




Abstract
The most common cause of primary autosomal recessive microcephaly (MCPH) appears to be mutations in the ASPM gene which is involved in the regulation of neurogenesis. The predicted gene product contains two putative N-terminal calponin-homology (CH) domains and a block of putative calmodulin-binding IQ domains common in actin binding cytoskeletal and signaling proteins. Previous studies in mouse suggest that ASPM is preferentially expressed in the developing brain. Our analyses reveal that ASPM is widely expressed in fetal and adult tissues and upregulated in malignant cells. Several alternatively spliced variants encoding putative ASPM isoforms with different numbers of IQ motifs were identified. The major ASPM transcript contains 81 IQ domains, most of which are organized into a higher order repeat (HOR) structure. Another prominent spliced form contains an in-frame deletion of exon 18 and encodes 14 IQ domains not organized into a HOR. This variant is conserved in mouse. Other spliced variants lacking both CH domains and a part of the IQ motifs were also detected, suggesting the existence of isoforms with potentially different functions. To elucidate the biochemical function of human ASPM, we developed peptide specific antibodies to the N- and C-termini of ASPM. In a western analysis of proteins from cultured human and mouse cells, the antibodies detected bands with mobilities corresponding to the predicted ASPM isoforms. Immunostaining of cultured human cells with antibodies revealed that ASPM is localized in the spindle poles during mitosis. This finding suggests that MCPH is the consequence of an impairment in mitotic spindle regulation in cortical progenitors due to mutations in ASPM.
INTRODUCTION
Primary autosomal recessive microcephaly (MCPH) is a genetic disorder in which an affected individual has a head circumference >3 standard deviations below the age- and sex-related mean. The affected individuals are born with a significantly smaller head circumference and are mentally retarded but have no other abnormal findings or neurological features in brain size (1–3). Although brain scans show that the whole brain is reduced in size, the cerebral cortex is most severely affected. Because the vast majority of neurons are generated by week 21 of fetal life, MCPH is likely due to a reduced production of neurons. MCPH is genetically heterogeneous, with six loci currently mapped (2–4). The most common cause of MCPH appears to be mutations in the ASPM gene (5,6).
The ASPM gene contains a 10 434 bp long coding sequence with 28 exons and spans 65 kb of genomic DNA at 1q31 (6). Expression studies in mice show that Aspm is specifically expressed in the cerebral cortical ventricular zone, the proliferative region of the lateral and medial ganglionic eminence and the ventricular zone of the dorsal diencephalon at embryonic day 14.5 (6). Expression diminished by E16.5 and was greatly reduced at birth. Aspm mRNA was also detected in other developing organs on day E16.7 as well as in testis, ovary and spleen in the adult mouse (7).
The predicted ASPM protein has two distinguished regions, a tandem pair of N-terminal putative calponin-homology (CH) domains, and a large block of IQ motifs (the single letter code for the amino acids isoleucine and glutamine), which mediate interaction with calmodulin and calmodulin-related proteins. CH domains are ∼100 residues long and are commonly involved in actin binding, but many other substrates have been found in vivo (8,9). IQ calmodulin-binding motifs comprise 20–25 amino acids, with the core fitting the consensus IQXXXRGXXXR (where X is any amino acid) (10,11). CH and IQ domains were first discovered in motor proteins such as unconventional myosins (12). It has been shown that calmodulin binding to IQ motifs induces a conformational change in proteins that regulate the binding of actin to the amino-terminal CH domains (13,14). Typically unconventional myosins contain between 2 and 6 IQ motifs. Even within a single protein these motifs are not identical, some of them have a higher affinity for the Ca+2‐free form of calmodulin, whereas others have a higher affinity for the Ca+2/calmodulin complex. The number of IQ motifs and their divergence seems to determine the length of the lever arm and hence the step size of the myosin motor.
In contrast to unconventional myosins, ASPM does not have the catalytic S1 motor domain, and therefore it cannot function as a mechanochemical protein. ASPM also differs from all known motor proteins by an extreme abundance of IQ motifs. The protein contains 81 putative calmodulin-binding motifs, most of which are encoded by exon 18, the largest exon in ASPM (the data herein).
The predicted ASPM protein is conserved between mammals, Drosophila, and nematodes, with a consistent correlation between the organism's complexity and protein length, principally involving an increase in the number of encoded IQ domains (6,15). For example, the ASPM homolog of Caenorhabditis elegans contains two IQ domains, Drosophila melonagaster contains 24 and in mammals IQ domains vary between 60 and 81. Interspecies comparisons also reveal that ASPM has experienced intense positive selection during recent human evolution, suggesting that the selection of specific segments of this gene may play a role in the evolutionary enlargement of the human brain (15–17).
The biochemical function of human ASPM has been proposed on the basis of the analysis of Drosophila mutants with a deficiency in the asp gene (6,18,19). Drosophila abnormal spindle (asp) mutants exhibit a mitotic metaphase checkpoint arrest with abnormal spindle poles, which reflect the requirement of Asp for the integrity of microtubule organizing centers. The Drosophila asp gene encodes a 220 kDa microtubule-associated protein found at the spindle poles and centrosomes from prophase through early telophase. Besides calmodulin-IQ-binding and CH domains, the Asp protein contains consensus phosphorylation sites for CDK1 and MAP kinases (20). Asp is co-purified with γ-tubulin from centrosomes and both are required for the organization of microtubules into asters (18). This activity is dependent on the phosphorylation of Asp by the kinase Polo (19,21). Together, these observations suggest that mutations in human ASPM may cause microcephaly due to the disregulation of mitotic spindle activity in neuronal progenitor cells.
In the present report, we demonstrate that human ASPM localizes to the spindle pole from prophase through telophase, similar to its homolog in the fly. We also show that ASPM encodes several alternatively spliced isoforms that are expressed in most adult tissues and upregulated in malignant cells.
