
LRRK2 and the Endolysosomal System in Parkinson’s Disease
Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) cause autosomal dominant familial Parkinson’s disease (PD), with pathogenic mutations enhancing LRRK2 kinase activity. There is a growing body of evidence indicating that LRRK2 contributes to neuronal damage and pathology both in familial and sporadic PD, making it of particular interest for understanding the molecular pathways that underlie PD. Although LRRK2 has been extensively studied to date, our understanding of the seemingly diverse functions of LRRK2 throughout the cell remains incomplete. In this review, we discuss the functions of LRRK2 within the endolysosomal pathway. Endocytosis, vesicle trafficking pathways, and lysosomal degradation are commonly disrupted in many neurodegenerative diseases, including PD. Additionally, many PD-linked gene products function in these intersecting pathways, suggesting an important role for the endolysosomal system in maintaining protein homeostasis and neuronal health in PD. LRRK2 activity can regulate synaptic vesicle endocytosis, lysosomal function, Golgi network maintenance and sorting, vesicular trafficking and autophagy, with alterations in LRRK2 kinase activity serving to disrupt or regulate these pathways depending on the distinct cell type or model system. LRRK2 is critically regulated by at least two proteins in the endolysosomal pathway, Rab29 and VPS35, which may serve as master regulators of LRRK2 kinase activity. Investigating the function and regulation of LRRK2 in the endolysosomal pathway in diverse PD models, especially in vivo models, will provide critical insight into the cellular and molecular pathophysiological mechanisms driving PD and whether LRRK2 represents a viable drug target for disease-modification in familial and sporadic PD.
INTRODUCTION
Parkinson’s disease (PD) is a common neurodegenerative disorder that causes a progressive loss of dopaminergic neurons in the substantia nigra pars compacta, resulting in the depletion of striatal dopamine [1, 2]. This decrease in dopamine levels primarily causes resting tremor, muscle rigidity, and bradykinesia, and frequently postural instability [1, 2]. PD is the second most prevalent neurodegenerative disorder, affecting 1–2% of people over the age of 65 worldwide [1, 2]. Currently, therapies for PD patients alleviate motor symptoms but do not slow disease progression [1, 2]. The etiology of PD is unknown and the molecular pathways that precipitate disease onset and contribute to disease progression remain incompletely understood. Identifying and characterizing these pathways has been the focus of PD research for the last 20 years, as this is one of the most promising strategies for developing effective disease-modifying treatments. A growing body of research indicates that risk for developing PD is influenced by a complex interaction between genetic and environmental factors together with age. Many genes have been linked to PD, providing insight into the cellular mechanisms that may underlie neuronal pathology. Most of the monogenic causes of PD lead to early-onset autosomal recessive PD or parkinsonism (Parkin, PINK1, DJ-1, ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1), or X-linked early-onset PD (RAB39B) [3–6]. Missense mutations have also been identified in a handful of genes that cause early or late-onset autosomal dominant PD, including SNCA, LRRK2, and VPS35 [3–6]. Additionally, common coding or non-coding variants at several distinct genomic loci have been discovered to modify risk of developing PD (i.e., GBA, LRRK2, MAPT, SNCA, SCARB2, GAK, SH3GL2, TMEM175, ATP6V0A1, GALC, CTSB) [7–18]. Many genes linked to PD or parkinsonism play roles in endocytosis (LRRK2, SNCA, DNAJC6, SYNJ1, GAK, SH3GL2) [19–26], vesicular trafficking/sorting (LRRK2, VPS35, RAB39B) [27–29], mitophagy (Parkin, PINK1) [30–32], or lysosomal function (ATP13A2, GBA, SCARB2, TMEM175, ATP6V0A1, GALC, CTSB) [33–42], broadly implicating the endolysosomal pathway in PD.
LRRK2
Mutations in leucine-rich repeat kinase 2 (LRRK2, PARK8) are the most common cause of familial PD [43]. Missense mutations in LRRK2 cause late-onset autosomal dominant PD, that is clinically indistinguishable from sporadic PD [44, 45]. Neuropathology in LRRK2 PD cases is also similar to sporadic PD, with LRRK2 subjects experiencing progressive neurodegeneration of the nigrostriatal pathway and often developing α-synuclein-positive Lewy bodies, as well as tau pathology in the brain [44, 46–48]. LRRK2 coding mutations have also been identified that are associated with sporadic PD whereas common non-coding variants at the LRRK2 locus are associated with increased risk for developing PD [15, 49–51]. These findings suggest that in addition to precipitating neuropathology in familial PD, LRRK2 also contributes to pathways that are involved in sporadic PD. Accordingly, the characterization of LRRK2-dependent molecular pathways is a promising approach for identifying novel therapeutic targets to treat familial and sporadic PD.
