
Abstract
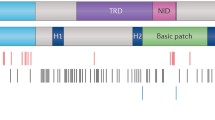
Genetic mutations of the X-linked gene MECP2, encoding methyl-CpG-binding protein 2, cause Rett syndrome (RTT) and other neurological disorders. It is increasingly recognized that MECP2 is a multifunctional protein, with at least four different functional domains: (1) a methyl-CpG-binding domain; (2) an arginine-glycine repeat RNA-binding domain; (3) a transcriptional repression domain; and (4) an RNA splicing factor binding region (WW group II binding domain).
There is evidence that MECP2 is important for large-scale reorganization of pericentromeric heterochromatin during differentiation. Studies in MECP2-deficient mouse brain have identified a diverse set of genes with altered levels of mRNA expression or splicing. It is still unclear how altered MECP2 function ultimately results in neuronal disease after a period of grossly normal development.
However, mounting evidence suggests that neuronal health and development depend on precise regulation of MECP2 expression. In genetically engineered mice, both increased and decreased levels of MECP2 result in a neurological phenotype. Furthermore, it was recently discovered that MECP2 gene duplications underlie a small number of atypical Rett cases and mental retardation syndromes. The finding that MECP2 levels are tightly regulated in neurons has important implications for the design of gene replacement or reactivation strategies for treatment of RTT, because affected individuals typically are somatic mosaics with one set of cells expressing a mutated MECP2 from the affected X, and another set expressing normal MECP2 from the unaffected X. Further studies are necessary to elucidate the molecular pathology of both loss-of-function and gain-of-function mutations in MECP2.
Similar content being viewed by others


References
Aber K. M., Nori P., MacDonald S. M., Bibat G., Jarrar M. H., and Kaufmann W. E. (2003) Methyl-CpG-binding protein 2 is localized in the postsynaptic compartment: an immunochemical study of subcellular fractions. Neuroscience 116, 77–80.
Akbarian S. (2003) The neurobiology of Rett syndrome. Neuroscientist 9, 57–63.
Akbarian S., Chen R. Z., Gribnau J., et al. (2001) Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol. Dis. 8, 784–791.
Amir R. E., Van den Veyver I. B., Wan M., Tran C. Q., Francke U., and Zoghbi H. Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Ariani F., Mari F., Pescucci C., et al. (2004) Real-time quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: Report of one case of MECP2 deletion and one case of MECP2 duplication. Hum. Mutat. 24, 172–177.
Armstrong D., Dunn J. K., Antalffy B., and Trivedi R. (1995) Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 54, 195–201.
Armstrong D. D., Dunn J. K., Schultz R. J., Herbert D. A., Glaze D. G., and Motil K. J. (1999) Organ growth in Rett syndrome: a postmortem examination analysis. Pediatr. Neurol. 20, 125–129.
Ballas N., Grunseich C., Lu D. D., Speh J. C., and Mandel G. (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657.
Balmer D., Goldstine J., Rao Y. M., and LaSalle J. M. (2003) Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J. Mol. Med. 81, 61–68.
Bauman M. L., Kemper T. L., and Arin D. M. (1995) Microscopic observations of the brain in Rett syndrome. Neuropediatrics 26, 105–108.
Bedford M. T., Chan D. C., and Leder P. (1997) FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. Embo. J. 16, 2376–2383.
Brero A., Easwaran H. P., Nowak D., et al. (2005) Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 169, 733–743.
Buschdorf J. P. and Stratling W. H. (2004) A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J. Mol. Med. 82, 135–143.
Carro S., Bergo A., Mengoni M., et al. (2004) A novel protein, Xenopus p20, influences the stability of McCP2 through direct interaction. J. Biol. Chem. 279, 25,623–25,631.
Chen R. Z., Akbarian S., Tudor M., and Jaenisch R. (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Chen W. G., Chang Q., Lin Y., et al. (2003) Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889.
Christodoulou J., Grimm A., Maher T., and Bennetts B. (2003) Rett BASE: The IRSA MECP2 variation database-a new mutation database in evolution. Hum. Mutat. 21, 466–472.
Cohen D., Lazar G., Couvert P., et al. (2002) MECP2 mutation in a boy with language disorder and schizophrenia. Am. J. Psychiatry 159, 148–149.
Colantuoni C., Jeon O. H., Hyder K., et al. (2001) Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol. Dis. 8, 847–865.
Collins A. L., Levenson J. M., Vilaythong A. P., et al. (2004) Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689.