RESULTS
ASPM is expressed in a variety of embryonic and adult tissues and is upregulated in cancer
Previously, using in situ hybridization, expression of mouse Aspm was detected in the cerebral cortical ventricular zone, the proliferative region of the lateral and medial ganglionic eminence and the ventricular zone of the dorsal diencephalon (6). In addition, Luers et al. (7) reported the presence of Aspm mRNA in other developing organs on day E16 as well as in some adult tissues. In our study, we re-investigated gene expression in the whole mouse embryo. We confirmed that in addition to the brain, Aspm is expressed in other organs during fetal development including the liver, heart, lung and kidney (Fig. 1A). Similar results were obtained for human ASPM using RT–PCR analysis. ASPM transcripts were detected in a variety of human embryo tissues (brain, bladder, colon, heart, liver, lung, skeletal muscle, skin, spleen and stomach) using two pairs of primers specific for exons 2 and 3 and exons 14 and 15 (FN2–3/RN2–3 and FN14–15/RN14–15, respectively) (Supplementary Material, Table S1). Bands of the expected size (258 and 280 bp for exon 2–3 and exon 14–15 sequences, correspondingly) were detected in all samples, indicating ubiquitous expression of ASPM (Fig. 1B). Given that the mouse Aspm homolog is expressed in some adult tissues, we examined whether human ASPM is also expressed in adult tissues (breast, lung, pancreas, uterus, colon, thyroid, liver, bladder, kidney, ovary, testis, stomach, lymph node, cervix, esophagus and brain). The same exon-specific products were detected in all tissues except for the adult brain (data not shown), although the level of expression was much lower than in fetal tissues. Thus, it is possible that ASPM has additional non-CNS (non-central nervous system) function. We also examined the expression data obtained from over 120 uterine cancers determined by high-density oligonucleotide microarrays and found ASPM to be one of several genes highly overexpressed when compared with the normal endometrium (C. Gadisetti and J. Risinger, manuscript in preparation). On the basis of this initial observation, we further examined ASPM expression in normal and cancerous tissues using quantitative real-time RT–PCR and confirmed that ASPM is more highly expressed in cancers of the uterus and ovary when compared with their normal tissue counterparts (Fig. 2). We expanded our analysis of ASPM expression in cancer by including several other tumor types and obtained similar results for normal and matching tumor tissues of the breast, colon, thyroid, testis, lymph node and stomach, as well as for 60 cancer cell lines (Supplementary Material, Fig. S1 and Table S3).
Thus, we conclude that the human ASPM gene is widely expressed in a variety of adult and embryonic tissues. Because ASPM is upregulated in malignancy, its expression presumably tracks a fraction of dividing cells in the tissue. In agreement with this, a good correlation was observed between ASPM expression and the expression of a common cell proliferation marker, PCNA (data not shown).
Alternatively spliced variants of human ASPM code for different numbers of IQ domains
The open reading frame (ORF) of ASPM was predicted, on the basis of the sequences of multiple small size ESTs deposited into GenBank covering 28 exons. To characterize the human transcript(s), we carried out an RT–PCR analysis using the primers ASPM1F and ASPM28R, which are present in the presumed first and last exons of ASPM (Supplementary Material, Table S1). As seen in Fig. 3, two predominant PCR products were detected with the RNA from fetal tissues. The largest of these migrated at a size consistent with the predicted full-length mRNA of 10 434 bp and was present in all fetal human tissues analyzed. The next most abundant band migrated at ∼5.6 kb. Additional smaller PCR products were also observed. Similar sized bands were detected in normal as well as in matching tumor tissues (Supplementary Material, Fig. S1A). Next, we directly sequenced the predominant PCR products and confirmed that the largest one corresponded to the predicted full-length ORF of 10 434 nucleotides. Further cloning and sequencing of the smaller RT–PCR fragments revealed three mRNAs with ORFs of 5678, 4259 and 3189 bp (GenBank accession nos AY971956, AY971957 and AY971955, respectively) (Supplementary Material, Table S2). Therefore, in addition to the predicted protein of 3477 amino acid residues, the ASPM gene may encode at least three isoforms containing 1892 (variant 1 corresponding to the abundant RT–PCR product), 1389 (variant 2) and 1062 (variant 3) amino acid residues. The difference between these shorter isoforms is primarily in the number of IQ domains (Fig. 4A). Specifically, variant 1 lacks exon 18 (carrying 67 IQ domains), leaving this isoform with 14 total IQ domains. Variants 2 and 3 utilize different splice junctions within exon 18 and truncate parts of the IQ array. As a result of this splicing, variant 2 has 41 complete IQ domains plus an incomplete one, and variant 3 has 27 complete IQ domains plus an incomplete one. In addition, both variants 2 and 3 lack exons 4–17 carrying two CH domains. Variant 2 also lacks exon 27 and has a stop codon in the intronic region after exon 26.
To investigate whether alternatively spliced ASPM isoforms are conserved in evolution, we performed a RT–PCR analysis of a panel of multiple mouse tissue cDNAs using a pair of primers from the first and last exons of the murine Aspm. We detected at least two predominant isoforms on ethidium bromide stained gels (Supplementary Material, Fig. S1B). Sequencing of these prominent bands indicated that the largest was the full-length Aspm of 9217 bp (containing 67 IQ domains), and the second was an exon 18 deleted isoform transcript of 5574 bp (15 IQ domains; GenBank accession no AY971958) (Supplementary Material, Table S2). Both transcripts are abundant in the testis and the embryo. The presence of identically spliced ASPM variants lacking exon 18 in the human and mouse suggests that this variant encodes an isoform essential for ASPM function.