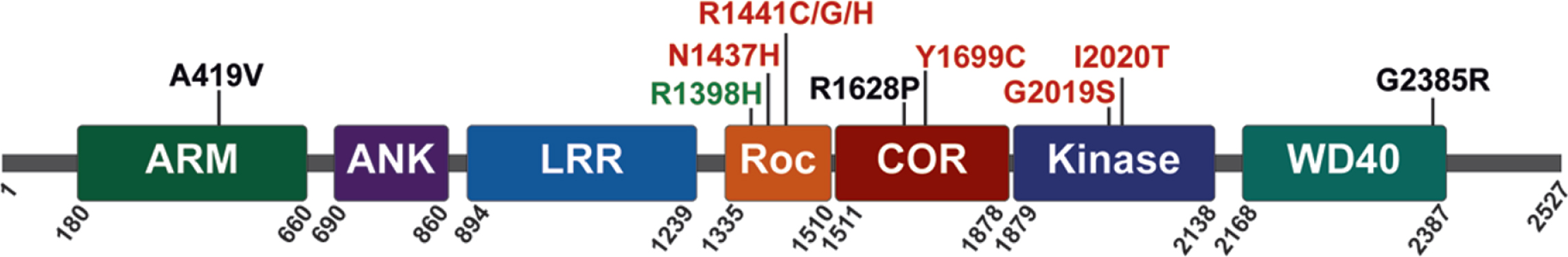
LRRK2 is a relatively large multidomain protein (2527 amino acids) that is expressed, at varying levels, in many diverse tissues and cell types. At its N-terminus, LRRK2 contains armadillo repeat, ankyrin repeat and leucine-rich repeat domains. The enzymatic domains of LRRK2 are located in the center of the protein; they include a Ras-of-Complex (ROC) GTPase, a C-terminal-of-Roc (COR) and kinase domain, followed by WD40 repeats at its C-terminus (Fig. 1). LRRK2 has been shown to form homodimers, and dimerization is likely important for regulating LRRK2 activity [52–54]. PD-linked familial mutations cluster within the central enzymatic domains (N1437H, R1441C, R1441G, R1441H, Y1699C, G2019S, and I2020T) [55–60], and enhance LRRK2 kinase activity [61–64]. Familial mutations commonly increase LRRK2 autophosphorylation at Ser1292 and the LRRK2-mediated phosphorylation of substrates that include a subset of Rab GTPases, suggesting that sustained increases in LRRK2 kinase activity can contribute to the initiation and progression of PD [61, 64–66].
Fig. 1
LRRK2 protein domain architecture with PD-linked familial mutations (red), sporadic PD risk variants (black), and sporadic PD protective variants (green), as indicated.
Recent studies suggest increased LRRK2 kinase activity and endolysosomal pathology in nigral dopa-minergic neurons from brains of sporadic PD subjects, including the accumulation of Rab5-positive early endosomes, depletion of late endosomes, depletion of lysosomes, and depletion of lysosomal GCase [67, 68]. Whether these vesicular abnormalities are dependent on LRRK2 activity is uncertain. In experimental neurotoxin models, systemic rotenone exposure in rats elevates the kinase activity of endogenous LRRK2, induces nigral dopaminergic neurodegeneration, accumulation of phosphorylated α-synuclein, and endolysosomal pathology that closely resembles sporadic PD brains [67, 68]. Additionally, rotenone-induced dopaminergic neurodegeneration and endolysosomal pathology are prevented by administration of the selective LRRK2 kinase inhibitor PF-360 [67, 68]. Although these studies are certainly persuasive, the increase in LRRK2 kinase activity in PD or rodent brain sections is monitored by novel proximity ligation assays that have not yet been independently replicated by other groups or validated by other methods. The precise mechanisms by which LRRK2 kinase activity modulates endocytosis, vesicle trafficking, and degradation pathways are areas of rapidly growing interest, as these pathways appear to be central contributors to the pathogenesis of familial and sporadic PD [69, 70]. This review will focus on the effects of pathogenic LRRK2 variants in endocytosis, vesicle trafficking, lysosomal homeostasis and Golgi maintenance, as these intersecting pathways are components of the endolysosomal system and have been implicated in disease pathology in PD subjects and in cellular and animal models of PD [69, 70].
THE ENDOLYSOSOMAL PATHWAY
The endolysosomal pathway is a multifaceted, highly regulated process that is critically important for many cellular functions including proteostasis, extracellular and intracellular signaling, organelle homeostasis, and membrane organization. The canonical endosomal pathway initiates with clathrin-mediated endocytosis, and vesicle uncoating followed by fusion of newly formed endocytic vesicles with early endosomes. Early endosomes are mildly acidic, which facilitates the uncoupling of ligands and membrane-bound receptors for recycling. The early endosome also acts as a sorting station, targeting cargo towards one of three destinations: 1) back to cell surface membranes, directly or via recycling endosomes, 2) for degradation through the endosomal sorting complexes required for transport (ESCRT) pathway or 3) for retrograde sorting to the trans-Golgi network (TGN) through pathways such as retromer complex sorting. Endosomes that are targeted to the ESCRT pathway mature into late endosomes where luminal pH decreases, and the concentration of lysosomal enzymes increases as endosomes move towards the nucleus. During late endosome maturation, the ESCRT complex also sequesters ubiquitinated proteins into intraluminal vesicles forming multivesicular bodies (MVBs). Mature late endosomes or MVBs then fuse with lysosomes to initiate the proteolytic degradation of cargo. (for review, see [71–73]).