Deguchi K., Antalffy B. A., Twohill L. J., Chakraborty S., Glaze D. G., and Armstrong D. D. (2000) Substance P immunoreactivity in Rett syndrome. Pediatr. Neurol. 22, 259–266.
Dintilhac A. and Bernues J. (2002) HMGB1 interacts with many apparently unrelated proteins by recognizing short amino acid sequences. J. Biol. Chem. 277, 7021–7028.
Georgel P. T., Horowitz-Scherer R. A., Adkins N., Woodcock C. L., Wade P. A., and Hansen J. C. (2003) Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 278, 32,181–32,188.
Guy J., Hendrich B., Holmes M., Martin J. E., and Bird A. (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Hagberg B. (1995) Rett syndrome: clinical peculiarities and biological mysteries. Acta Paediatr. 84, 971–976.
Hagberg B. (2002) Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Distabil. Res. Rev. 8, 61–65.
Harikrishnan K. N., Chow M. Z., Baker E. K. et al. (2005) Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 37, 254–264.
Hendrich B. and Tweedie S. (2003) The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 19, 269–277.
Horike S., Cai S., Miyano M., Cheng J. F., and Kohwi-Shigematsu T. (2005) Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37, 31–40.
Iwasato T., Erzurumlu R. S., Huerta P. T., et al. (1997) NMDA receptor-dependent refinement of somatotopic maps. Neuron 19, 1201–1210.
Jeffery L. and Nakielny S. (2004) Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 279, 49,479–49,487.
Jung B. P., Jugloff D. G., Zhang G., Logan R., Brown S., and Eubanks J. H. (2003) The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 55, 86–96.
Kaludov N. K. and Wolffe A. P. (2000) MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 28, 1921–1928.
Kaufmann W. E., Johnston M. V., and Blue M. E. (2005) MeCP2 expression and function during brain development: implications for Rett syndrome's pathogenesis and clinical evolution. Brain Dev. 27(Suppl. 1), S77-S87.
Kerr A. M., Nomura Y., Armstrong D., et al. (2001) Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev. 23, 208–211.
Kimura H. and Shiota K. (2003) Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 278, 4806–4812.
Klose R. J., Sarraf S. A., Schmiedeberg L., McDermott S. M., Stancheva I., and Bird A. P. (2005) DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 19, 667–678.
Kokura K., Kaul S. C., Wadhwa R., et al. (2001) The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 276, 34,115–34,121.
Kriaucionis S. and Bird A. (2004) The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 32, 1818–1823.
Krithivas A., Fujimuro M., Weidner M., Young D. B., and Hayward S. D. (2002) Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76, 11,596–11,604.
Lee S. S., Wan M., and Francke U. (2001) Spectrum of MECP2 mutations in Rett syndrome. Brain Dev. 23(Suppl. 1), S138-S143.
Lehnertz B., Ueda Y., Derijck A. A., et al. (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13, 1192–1200.
Lewis J. D., Meehan R. R., Henzel W. J., et al. (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914.
Luikenhuis S., Giacometti E., Beard C. F., and Jaenisch R. (2004) Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. USA 101, 6033–6038.
Macias M. J., Wiesner S., and Sudol M. (2002) WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 513, 30–37.
Manuelidis L. (1985) Indications of centromere movement during interphase and differentiation. Ann. NY Acad. Sci. 450 205–221.
Martinowich K., Hattori D., Wu H., et al. (2003) DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893.
Martou G. and De Boni U. (2000) Nuclear topology of murine, cerebellar Purkinje neurons: changes as a function of development. Exp. Cell Res. 256, 131–139.
Matarazzo V. and Ronnett G. V. (2004) Temporal and regional differences in the olfactory proteome as a consequence of MeCP2 deficiency. Proc. Natl. Acad. Sci. USA 101, 7763–7768.
Meins M., Lehmann J., Gerresheim, F., et al. (2005) Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J. Med. Genet. 42, e12.
Miltenberger-Miltenyi G. and Laccone F. (2003) Mutations and polymorphisms in the human methyl CpG-binding protein MECP2. Hum. Mutat. 22, 107–115.
Mnatzakanian G. N., Lohi H., Munteanul I., et al. (2004) A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 36, 339–341.
Moog U., Roozendaal K. V., Smeets E., et al. (2005) MECP2 mutations are an infrequent cause of mental retardation associated with neurological problems in male patients. Brain Dev. (e-pubahead of print)
Moretti P., Levenson J. M., Battaglia F., et al. (2006) Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327.
Mullaney B. C., Johnston M. V., and Blue M. E. (2004) Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience 123, 939–949.