IQ motifs in the ASPM protein are organized into a HOR structure
More than half of the human ASPM protein consists of repeated calmodulin-binding IQ domains. This IQ array, found at positions 1273–3234, is formed by 81 distinct IQ motifs of variable length (Fig. 5A). In comparison, the IQ array present in the mouse and rat exhibits one large deletion corresponding to a region between IQs 57 and 70 in the human along with several smaller insertions and deletions. In total, 67 IQ repeats are found in the mouse Aspm (Supplementary Material, Fig. S2). The number of IQ repeats is slightly higher than previously reported (74 and 61 repeats for human and mouse, respectively) (6,22), and the difference can be attributed to the very sensitive hidden Markov profile search used in our analysis.
We further examined the IQ repeat region of ASPM and found that the length of individual IQ domains is variable ranging from 14 (IQ 78) to 38 (IQ 76) amino acids. The distribution of variably sized IQ repeats is not random (Fig. 5A). The central part of the array between IQs 4 and 54 displays a striking periodicity, with long (27 amino acid) IQ repeats being dispersed among short (23 amino acid) IQ repeats. The only exceptions are IQ 43 with 26 amino acids instead of 27, and IQs 7–8 with 22 amino acids instead of 23. The spacing between long and short units is highly regular. The long IQ 6 repeat is followed by four short units; IQ 11 and IQ 15 are followed by three units. The next 12 long units are in each case followed by two short units. This region forms a highly regular array of 63 amino acid long superunits, composed of one long 27 amino acid unit and two short 23 amino acid units. The N- and C-terminal parts of the IQ repeat region are less regular and contain IQ units of variable length. The mouse Aspm protein displays the same periodicity 27–23–23 in the central region; the terminal parts are less organized as in the case of human ASPM (Supplementary Material, Fig. S2).
We were also interested in the sequence conservation of the long (Fig. 5B) and short units (Fig. 5C). Our data indicates the consensus sequence is (I,l,v)QX2(Y,F,w)(k,r)aX10(y,f)X3(k,r)X3(i,l,v)X for the long unit and (I,l,v)QsX(Y,F,w)(R)X15(i,l,v)X for the short unit. Strongly conserved amino acids are in uppercase letters, less preserved/minor residues are in lowercase, and ‘X’ denotes non-conserved positions. Physico-chemical properties of individual positions are shown in Figure 5B and C.
Finally, we analyzed the pattern of amino acid replacements during primate evolution (Fig. 5A). The conserved positions are mostly intact, as expected, and the majority of the changes are in non-conserved sites. Both conservative and non-conservative substitutions are found in all long, short and unordered repeats. A similar situation is found in the mouse and rat (Supplementary Material, Fig. S2). Interestingly, IQ repeats 66–69, which are deleted in mouse and rat, are almost identical in the examined primates. The presence of higher-order repeat structure of IQ domains in all mammalian ASPM proteins suggests the structure is of functional significance. It is worth noting that because the IQ consensus includes two positively charged amino acids, the IQ-containing region that spans ∼2000 amino acid is highly positively charged. Among the 530 charged residues, 467 are arginine and lysine. Although such a high density of basic residues may be required for the binding of calmodulin, it may also facilitate the interaction of ASPM with other acidic proteins.
Identification of a novel ASNP repeat region within ASPM
The analysis of ASPM revealed two novel repeats in the N-terminal part of the protein. The repeats are 32 and 35 amino acids long and localized to positions 316–347 and 366–400, respectively (Fig. 4B). These repeats are highly conserved near the termini, with the central part being more variable. Similar repeats were found in all mammalian ASPM homologs. A single repeat was identified in the chicken gene homolog. The interspecies conservation is similar to intraspecies comparisons, i.e. the termini are highly similar: the N-terminus is almost identical, but the central part is variable. We named the repeats ASNP (for ASPM N-proximal). We did not detect any significant similarity of this motif to other proteins even when using very sensitive searches (psiblast, HMMER; data not shown). Yet the conservation in the compared species suggests that strong selective pressure preserves the repeats, indicating their importance for ASPM function.
ASPM is found at the spindle poles during mitosis in human cell cultures
The role of Drosophila Asp in nucleating microtubules at centrosomes is consistent with its localization at the spindle poles during mitotic periods (20). In order to understand the functional property of human ASPM, we have generated polyclonal antibodies against three epitopes of the human protein (see Materials and Methods). The polyclonal antibodies showed a high degree of specificity that allowed us to confirm the existence of several ASPM isoforms that were predicted on the basis of the analysis of alternatively spliced mRNA variants. In the western analysis of proteins from HT1080 human cells, the affinity-purified VTRK and QSPE antibodies detected two bands: one with the mobility predicted for the full-size ASPM protein (410 kDa) and another with the mobility predicted for the prominent spliced form, which contains an in-frame deletion of exon 18 (218 kDa). In addition, the antibodies detect at least four other bands in the interval between 110 and 150 kDa (Fig. 6A). On the basis of their mobility, two of them presumably correspond to the predicted alternatively spliced variants 2 and 3 (Fig. 4). Because the epitopes were chosen from conserved regions of the ASPM protein, the developed antibodies also detect mouse ASPM. Two major bands with molecular weights 364 and 212 kDa, corresponding to the full-size mouse ASPM and its isoform lacking the exon 18 coding sequence, can be seen on westerns (Fig. 6B). To further characterize the ASPM isoforms, we analyzed proteins in the MM10458 cell line which was derived from a patient with primary microcephaly. The ASPM gene in this cell line carries a frameshift mutation in exon 24 and therefore is certainly non-functional (22). The position of the frameshift mutation suggests that nonsense-mediated RNA decay (NMD) would prevent the formation of a truncated protein (23). However, western analysis detected bands with motilities very close to those observed for the full-size ASPM and its prominent isoform (Fig. 6A). Because the calculated molecular weights of the bands correspond to the predicted truncated isoforms (385 and 183 kDa), they seem to represent real truncated products rather than new ASPM isoforms generated by alternative splicing. This is also supported by the fact that the SRKL antibody raised against the C-terminus of ASPM does not recognize these bands (data not shown). This finding indicates that the mutated ASPM mRNA is not sensitive to NMD, as has been shown for some other mRNAs (23,24). It is worth noting that three of the four bands identified in HT1080 cells with an apparent molecular weight ranging from 110 to 140 kDa are missing in MM10458 cell extracts (Fig. 6A). This observation may be explained by a specific effect of the mutation in exon 24 on ASPM mRNA splicing or a different pattern of splice variants produced in HT1080 and MM10458 cells.