The endolysosomal pathway also intersects with the phagocytosis and autophagy pathways. Phagocytosis occurs when immune-related cells engulf extracellular material such as apoptotic cells, cellular debris, or infectious agents. Cargo in the phagocytic pathway is recycled or degraded via the endolysosomal pathway when phagosomes fuse with late endosomes or lysosomes. Autophagy occurs when cells digest intracellular components through one of three pathways: 1) macroautophagy, 2) microautophagy, or 3) chaperone-mediated autophagy. Macroautophagy is regulated by ULK1, which induces the formation of a double-membraned autophagosome, around large intracellular components, that are marked for degradation. After formation, autophagosomes undergo a series of maturation steps, including lipidation of LC3-I to LC3-II before fusing with late endosomes or lysosomes. Microautophagy occurs through the direct lysosomal engulfment of small, unlabeled cellular components. Chaperone-mediated autophagy is a process through which chaperone proteins, such as Hsc70, deliver small cellular components that are marked for degradation, usually by ubiquitination, in cooperation with the LAMP2A receptor to lysosomes for degradation. Phagocytosis, autophagy and vesicle trafficking through the endolysosomal pathway are highly coordinated processes that are regulated by many factors (for review, see [71–73]).
Recent findings show that LRRK2 physically or functionally interacts with numerous proteins throughout the endolysosomal pathway, and that manipulating LRRK2 expression levels or kinase activity leads to pathological disruptions in vesicle trafficking and protein degradation (Fig. 2; Table 1). Additionally, brain tissue from sporadic PD subjects has shown accumulation of LRRK2 in enlarged granules that colocalizes with the late endosome protein Rab7b and lysosomal protein LAMP2 [74]. Disrupted trafficking or degradation of LRRK2 in the brain of sporadic PD subjects highlights the endolysosomal system in PD pathogenesis and suggests that LRRK2 may contribute to this pathology.
Fig. 2
LRRK2 in the endolysosomal pathway. Key aspects of the endolysosomal pathway highlighting LRRK2-interacting proteins and kinase substrates. *The specific subcellular location of VPS35-induced activation of LRRK2 and subsequent Rab10 phosphorylation not been established. As VPS35 resides in the retromer complex, and the retromer complex primarily localizes to endosomal and vesicular membranes in the retrograde trafficking pathway, it most likely occurs on endosomal membranes, however this has not been directly assessed.
