881–24885
Okamura K, Kimata Y, Higashio H, Tsuru A and Kohno K (2000) Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 279: 445–450
Harding HP, Zhang Y, Bertolotti A, Zeng H and Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5: 897–904
Fawcett TW, Martindale JL, Guyton KZ, Hai T and Holbrook NJ (1999) Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 339: 135–141
Fernandez J, Yaman I, Sarnow P, Snider MD and Hatzoglou M (2002) Regulation of internal ribosomal entry site-mediated translation by phosphorylation of the translation initiation factor eIF2alpha. J. Biol. Chem. 277: 19198–19205
Fernandez J, Yaman I, Merrick WC, Koromilas A, Wek RC, Sood R, Hensold J and Hatzoglou M (2002) Regulation of internal ribosome entry site-mediated translation by eukaryotic initiation factor-2alpha phosphorylation and translation of a small upstream open reading frame. J. Biol. Chem. 277: 2050–2058
Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A and Terada N et al. (1998) Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells 3: 801–810
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S and Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7: 1165–1176
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM and Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11: 619–633
Ma Y, Brewer JW, Diehl JA and Hendershot LM (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318: 1351–1365
Luo S, Baumeister P, Yang S, Abcouwer SF and Lee AS (2003) Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 278: 37375–37385
Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM and Ron D (1996) Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 16: 4273–4280
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT and Remotti H et al. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12: 982–995
Jousse C, Bruhat A, Carraro V, Urano F, Ferrara M, Ron D and Fafournoux P (2001) Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5′UTR. Nucleic Acids Res. 29: 4341–4351
Ubeda M, Wang XZ, Zinszner H, Wu I, Habener JF and Ron D (1996) Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol. Cell. Biol. 16: 1479–1489
Ubeda M, Vallejo M and Habener JF (1999) CHOP enhancement of gene transcription by interactions with Jun/Fos AP- 1 complex proteins. Mol. Cell. Biol. 19: 7589–7599
Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18: 3066–3077
Brush MH, Weiser DC and Shenolikar S (2003) Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 23: 1292–1303
Novoa I, Zeng H, Harding HP and Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 153: 1011–1022
Aggen JB, Nairn AC and Chamberlin R (2000) Regulation of protein phosphatase-1. Chem. Biol. 7: R13–R23
Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR and Wek RC (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 24: 1365–1377
Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP and Ron D (2003) Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22: 1180–1187
Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K, Takeda K, Akira S, Shimokata K and Isobe K (2003) The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 17: 1573–1575
Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP and Ron D (2003) Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol. 163: 767–775
Kebache S, Cardin E, Nguyen DT, Chevet E and Larose L (2004) Nck-1 antagonizes the endoplasmic reticulum stress-induced inhibition of translation. J. Biol. Chem. 279: 9662–9671
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ and Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23: 7198–7209
Venugopal R and Jaiswal AK (1998) Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 17: 3145–3156
Venugopal R and Jaiswal AK (1996) Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 93: 14960–14965
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M and Nabeshima Y (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236: 313–322
Urano F, Bertolotti A and Ron D (2000) IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 113 (Part 21): 3697–3702
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP and Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666
Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K and Ichijo H (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 2: 389–395
Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K and Inoue K et al. (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16: 1345–1355
Nakanishi K, Sudo T and Morishima N (2005) Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 169: 555–560
Matsumoto M, Minami M, Takeda K, Sakao Y and Akira S (1996) Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. 395: 143–147
McCullough KD, Martindale JL, Klotz LO, Aw TY and Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21: 1249–1259
Yamaguchi H and Wang HG (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279: 45495–45502
Schneider P and Tschopp J (2000) Apoptosis induced by death receptors. Pharm. Acta. Helv. 74: 281–286
Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D and Harding HP (2004) Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 23: 169–179
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D and Yuan J (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307: 935–939
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W and Reed JC (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 53: 4701–4714
Chen-Levy Z, Nourse J and Cleary ML (1989) The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol. Cell. Biol. 9: 701–710
Kuwana T and Newmeyer DD (2003) Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 15: 691–699