Anti-ASPM antibodies for three different epitopes, VTRK, QSPE and SRLK, were used for indirect immunostaining of human HT1080 cultured cells. During prometaphase, metaphase and anaphase, the VTRK and QSPE antibodies showed a strong and similar typical spindle pole-staining pattern (Fig. 7). A similar staining pattern was observed with the SRLK antibody during mitotic periods. During interphase, the antibody revealed the focal staining external to the nucleus. The observed mitotic subcellular localization of ASPM and the highly conserved structural homology between Drosophila Asp and human protein together suggest that ASPM may also be involved in the common spindle function conserved between the two organisms.
DISCUSSION
The results reported herein suggest that human ASPM is a spindle pole/centrosome protein and therefore appears to be a functional ortholog of the Drosophila asp protein, as it has been previously proposed on the basis of the protein's similarity (6). This idea is supported by a recent analysis of proteins from purified human mitotic spindles (25) and provides more evidence that MCPH is a disorder of neurogenic mitosis. Being a component of the mitotic spindle, ASPM may control the proliferative symmetry of progenitors that appears to be pivotal for the expansion of cerebral cortical size (26). Alternatively, the lack of a functional ASPM may affect the fidelity of chromosome segregation, resulting in a high incidence of chromosome aneuploidy that leads to a reduced ability of fetal stem cells to produce neurons. In a recently published paper, Bond et al. (4) identified two additional MCPH genes, MCPH3 and MCPH6. It is notable that both also encode for spindle pole proteins. This discovery and our findings together indicate a key role for the centrosome in regulating the number of neurons generated by neural precursor cells.
Further experiments are required to elucidate the molecular mechanisms of such regulation by ASPM. At this time, the role of ASPM in non-CNS tissues remains obscure. The fact that ASPM localizes at the spindle poles in cultivated cells do not prove that it is essential for mitosis, because no defects other than microcephaly are found in patients carrying mutations in this gene. So far, over 20 different mutations causing brain size reduction have been reported, with all mutations predicted to be protein-truncating (22). Formally, this observation indicates that ASPM is essential only for neurogenic mitosis. However, we cannot exclude the possibility that indeed ASPM encodes two different gene products, one of which is required for mitosis by all dividing cells, whereas the function of the other is restricted to the developing brain. If true, then the ASPM mutations reported so far would result in the loss of only one protein isoform. The other isoform's expression would not be affected because it is encoded by one of the multiple alternatively spliced mRNAs, as is common for brain-expressed genes (27). The brain-specific isoform is likely to correspond to ∼410 kDa protein detected by western blotting. One of the smaller proteins detected in cell extracts may be a major ASPM product required for mitosis by all dividing cells.
Although further analysis is required to clarify the role of the identified ASPM isoforms in non-CNS tissues, detection of the ASPM protein in spindle poles of cultivated cells is an important step in understanding ASPM's function in neurogenic mitosis. For example, we can identify molecular partners of human ASPM using cell extracts, as has been done for the fly Asp protein (18,19,21). Cultivated cells can also be used to characterize alternatively spliced variants that encode ASPM isoforms with different numbers of IQ motifs. The main ASPM isoform corresponds to a 3477 amino acid residue protein containing 81 IQ motifs. Most IQ motifs are organized into a higher-order trimer repeat (HOR) containing two 23 and one 27 amino acid residue units. The HOR structure of the IQ array is conserved in primate and mouse ASPM proteins, suggesting that such a structure is essential for the protein's function. Interestingly, five IQ repeats of myosin V are also organized into a higher-order structure, but the IQ domain is much shorter and forms a 25–23–25–23–25 amino acid residue array. Myosin V is an unconventional myosin that transports cellular cargos such as vesicles, melanosomes or mRNA on actin filaments. The IQ periodicity seems to be necessary for an efficient interaction between myosin V IQ domains and actin half-repeats, which is 36 nm long (28). Analogously, the ASPM IQ periodicity may provide the exact spacing required for interactions with polymeric periodical proteins such as actin.
The major alternatively spliced form contains an in-frame deletion of the entire exon 18 sequence and encodes a 1892 amino acid protein that is predicted to harbor 14 flanking IQ domains not exhibiting any periodicity. Similarly, two predominant transcripts are also present in the mouse. In addition, alternatively spliced variants lacking both CH domains and more than half of the mostly periodical IQ domains were also detected, suggesting that ASPM may encode several proteins with different functions.
The presence of a long array of putative calmodulin-binding IQ domains is a unique feature of ASPM (13). Of the IQ-containing proteins identified so far (>100), ASPM has the highest number of IQ domains. Even if some of these domains are non-functional, ASPM could potentially bind more calmodulin molecules than any other known IQ-containing protein. Calmodulin appears to regulate protein function by modulating its ability to bind to different targets. This can occur by two mechanisms, either by altering the conformation of the protein or by influencing its subcellular location. Both the copy number and HOR structure of the calmodulin-binding domains in ASPM can greatly affect both mechanisms. The presence of multiple domains (IQ, CH and ASNP identified in this work) within ASPM also suggests additional functions such as serving as a scaffolding protein that assembles multiprotein complexes.