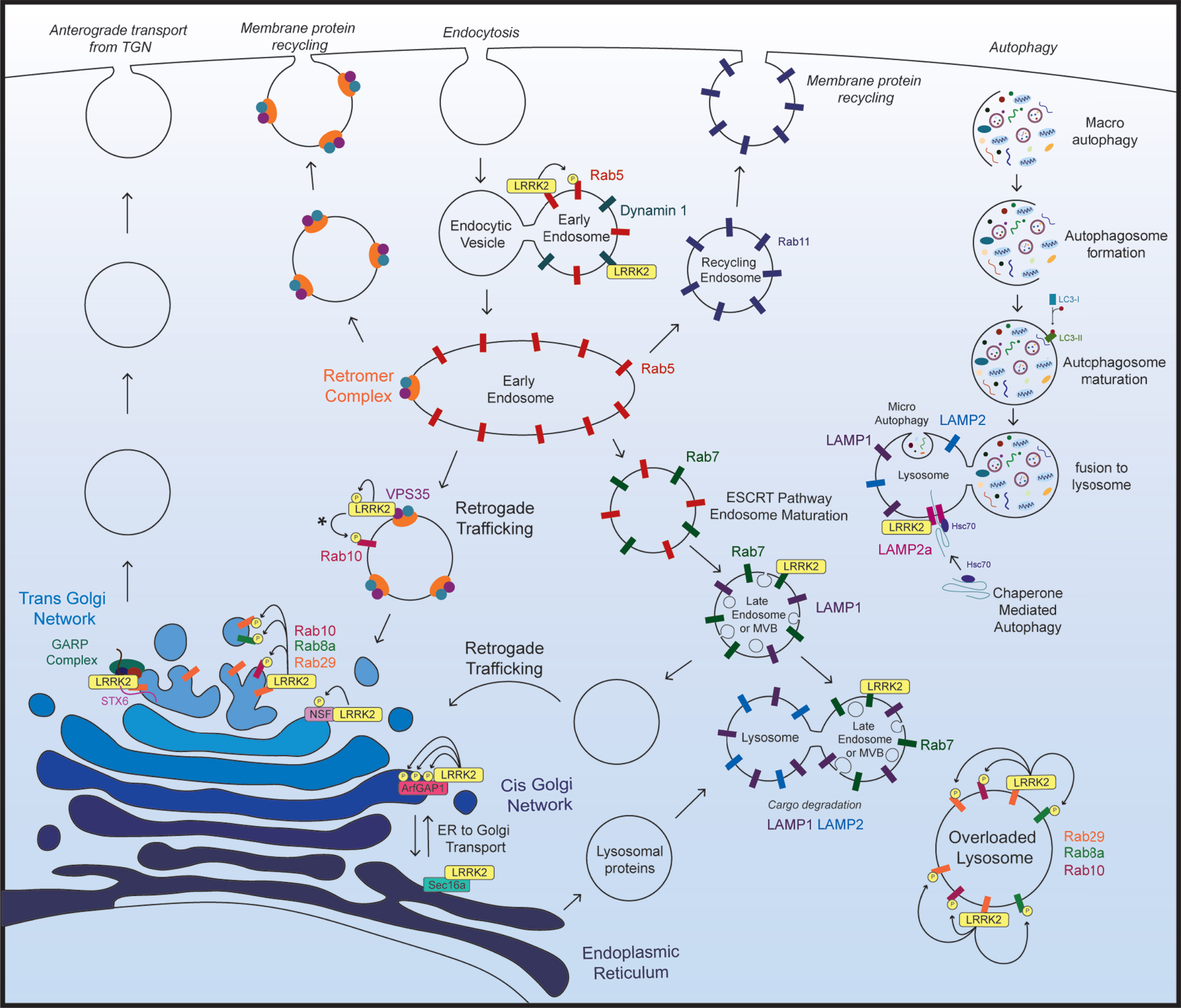
Table 1
LRRK2-interacting proteins in the endolysosomal pathway
| Gene | Relationship to LRRK2 | Function in endolysosomal pathway | Reference |
| AP3B1 | LRRK2-interacting protein | AP3 protein complex component | [91, 98, 99] |
| LRRK2 and AP3B1 complex together to recycle lysosomal membrane proteins LAMP1 and LAMP2 | Localized to endosomal membranes | ||
| Regulates endosome maturation and vesicle exocytosis, and recycling of lysosomal membrane proteins | |||
| Generation of new synaptic vesicles | |||
| ArfGAP1 | GAP-like protein for LRRK2 | Golgi to ER retrograde vesicular sorting | [125, 131, 132] |
| LRRK2 genetic modifier | |||
| LRRK2 kinase substrate | |||
| ATP13A2 | Increased protein expression in G2019S LRRK2 brain tissue | P5-type ATPase localized to late endosomal and lysosomal membranes | [96] |
| Auxilin 1 (DNAJC6) | LRRK2 kinase substrate | Synaptic Vesicle Endocytosis | [110] |
| LRRK2-interacting protein | |||
| Auxilin 2 (GAK) | LRRK2-interacting protein | Clathrin uncoating | [24, 126] |
| Facilitates clathrin receptor binding at Golgi and plasma membrane | |||
| Dynamin 1, 2, 3 | LRRK2-interacting proteins | Membrane scission in clathrin-mediated endocytosis | [115] |
| Endophilin A | LRRK2 kinase substrate | Synaptic Vesicle Endocytosis | [19, 100, 117] |
| Autophagosome formation at neuromuscular junction synapses | |||
| GBA | LRRK2 familial mutants decrease lysosomal GCase activity | Lysosomal hydrolase | [110, 111] |
| LAMP2a | LRRK2-binding partner | Lysosomal membrane protein; chaperone-mediated autophagy receptor | [158] |
| NSF | LRRK2 kinase substrate | Golgi disassembly | [104, 127, 137] |
| Sec16a | LRRK2-interacting protein | ER to Golgi anterograde transport | [138] |
| Syntaxin-6 (STX-6) | LRRK2-interacting protein | SNARE protein at TGN | [94] |
| SYNJ1 | LRRK2 kinase substrate | Synaptic vesicle endocytosis | [101, 116] |
| VPS35 | LRRK2-interacting protein | Retromer complex subunit | [79, 147, 148] |
| D620N mutation enhances phosphorylation of LRRK2 kinase substrates | |||
| VPS52 | LRRK2-interacting protein | Golgi-associated retrograde protein (GARP) complex subunit | [94] |
LRRK2 regulates the endolysosomal pathway at multiple steps, through mechanisms that are not yet well defined. One way that LRRK2 likely impacts this pathway is through interaction with or direct phosphorylation of multiple Rab proteins localized throughout the endolysosomal system [64, 66, 75] (Table 2).