Nagai K., Miyake K., and Kubota T. (2005) A transcriptional repressor MeCP2 causing Rett syndrome is expressed in embryonic non-neuronal cells and controls their growth. Brain. Res. Dev. Brain Res. 157, 103–106.
Naidu S., Bibat G., Kratz L., et al. (2003) Clinical variability in Rett syndrome. J. Child Neurol. 18, 662–668.
Nan X., Meehan, R. R., and Bird A. (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 21, 4886–4892.
Nan X., Tate P., Li E., and Bird A. (1996) DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell Biol. 16, 414–421.
Nan X., Ng H. H., Johnson C. A., et al. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389.
Nuber U. A., Kriaucionis S., Roloff T. C., et al. (2005) Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum. Mol. Genet. 14, 2247–2256.
Paterson D. S., Thompson E. G., Belliveau R. A., et al. (2005) Serotonin transporter abnormality in the dorsal motor nucleus of the vagus in Rett syndrome: potential implications for clinical autonomic dysfunction. J. Neuropathol. Exp. Neurol. 64, 1018–1027.
Percy A. K. (1997) Neurobiology and neurochemistry of Rett syndrome. Eur. Child Adolesc. Psychiatry 6(Suppl. 1), 80–82.
Percy A. K. and Lane J. B. (2004) Rett syndrome: clinical and molecular update. Curr. Opin. Pediatr. 16, 670–677.
Samaco R. C., Hogart A., and LaSalle J. M. (2005) Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492.
Saxena A., de Lagarde D., Leonard H., et al. (2005) Lost in translation: Translational interference from a recurrent mutation in exon 1 of MECP2. J. Med. Genet. (e-pub ahead of print)
Schanen C., Houwink E. J., Dorrani N., et al. (2004) Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am. J. Med. Genet. A 126, 129–140.
Schultz R. J., Glaze D. G., Motil K. J., et al. (1993) The pattern of growth failure in Rett syndrome. Am. J. Dis. Child 147, 633–637.
Shahbazian M. D., Antalffy B., Armstrong D. L., and Zoghbi H. Y. (2002) Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11, 115–124.
Shi J., Shibayama A., Liu Q., et al. (2005) Detection of heterozygous deletions and duplications in the MECP2 gene in Rett syndrome by Robust Dosage PCR (RD-PCR). Hum. Mutat. 25, 505.
Skabkin M. A., Kiselyova O. I., Chernov K. G., et al. (2004) Structural organization of mRNA complexes with major core mRNP protein YB-1. Nucleic Acids Res. 32, 5621–5635.
Stickeler E., Fraser S. D., Honig A., Chen A. L., Berget S. M., and Cooper T. A. (2001) The RNA binding protein YB-1 binds A/C-rich exon enhancers and stimulates splicing of the CD44 alternative exon v4. Embo. J. 20, 3821–3830.
Subramaniam B., Naidu S., and Reiss A. L. (1997) Neuroanatomy in Rett syndrome: cerebral cortex and posterior fossa. Neurology 48, 399–407.
Suzuki M., Yamada T., Kihara-Negishi F., Sakurai T., and Oikawa T. (2003) Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU.1-mediated transcriptional repression. Oncogene 22, 8688–8698.
Tudor M., Akbarian S., Chen R.Z., and Jaenisch R. (2002) Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transciptional changes in the brain. Proc. Natl. Acad. Sci. USA 99, 15,536–15,541.
Van den Veyver I. B. and Zoghbi H. Y. (2002) Genetic basis of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 8, 82–86.
Van Esch H., Bauters M., Ignatius J., et al. (2005) Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 77, 442–453.
Viemari J. C., Roux J. C., Tryba A. K., et al. (2005) Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J. Neurosci. 25, 11,521–11,530.
Watson P., Black G., Ramsden S., et al. (2001) Angelman syndrome phenotype associated with mutations in MECP2 a gene encoding a methyl CpG binding protein. J. Med. Genet. 38, 224–228.
Young J. I., Hong E. P., Castle J. C., et al. (2005) Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 102, 17,551–17,558.
Zeev B. B., Yaron Y., Schanen N. C., et al. (2002) Rett syndrome: clinical manifestations in males with MECP2 mutations. J. Child Neurol. 17, 20–24.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Akbarian, S., Jiang, Y. & Laforet, G. The molecular pathology of rett syndrome. Neuromol Med 8, 485–494 (2006). https://doi.org/10.1385/NMM:8:4:485
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1385/NMM:8:4:485