If human ASPM is involved in nucleating microtubules at centrosomes, similar to the Drosophila ortholog (18–20), the specific role of IQ repeats may be to accumulate Ca+2/calmodulins at the central region of the protein. One molecule of ASPM protein can potentially bind several hundred calcium ions through calmodulin. The release of bound Ca+2 may signal microtubule polymerization. Future studies are required to check this hypothesis and to elucidate why mutations in ASPM affect brain development without effecting the development and function of non-CNS tissues.
Our results indicate that ASPM is widely expressed in adult tissues and is upregulated in cancer cells. At present, we do not know whether ASPM plays any role in mitosis of non-CNS cells or whether its transcription simply reflects the proliferation of a tissue. Nevertheless, the localization of the gene product(s) at the mitotic spindle suggests that some mutations (or polymorphic variants) may not affect ASPM function in neural progenitors but may instead compromise the fidelity of cell division and produce chromosome instability in adult tissues. Intriguingly, centrosome defects have been found in numerous forms of cancer (29,30). Therefore, further studies of ASPM polymorphism and isoform function are required not only to clarify the molecular mechanisms of microcephaly but also to check the possible involvement of the spindle protein in the predisposition to cancer.
MATERIALS AND METHODS
Mouse embryo in situ hybridization
Non-radioactive in situ hybridization was performed using a digoxigenin-labeled cRNA probe. The antisense probe was generated from a mouse EST clone (GenBank accession no. AW558815) using standard methods, and frozen sections were hybridized and visualized using methods described previously (31).
Analysis of fetal and adult tissues by RT–PCR
Total RNA from human fetal tissues (brain, bladder, colon, heart, liver, lung, skeletal muscle, skin, spleen and stomach) (Stratagene, La Jolla, CA, USA), adult human tissues (brain, breast, lung, colon, thyroid, ovary, testis, stomach and lymph node), matching adult human tumor tissues and normal adult mouse tissues (brain, testis, liver, heart) (Ambion Inc., Austin, TX) was used for screening ASPM expression with the primers described in Supplementary Material, Table S1. cDNA was made from 1 µg of total RNA using the Superscript first strand system kit (Invitrogen, Carlsbad, CA, USA) and priming with oligo dT per their standard protocol. Human β-actin primers (BD Biosciences Clontech, Mountain View, CA, USA) were used as positive controls for both human and mouse RT–PCR. NCI-60 cancer cell lines were from the National Cancer Institute, NIH. RT–PCR was performed using 1 µl of cDNA in a 50 µl reaction volume. Standard reaction conditions were 94 C 5 m, (94 C 1 m, 55 C 1 m, 72 C 1 m×35 cycles), 72 C 7 m, 4 C hold. Sequencing of the RT–PCR products confirmed that they were indeed ASPM transcripts.
Analysis of cancer tissues by real-time PCR
RNA levels of ASPM were assessed in clinical specimens. The relative expression of ASPM was measured by quantitative PCR using FAM-labeled TaqMan® Gene Expression Assays purchased from Applied Biosystems, (Foster City, CA, USA) with VIC-labeled ββ-actin (4326315E) as the reference. Samples were run on an ABI Prism® 7700 Sequence Detection System according to the manufacturer's suggested protocols. The relative quantitation was calculated for each sample using the comparative CT method.
Sequencing of ASPM splice variants
The full-length ASPM transcripts (10 434 bp for human and 9846 bp for mouse) were sequenced after cloning of RT–PCR products into a TA vector. The RT–PCR products corresponding to human ASPM alternatively spliced variants of 5679, 4259, 3189 bp as well as the mouse splice variant of 5574 bp were also sequenced after cloning into a TA vector. Forward and reverse sequence reactions were run on a PE-Applied Biosystem 3100 Automated Capillary DNA Sequencer. Comparative analyses were performed against the wild-type cDNA sequences (GenBank accession nos gi:24211028 and gi:36031058 for human and mouse) using the GCG DNA Analysis Wisconsin Package and NCBI BLAST. All sequenced clones were named and numbered according to the clone/accession identifier (Supplementary Material, Table S2).
Sequence analysis and identification of IQ repeats
Sequences were aligned by Dialign2.1 (32) (http://bibiserv.techfak.uni-bielefeld.de/dialign/). Protein alignments were visualized by GeneDoc (33) (http://www.psc.edu/biomed/genedoc) and WAVis (34) (http://wavis.img.cas.cz). Conserved domains in the ASPM protein were detected by Rpsblast using the default parameters (35) (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). The human ASPM protein was initially screened for IQ repeats by Radar (http://www.ebi.ac.uk/Radar/). The detected IQ repeats were used to build a hidden Markov profile using HMMER version 2.2 (36) (http://hmmer.wustl.edu/). Next, the obtained profile was used to search for the remaining IQ repeats. The IQ sequences were manually edited to obtain the exact boundaries of all IQ units. We used this complete set to build a new hidden Markov profile, which subsequently served to detect IQ repeats in mouse Aspm. Other ASPM repeats, including newly detected N-terminal repeats, were identified by Radar.