Table 2
Rab proteins that interact with LRRK2
| Gene | Relationship to LRRK2 | Function in endolysosomal pathway | Reference |
| Rab3A/B/C/D | LRRK2 kinase substrate | Ca2 +-triggered synaptic vesicle release | [66, 165] |
| Rab5B | LRRK2-binding partner | Endocytic vesicle fusion to early endosomes | [102, 166, 167] |
| putative LRRK2 kinase substrate | Synaptic Vesicle Endocytosis | ||
| Rab7A | LRRK2-interacting protein | Late endosome and lysosome membrane protein | [166] |
| Endocytic trafficking and lysosome biogenesis | |||
| Rab8A | LRRK2 kinase substrate | Trafficking and recycling of epidermal growth factor (EGF) and EGF receptors from cell surface membrane | [66, 80, 168] |
| LRRK2-mediated recruitment to overloaded lysosomes | Lysosomal overloading response | ||
| Rab8B | LRRK2 kinase substrate | Endocytosis and secretion | [66] |
| Rab9 | LRRK2-interacting protein | Retrograde trafficking | [169] |
| Lysosome biogenesis | |||
| Rab10 | LRRK2 kinase substrate | Lysosomal overloading response | [66, 80] |
| LRRK2-mediated recruitment to overloaded lysosomes | |||
| Rab29 (also known as Rab7L1) | LRRK2 kinase substrate Recruits LRRK2 to the TGN and to overloaded lysosomes | TGN maintenance, retrograde trafficking from late and recycling endosomes to TGN Lysosomal overloading response | [78, 80, 81, 128] |
| Activates LRRK2 | |||
| Rab35 | LRRK2 kinase substrate | Endosome to plasma membrane recycling | [75, 80, 170] |
| Recruited to LRRK2-positive overloaded lysosomes | Plasma membrane receptor recycling | ||
| Rab32 | LRRK2-interacting protein | Retrograde trafficking | [149, 150] |
| Rab43 | Putative LRRK2 kinase substrate | Anterograde trafficking of GPCRs from ER to Golgi | [66, 171] |
LRRK2 AND RABS
Rabs are small GTPases that reside on specific intracellular compartments, many of which are in the endolysosomal pathway. The highly specific subcellular localization of Rabs is important for their regulatory functions in this pathway and is controlled by several distinct Rab-interacting proteins. For example, binding to Rab escort proteins (REPs) promotes prenylation of Rabs via geranylgeranyl transferase, which subsequently tethers Rabs to compartment-specific membranes. Membrane-bound Rabs can then be activated by guanine nucleotide exchange factors (GEFs) that catalyze the exchange of GDP for GTP. GTP-bound active Rabs recognize specific effector proteins to facilitate docking and fusion of Rab-positive membranes to target membranes. Rabs are also regulated by GTPase-activating proteins (GAPs) that catalyze the hydrolysis of GTP to GDP, switching their target Rabs to an inactive state. GDP dissociation inhibitor (GDI) proteins bind inactive Rabs and remove them from membranes. Individual Rabs move onto and off specific subcellular membrane compartments throughout the endolysosomal pathway, in a highly controlled manner to regulate signaling and sorting throughout this pathway (for review, see [69, 76, 77]).
LRRK2 complexes with, or directly phosphorylates multiple Rab proteins in the endolysosomal pathway, including Rab29 [66, 75, 78, 79] (Table 2). Recent studies have found that Rab29 can recruit LRRK2 to specific cellular compartments, including the TGN and overloaded lysosomes [79–84]. Recruitment of LRRK2 by Rab29 subsequently enhances LRRK2 kinase activity and promotes LRRK2-mediated recruitment of additional Rab proteins [79–84]. Surprisingly, LRRK2 kinase activity is also enhanced by Rab8a, Rab12 and Rab38, but not to the extent of LRRK2 activation by Rab29 [82]. LRRK2 activation by other Rabs has not yet been fully investigated, and the functional implications of these pathways are currently unclear. However, emerging data suggest intriguing roles for LRRK2-mediated phosphorylation of Rab8a and Rab10 in regulating ciliogenesis and centrosomal cohesion [66, 83, 85–87]. LRRK2-Rab interactions will be further discussed in the following sections focusing on individual components of the endolysosomal pathway.
LRRK2 DELETION DISRUPTS THE ENDOLYSOSOMAL PATHWAY
LRRK2 knockout in rodent models
LRRK2 is expressed in many tissues and cell types, with highest expression in lung, kidney, and immune cells and relatively low expression in the brain [88]. LRRK2 knockout mice and rats have discolored kidneys with lysosomal pathology that progresses with age [88–94]. For example, LRRK2 knockout leads to the formation of large vacuoles, the accumulation of lipofuscin granules, and formation of high-molecular weight ubiquitin-positive protein aggregates in the kidneys [88–93]. Specific perturbations in the endolysosomal pathway have also been observed in LRRK2 knockout kidneys including increased levels of lysosomal proteins LAMP1, LAMP2 and cathepsin B, as well as enlarged LAMP1-positive vesicles [89, 91, 93, 94]. Additionally, LRRK2 knockout mice are more susceptible to lysosomal overloading, specifically in kidney tissue, in response to intraperitoneal chloroquine injections, exhibiting increased LAMP1 staining, vacuolization, and lipofuscin in renal proximal tubule cells [80]. Perturbations in autophagy have also been observed in LRRK2 knockout kidneys, as changes in LC3-II/LC3-I protein levels, however it is currently unclear whether LRRK2 knockout enhances autophagosome formation or impairs autophagic flux in kidney tissue [88, 92].