Annis MG, Yethon JA, Leber B and Andrews DW (2004) There is more to life and death than mitochondria: Bcl-2 proteins at the endoplasmic reticulum. Biochim. Biophys. Acta 1644: 115–123
Thomenius MJ and Distelhorst CW (2003) Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. J. Cell Sci. 116: 4493–4499
Wang NS, Unkila MT, Reineks EZ and Distelhorst CW (2001) Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem. 276: 44117–44128
Thomenius MJ, Wang NS, Reineks EZ, Wang Z and Distelhorst CW (2003) Bcl-2 on the endoplasmic reticulum regulates Bax activity by binding to BH3-only proteins. J. Biol. Chem. 278: 6243–6250
Morishima N, Nakanishi K, Tsuchiya K, Shibata T and Seiwa E (2004) Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem. 279: 50375–50381
Germain M, Mathai JP, McBride HM and Shore GC (2005) Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 24: 1546–1556
Germain M, Mathai JP and Shore GC (2002) BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 277: 18053–18060
Mathai JP, Germain M, Marcellus RC and Shore GC (2002) Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene 21: 2534–2544
Mathai JP, Germain M and Shore GC (2005) BH3-only Bik regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J. Biol. Chem. 280: 23829–23836
Reimertz C, Kogel D, Rami A, Chittenden T and Prehn JH (2003) Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 162: 587–597
Luo X, He Q, Huang Y and Sheikh MS (2005) Transcriptional upregulation of PUMA modulates endoplasmic reticulum calcium pool depletion-induced apoptosis via Bax activation. Cell Death Differ. 12: 1310–1318
Zong WX, Lindsten T, Ross AJ, MacGregor GR and Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15: 1481–1486
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V and Ross AJ et al. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, O'Neil RG and McConkey DJ (2002) Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J. Biol. Chem. 277: 20301–20308
Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ and Swisher SG (2002) Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277: 9219–9225
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M and Pozzan T et al. (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102: 105–110
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD and Pozzan T et al. (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J and Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 162: 59–69
Klee M and Pimentel-Muinos FX (2005) Bcl-X(L) specifically activates Bak to induce swelling and restructuring of the endoplasmic reticulum. J. Cell Biol. 168: 723–734
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA and Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103
Nakagawa T and Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150: 887–894
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T and Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276: 13935–13940
Rao RV, Poksay KS, Castro-Obregon S, Schilling B, Row RH, del Rio G, Gibson BW, Ellerby HM and Bredesen DE (2004) Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J. Biol. Chem. 279: 177–187
Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE and Ellerby HM (2002) Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J. Biol. Chem. 277: 21836–21842
Morishima N, Nakanishi K, Takenouchi H, Shibata T and Yasuhiko Y (2002) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 277: 34287–34294
Chandler JM, Cohen GM and MacFarlane M (1998) Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J. Biol. Chem. 273: 10815–10818
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM and Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J. Biol. Chem. 276: 33869–33874
Song L, De Sarno P and Jope RS (2002) Central role of glycogen synthase kinase-3b inendoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 12: 12
Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT, Hotchkiss RS, Buchman TG, Zehnbauer BA, Hayden MR, Farrer LA, Roy S and Nicholson DW (2004) Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429: 75–79
Fischer H, Koenig U, Eckhart L and Tschachler E (2002) Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Commun. 293: 722–726
Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y and Tohyama M (2004) Involvement of caspase-4 in endoplasmic reticulum st. J. Cell Biol. 165: 347–356
Acknowledgements
We thank G Hotamisligil, T Rapoport and J Tschöpp for helpful comments and discussion. This work was supported in part by a grant (R37 AG012859) from NIA to JY.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Boyce, M., Yuan, J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 13, 363–373 (2006). https://doi.org/10.1038/sj.cdd.4401817
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401817
Keywords
This article is cited by
-
Targeting and regulation of autophagy in hepatocellular carcinoma: revisiting the molecular interactions and mechanisms for new therapy approaches
Cell Communication and Signaling (2023)
-
Prolyl 4-hydroxylase subunit beta (P4HB) could serve as a prognostic and radiosensitivity biomarker for prostate cancer patients
European Journal of Medical Research (2023)
-
Involvement of histone methylation in the regulation of neuronal death
Journal of Physiology and Biochemistry (2023)
-
Association between protein undernutrition and diabetes: Molecular implications in the reduction of insulin secretion
Reviews in Endocrine and Metabolic Disorders (2023)
-
A Review on Caspases: Key Regulators of Biological Activities and Apoptosis
Molecular Neurobiology (2023)