Peptide specific antibodies and western blot analysis
Three synthetic peptides representing amino acids 422–441 (QSPEDWRKSEVSPRIPE), 497–520 (VTKRKATCTRENQTEINKPKAKR) and 3443–3463 (SRLKPDWVLRRDNMEEITNPL) from the ASPM sequence were conjugated to Keyhole Limpet Hemocyanin and used as immunogens as previously described (37). The resulting antisera were affinity-purified over columns of peptides conjugated to Affigel 15 (Bio-Rad, Hercules, CA, USA) and concentrated in stirred cells with YM30 membranes (Millipore, Billerica, MA, USA). The concentrates were then subjected to gel filtration chromatography using 2.6×60 cm2 Superdex 200 columns (GE Healthcare, Piscataway, NJ, USA), and the monomeric IgG fractions were pooled and concentrated. The protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). These peptides are identical to both human and mouse ASPM proteins. The antibody specificities were first confirmed using recombinant ASPM fragments (Fig. 6C and D). Plasmids for ASPM expression in Escherichia coli cells were constructed by inserting a 1353 bp fragment containing QSPE and VTKR epitopes and a 705 bp fragment containing an SRLK epitope. The fragments were PCR amplified from the full-size ASPM cDNA (positions 346–1695 and 9730–10434) and cloned into a BamHI site of the pMAL-p2X expression vector (New England BioLabs Inc., Beverly, MA, USA). To analyze the ASPM protein in mammalian cells, human (fibrosarcoma HT1080 and MM10458) and mouse (primary mouse embryonic, PMEC) cells were used. MM10458 is a lymphoblast cell line derived from a patient with primary microcephaly carrying a frameshift mutation in ASPM (2). The cells were mixed with SDS sample buffer containing a protease inhibitor cocktail (Sigma-Aldrich Corp., St. Louis, MO, USA), homogenized using a 27 Ga needle and resolved in a 3.4 or 4% acrylamide/bis-acrylamide (29:1) gel. Following electrophoresis, the proteins were transferred to PVDF membranes (Millipore, Billerica, MA, USA) for 40 min at 15V in transfer buffer (50 mm Tris, 380 mm glycine, 0.1% SDS and 20% methanol) by the semi-dry method. All subsequent steps were carried out in PBS containing 0.05% Tween-20 (TPBS). After blocking for 30 min with 10% non-fat milk–TPBS, the membranes were exposed to 1/5000 diluted anti-QSPE or anti-SRLK antibodies for 1.5 h. The PVDF membrane was washed three times with TPBS, incubated for 30 min with 1/5000 diluted HRP conjugated anti-rabbit IgG and then washed as in the previous step. The membranes were incubated for 1 min with ECL plus reagents (GE Healthcare, Piscataway, NJ, USA) and exposed to Hyperfilm ECL (GE Healthcare, Piscataway, NJ, USA). Immunoblotting of proteins from human and mouse cells revealed ASPM isoforms corresponding to the predicted alternatively spliced variants. No bands were detected with the pre-immune serum. Pre-absorption of the VTKR and SRLK antibodies with excess antigenic peptide (100 µm) abolished the signal (data not shown).
Indirect immunofluorescence staining with anti-ASPM antibodies
To detect the endogenous ASPM protein, two different fixation methods were employed with HT1080 cells grown on poly-d-lysine coated coverslips: 1) cultured cells were fixed by incubating for 15 min with 2% paraformaldehyde, washed twice and treated for 5 min with 0.5% Triton X-100 (Sigma-Aldrich Corp., St Louis, MO, USA) followed by 5 min with 0.1 m Glycine (Sigma-Aldrich Corp.); 2) cultured cells were fixed by treating for 30 min with 100% methanol (Mallinckrodt Baker Inc., Phillipsburg, NJ, USA). Fixed samples were incubated for 1 h at 37°C with anti-ASPM antibodies (1:200). After three 5 min PBS washes, samples were incubated for 1 h at 37°C with Alexa Fluor 594 goat anti-rabbit IgG (Invitrogen). After three 5 min PBS washes, samples were stained with 1 µg/ml of DAPI and rinsed again with PBS. Samples were mounted using VECTASHIELDR mounting medium (Vector Laboratories, Burlingame, CA, USA). Images were captured using a Zeiss microscope (Axiophoto) equipped with a cooled-CCD camera (Cool SNAP HQ, Photometrics, Tucson, AZ, USA) and analyzed by IPLab software (Scanalytics Inc., Fairfax, VA, USA).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank Dr Urs Berger for his help with in situ hybridization and Ms Nina Kouprina for professional editing of this manuscript.
Conflict of Interest statement. None declared.
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.
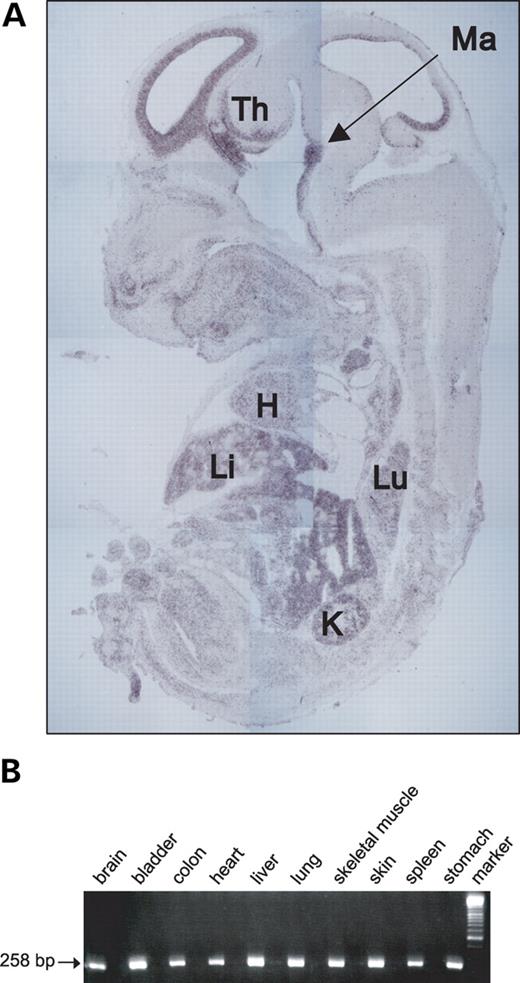
Figure 1. (A) Expression of Aspm in E14 whole mouse embryo detected by hybridization to the antisense probe. Embryonic parasagittal sections for in situ hybridization were prepared using a standard procedure. Th, thalamus; Ma, mammilary region; H, heart; Li, liver; Lu, lung; K, kidney. (B) Expression of human ASPM in embryonic tissues. The ASPM transcripts in various tissues were analyzed by RT–PCR. Expression analyses of mRNA were performed using human multiple fetal tissue cDNAs. The housekeeping gene, β-actin, was used as the internal control. β-Actin band of ∼838 bp was observed in all tissues (data not shown).