LRRK2 knockout also causes lysosomal pathology in lung and liver tissues, manifesting as vacuolization in both tissues, accumulation of lamellar bodies in type II pneuomocytes in the lungs and formation of lipid droplets in the liver [89, 90]. Notably, tissues that express lower levels of LRRK2, including the heart and brain, exhibit no apparent endolysosomal pathology in LRRK2 knockout animals [88–92]. LRRK2 knockout causes mild behavioral changes in mice without causing dopaminergic neuronal degeneration or overt neuropathology [88, 90–92]. LRRK2 knockout may be compensated for by the expression of its homolog LRRK1 in the brain. Interestingly, LRRK1/2 double knockout mice have shortened lifespan, as well as modest age-dependent dopaminergic neuronal degeneration and formation of large autophagic vacuoles in the SN [95].
LRRK2 knockout in non-mammalian models
In non-mammalian models and primary cell cultures, depletion of LRRK2 disrupts endocytosis, lysosomal function, and autophagosome formation [91, 96, 97]. In C. elegans, mutation of the LRRK2 homolog lrk-1 leads to depletion of endosomes in motor neuron axons [91]. In this system, lrk-1 acts downstream of Glo-1, the C. elegans homologue of Rab29, and upstream of ABP-3, the C. elegans ortholog of AP3B1, to regulate endocytosis [91]. Interestingly, Kuwahara et al. also observed a functional interaction between LRRK2 and Rab29 in mice [91]. Rab29 homozygous knockout mice have endolysosomal kidney pathology that closely resembles kidney pathology in LRRK2 knockout mice, and Rab29/LRRK2 double knockout mice do not have additional kidney pathology compared to single knockout mice [91]. These results suggest that Rab29 and LRRK2 operate in a single common pathway to maintain endolysosomal function [91]. AP3B1 is a component of the Adaptor Protein 3 (AP-3) complex, which regulates vesicle trafficking through the endolysosomal system and synaptic vesicle generation from endosomes [91, 98, 99]. In cultured mammalian cells, LRRK2 complexes with AP3B1 to regulate recycling of lysosomal membrane protein receptors LAMP1 and LAMP2 [91]. Knockdown of either LRRK2 or AP3B1, in these cells, causes mislocalization of LAMP proteins from lysosomal membranes to cell surface membranes [91].
LRRK2 also regulates lysosome function and morphology in primary cultures. In primary astrocytes, LRRK2 knockout causes an increase in lysosome number and a corresponding decrease in lysosome size, as well as increased LAMP1 and LAMP2 protein levels [96]. LRRK2 knockout in primary cortical neurons leads to increased lysosomal protein degradation and increased LC3-II protein levels, indicative of increased autophagosome formation [97].
LRRK2 knockout disrupts synaptic vesicle endocytosis
LRRK2 knockout or knockdown also disrupts synaptic vesicle endocytosis (SVE) in animals and primary neuronal cultures [19, 100–104]. Knockout of the Drosophila melanogaster LRRK2 homolog Lrrk, impairs SVE at the neuromuscular junction (NMJ) via decreased EndoA phosphorylation [100]. In this system, non-phosphorylated EndoA binds to synaptic membranes to initiate SVE [100]. LRRK2-mediated phosphorylation of EndoA then causes it to dissociate from membranes, which is a critical step in completing the SVE cycle [100]. In mouse primary striatal neurons LRRK2 knockout has been observed to impair or enhance compensatory SVE in response to neuronal stimulation [19, 103]. Mass et al. also observed that LRRK2 knockout did not cause SVE changes in primary hippocampal neurons or changes in neurotransmission in acute hippocampal slice recordings [103]. Additionally, LRRK1/2 double knockout did not affect SVE in primary hippocampal neurons or neurotransmission in hippocampal slices, indicating that LRRK1 expression most likely does not compensate for LRRK2 knockout in SVE pathways in these neurons [103]. In primary cortical neurons, acute knockdown of LRRK2 expression via siRNA gene silencing results in altered neurotransmission accompanied by changes in synaptic vesicle recycling [104]. LRRK2 knockdown in these neurons increases the number of recycling synapses under basal conditions, and in response to extracellular calcium, but decreases the number of recycling synapses in response to potassium-evoked depolarization [104].
As previously noted, LRRK2 knockout in mice does not result in dopaminergic neuronal degeneration in the SN, but does cause mild behavioral changes, including altered locomotor activity in the open field test [92]. Changes in neurotransmission observed in LRRK2 knockout or gene silencing experiments in vitro appear to be relatively mild, vary between neuronal subtypes, and do not result in complete loss of neurotransmission or changes in synaptic number [104]. These observations indicate that LRRK2 expression is not required for SVE or neurotransmission and support a model in which LRRK2 fine tunes this process to maintain optimal levels of SVE. There may also be redundancy in the SVE pathway that compensates for the absence of LRRK2 in acute knockdown studies and in LRRK2 knockout animals.
LRRK2 AND LYSOSOMES
Expression of familial PD LRRK2 mutations alters lysosome morphology, distribution, pH and degradative capacity in many distinct cell types including fibroblasts, striatal neurons, primary neurons, and primary astrocytes [79, 96, 105, 106]. Fibroblasts derived from G2019S LRRK2 PD subjects have enlarged lysosomes and altered distribution of lysosomes, where they are tightly clustered in perinuclear regions [105]. Aged R1441G LRRK2 knockin mice also have perinuclear clustering of LAMP1-positive lysosomes, and enlarged LAMP2a puncta in striatal neurons [106]. Additionally, mouse embryonic fibroblasts (MEFs) from these R1441G LRRK2 mice have impaired lysosomal degradative capacity [106]. Overexpression of G2019S LRRK2 in primary astrocytes leads to enlarged lysosomes with perinuclear clustering, as well as decreased LAMP1 and LAMP2 protein levels, increased lysosomal pH and impaired lysosomal degradation capacity [96]. Interestingly, a similar decrease in LAMP1 protein has been observed in the prefrontal cortex of G2019S LRRK2 PD subjects [96].
The effects of G2019S LRRK2 expression in primary neurons appear to vary depending on timing and LRRK2 protein levels. Similar to primary astrocytes, transient transfection of G2019S LRRK2 in primary cortical neurons causes lysosomal swelling [79]. In these neurons, G2019S LRRK2 overexpression also leads to a reduction of mannose-6-phosphate receptor (M6PR) at lysosomes and at the Golgi, which could indicate a defect in M6PR endosomal sorting [79]. Conversely, primary cortical neurons from G2019S LRRK2 knockin mice experience an opposing shift in lysosome morphology, generating both smaller lysosomes and an increase in total lysosome number without perinuclear clustering [107]. Like G2019S LRRK2-expressing primary astrocytes, G2019S LRRK2 knockin neurons have decreased LAMP1 protein levels and increased lysosomal pH [96, 107]. G2019S LRRK2 knockin mice likely express lower levels of pathogenic LRRK2 compared to transiently transfected neurons. Additionally, germline expression of G2019S LRRK2 could potentially result in developmental compensation within the endolysosomal pathway that modulates the toxicity of this pathogenic LRRK2 variant. These putative compensatory pathways are unlikely to be engaged in experiments where G2019S LRRK2 is transiently expressed for short periods and could contribute to the differences observed in lysosomal morphology.
Several recent studies have shown that LRRK2 is recruited to vesicular compartments in the endolysosomal pathway, often in response to stress [80, 108, 109]. LRRK2 subsequently phosphorylates and stabilizes Rab proteins at these compartments, which then recruit downstream effector proteins [80, 108, 109]. Eguchi et al. found that Rab29 recruits LRRK2 to enlarged lysosomes in response to chloroquine treatment, and that LRRK2 recruitment counteracts chloroquine-induced lysosomal overloading in RAW264.7 mouse macrophages and in HEK-293 cells [80]. LRRK2 recruitment initiates the recruitment of four additional Rab proteins, two of which, Rab8a and Rab10, are well-characterized LRRK2 kinase substrates [64, 66, 80]. Phosphorylation of Rab8a and Rab10 by LRRK2 subsequently stabilizes these proteins on lysosomal membranes allowing them to recruit EHBP1 and EHBP1L1, which promote lysosomal secretion and counteract lysosomal overloading [80]. Interestingly, LRRK2 kinase inhibition or LRRK2 knockdown exacerbates lysosomal enlargement in chloroquine-treated cells, and overexpression of PD-linked LRRK2 mutants protects cells from chloroquine-induced lysosomal enlargement [80]. Upregulation of this pathway may have beneficial effects in tissues that express high levels of LRRK2, such as lung and kidney, but could have adverse effects in cells that express low levels of LRRK2. For example, upregulating LRRK2-mediated lysosomal secretion in neurons could increase extracellular levels of proteins that would otherwise be degraded by the lysosomal pathway, such as α-synuclein. If this is the case, then LRRK2-mediated increases in extracellular α-synuclein could contribute to propagation of pathological α-synuclein in PD.