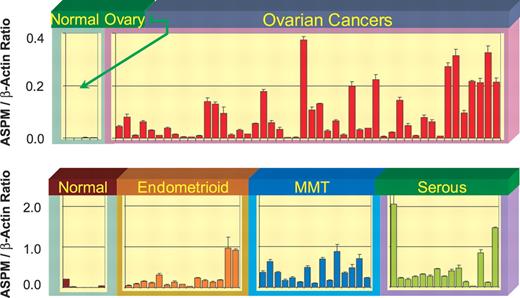
Figure 2. Expression of ASPM in ovarian and uterine cancers. ASPM expression was determined in gynecologic tissue biopsies using real-time PCR. Samples of normal ovary (n=4), ovarian cancer (n=48), normal endometrium (n=6), endometrioid endometrial cancer (n=15), mixed mesodermal tumors of the uterus, MMT, (n=15) and serous endometrial carcinomas (n=15) were analyzed. ‘Normal’ for uterine cancers is normal endometrium.
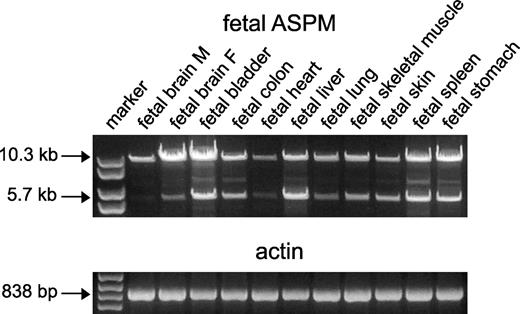
Figure 3. Alternatively spliced ASPM variants in fetal tissues. Two major ASPM transcripts with sizes of ∼10.3 and 5.7 kb were identified in all tissues analyzed. Additional spliced variants of variable sizes were also seen. β-Actin was used as the internal control.
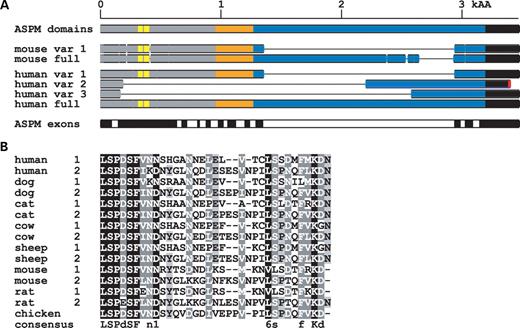
Figure 4.ASPM spliced forms in the human and mouse. (A) The upper scheme shows the positions of major domains in the ASPM protein. The putative microtubule-binding domain is marked in gray, the CH domain in orange (positions 960–1056 and 1114–1174), IQ repeats in blue and the terminal domain in black. Yellow marks the positions of the newly identified 32 and 35 amino acids long repeats in the N-terminus. The middle part shows major splice variants in comparison with the full-length ASPM protein. The bottom part shows parts of the human ASPM protein encoded by individual exons. Splice variant 2 contains exon 26 followed by part of intron 26 where the ORF is disrupted by a stop codon. To better separate individual exons, the odd numbered exons are colored in black and the even numbered exons in white. (B) Identification of two new ASPM repeats and their comparison with other species. The human ASPM protein contains two repeats in positions 316–347 and 366–400. These repeats are found in other mammals, and one is also preserved in chicken.

Figure 5. Structure and evolution of human ASPM IQ repeats. (A) Organization of IQ repeats in the human ASPM protein. The left column shows the number of IQ repeats and their positions in the full-length ASPM protein; the right column shows the length of the individual repeat. The main human alternatively spliced variants are highlighted in orange. Note that variants 2 and 3 contain large deletions that extend toward the C-terminus of ASPM. The blue box displays the region deleted in the mouse and rat. We use color to mark variable positions in the analyzed primates (green monkey, rhesus monkey, orangutan, gorilla, chimpanzee and human) as well as changes specific to African hominoids. On the basis of the Gonnet PAM250 matrix, substitutions were classified as non-conservative (P<0.5) or conservative (the rest). The IQ repeats 4–54 form an organized array of longer (∼27 amino acid) and shorter (∼23 amino acid) units. Alignment and conservation is separately shown for long (B) and short repeats (C) from the IQ 4–54 regions. For both alignments, we show the basic properties of the most conserved amino acid positions.
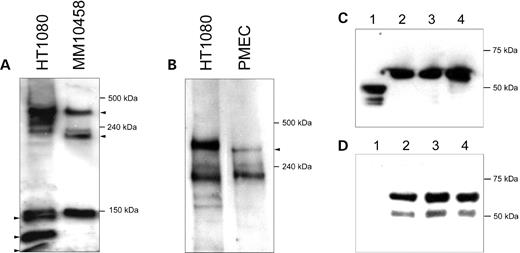
Figure 6. Antibodies to ASPM reveal several isoforms of the protein. (A) The protein extract (100 µg) from normal human cells (HT1080) and MM10458 cells carrying a frameshift mutation in ASPM was analyzed by SDS–PAGE, followed by immunoblotting using an anti-VTKR antibody. The sizes of the largest immunoreactive bands in HT1080 correspond to two predicted ASPM proteins (a full-size 410 kDa protein and a 218 kDa isoform lacking the exon 18-encoding segment). Because ASPM is truncated in MM10458 by a frameshift in exon 24, the predicted isoforms are 35 kDa shorter (i.e. 385 and 183 kDa, correspondingly). The anti-VTKR antibody also visualizes at least three additional bands in the HT1080 extract. Two of them are missing in the MM10458 extract. These bands may correspond to the predicted 164 and 124 kDa ASPM isoforms (Fig. 4). (B) Immunoblot analysis of mouse Aspm proteins using an anti-VTKR antibody. The largest immunoreactive band in the mouse cell extract seems to correspond to the predicted full-size 364 kDa Aspm protein that is 46 kDa shorter than the human ASPM. The second major band seems to correspond to the predicted 212 kDa mouse Aspm isoform, which lacks the exon 18-encoding segment and is approximately the same size as the human ASPM isoform (218 kDa). Note that the small size bands visible in Fig. 3A (i.e. <150 kDa) were run off the gel in order to see the difference between the full-size ASPM and Aspm proteins. (C and D) Western blot analysis showing the specificity of the affinity-purified anti-VTKR antibody. The antibody recognizes a recombinant MBP–ASPM fusion protein expressed in the pMAL-p2X expression vector. The protein extract (100 µg) from bacterial cells was analyzed by SDS–PAGE, followed by immunoblotting using either an anti-MBP antibody (C) or an anti-VTKR antibody (D). Lane 1 corresponds to the vector without the insert. The predicted ∼60 kDa band corresponding to the fusion protein was detected. Lanes 2, 3 and 4 correspond to individual transformants.
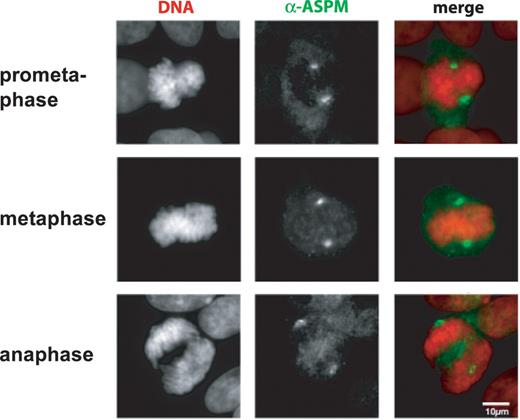
Figure 7. Cytological analysis of ASPM proteins. Mitotic HT1080 cells grown on coverslips were analyzed by indirect immunofluorescent staining using antibodies against human ASPM (green) Chromosomal DNA was counterstained with DAPI (Red). Both anti-ASPM antibodies showed similar spindle pole-staining patterns during mitotic periods, prometaphase (VTRK), metaphase (QSPE) and anaphase (VTRK). The scale bar indicates 10 µm.
References
McCreary, B.D., Rossiter, J.P. and Roberston, D.M. (
Mochida, G.H. and Walsh, C.A. (
Bond, J., Roberts, E., Springell, K., Lizarraga, S.B., Scott, S., Higgins, J., Hampshire, D.J. Morrison, E.E., Leal, G.F., Silva, E.O. et al. (
Roberts, E., Hampshire, D.J., Pattison, L., Springell, K., Jafri, H., Corry, P., Mannon, J., Rashid, Y., Crow, Y., Bond, J. and Woods, C.G. (
Bond, J., Roberts, E., Mochida, G.H., Hampshire, D.J., Scott, S., Askham, J.M., Springell, K., Mahadevan, M., Crow, Y.J., Markham, A.F. et al. (
Luers, G.H., Michels, M., Schwaab, U. and Franz, T. (
Gimona, M., Djinovic-Carugo, K., Kranewitter, W.J. and Winder, S.J. (
Korenbaum, E. and Rivero, F. (
Mooseker, M.S. and Cheney, R.E. (
Houdusse, A., Silver, M. and Cohen, C. (
Martin, S.R. and Bayley, P.M. (
Winder, S.J. and Kendrick-Jones, J. (
Kouprina, N., Pavlicek, A., Mochida, G.H., Solomon, G., Gersch, W., Yoon, Y.H., Collura, R., Ruvolo, M., Barrett, J.C., Woods, C.G. et al. (
Zhang, J. (
Evans, P.D., Anderson, J.R., Vallender, E.J., Gilbert, S.L., Malcom, C.M., Dorus, S. and Lahn, B.T. (
do Carmo Avides, M. and Glover, D.M. (
do Carmo Avides, M., Tavares A. and Glover, D.M. (
Saunders, R.D., Avides, M.C., Howard, T., Gonzalez, C. and Glover, D.M. (
Glover, D.M. (
Bond, J., Scott, S., Hampshire, D.J., Springell, K., Corry, P., Abramowicz, M.J., Mochida, G.H., Hennekam, R.C., Maher, E.R., Fryns, J.P. et al. (
Maquat, L.E. (
Denecke, J., Kranz, C., Kemming, D., Koch, H.G., Marquardt, T. (
Sauer, G., Korner, R., Hanisch, A., Ries, A., Nigg, E.A. and Sillje, H.H. (
Chenn, A. and Walsh, C.A. (
Andreadis, A. (
Sakamoto, T., Wang, F., Schmitz, S., Xu, Y., Xu, Q., Molloy, J.E., Veigel, C. and Sellers, J.R. (
Wang, Q., Hirohashi, Y., Furuuchi, K., Zhao, H., Liu, Q., Zhang, H., Murali, R., Berezov, A., Du, X., Li, B. and Greene, M.I. (
Marumoto, T., Zhang, D. and Saya, H. (
Berger, U.V. and Heidiger, M.A. (
Morgenstern, B. (
Nicholas, K.B., Nicholas, H.B. Jr and Deerfield, D.W. II. (
Zika, R., Paces, J., Pavlicek, A. and Paces, V. (
Marchler-Bauer, A. and Bryant, S.H. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}