LRRK2 is also recruited to late endosomes and lysosomes in RAW264.7 mouse macrophages in response to endolysosomal membrane damage via pathogen infection or L-leucyl-L-leucine methyl ester (LLOME) treatment [108]. At damaged endolysosomal membranes LRRK2 phosphorylates Rab8a and stabilizes it on these membranes [108]. Phosphorylated Rab8a then recruits the membrane damage marker Galectin-3 and the ESCRT-III pathway component CHMP4B [108]. LRRK2 KO or Rab8a KO enhances lysosomal membrane damage and increases lysophagy in response to LLOME treatment, indicating that this LRRK2-Rab8a pathway stabilizes endolysosomal membrane integrity and promotes endolysosomal membrane repair through the ESCRT pathway [108]. LRRK2 is also recruited to late phagosomes in activated human iPSC-derived macrophages [109]. Like LRRK2 recruitment to endolysosomal membranes, LRRK2 recruitment to late phagosomes promotes the recruitment, phosphorylation and stabilization of Rab8a and Rab10 [109]. LRRK2 does not appear to affect initiation or rate of phagocytosis in these cells, and the downstream effects of LRRK2-Rab recruitment to late phagosomes is currently unknown [109]. Likewise, whether Rab29 is required for LRRK2 recruitment to damaged endolysosomal membranes or late phagosomes has not yet been assessed.
Expression of PD-linked LRRK2 variants also appears to impair lysosomal function by negatively regulating lysosomal glucocerebrosidase (GCase) activity [110, 111]. iPSC-derived dopaminergic neurons from PD subjects harboring R1441C or R1441G LRRK2 mutations have increased accumulation of oxidized dopamine, decreased lysosomal GCase activity, and increased α-synuclein protein levels [110, 111]. Similarly, knockdown of Rab10, a well-characterized substrate of LRRK2 kinase activity, also reduces lysosomal GCase activity in human fibroblasts and iPSC-derived dopaminergic neurons [64, 111]. Surprisingly, overexpression of wild-type Rab10 is sufficient to restore lysosomal GCase activity in iPSC-derived dopaminergic neurons carrying familial LRRK2 mutations (G2019S or R1441C) or GBA mutations (N370S or E326K) [111]. Recent findings also indicate that pharmacological LRRK2 kinase inhibition can restore lysosomal GCase activity, and reduce oxidized dopamine and phosphorylated α-synuclein levels, in iPSC-derived dopaminergic neurons harboring GBA mutations (N370S or E326K) [111]. LRRK2 kinase inhibition also restores lysosomal defects, including altered lysosomal morphology and pH and cathepsin B/L activity, in primary astrocytes derived from D409V GBA knockin mice [112] and human iPSC-derived neurons heterozygous for GBA. LRRK2 appears to negatively regulate lysosomal GCase activity, in a kinase-dependent manner, at least in part through phosphorylation and possible inactivation of Rab10. While LRRK2 and Rab10 can clearly impact lysosomal function, the precise mechanistic details remain to be elucidated. These findings illuminate a novel functional relationship between LRRK2 and lysosomes and suggest that directly inhibiting LRRK2 kinase activity could be an effective approach to correct lysosomal abnormalities in PD subjects.
LRRK2 AND SYNAPTIC VESICLE ENDOCYTOSIS
G2019S LRRK2 overexpression impairs SVE in primary midbrain neurons, primary hippocampal neurons and in Drosophila motor neurons [100–102]. iPSC-derived dopaminergic neurons from PD subjects harboring R1441G LRRK2 mutations also have impaired SVE, reduced expression of SVE proteins, a reduction of small synaptic vesicles and accumulation of enlarged vesicles at presynaptic terminals [110]. LRRK2 interacts physically or functionally with many proteins that are critical facilitators of SVE, including Auxilin (DNAJC6), Dynamin 1, 2, and 3, Synaptojanin-1 (SYNJ1) and EndoA [19, 100, 114–117].
Auxilin regulates a key step in SVE by recruiting Hsc70 to clathrin-coated synaptic vesicles to initiate their uncoating, which is followed by vesicle repackaging and recycling [114]. LRRK2 is suggested to phosphorylate auxilin at Ser627 within its clathrin-binding domain [110]. Nguyen and Krainc show that phosphorylation status at S627 regulates auxilin-clathrin binding and propose that 1) LRRK2-mediated phosphorylation of auxilin at this residue inhibits its binding to clathrin and 2) inhibiting auxilin-clathrin interaction impairs recycling of synaptic vesicles, causing a reduction in synaptic vesicle number and an increase in cytosolic dopamine levels [110]. This hypothesis is supported by the observation that overexpression of wild-type auxilin partially protects R1441G LRRK2 iPSC-derived dopaminergic from the pathogenic effects of mutant LRRK2 (reduces oxidized dopamine, restores lysosomal GCase activity and reduces α-synuclein protein levels) [110]. However, whether overexpression of WT auxilin also restores SVE in dopaminergic neurons harboring pathogenic LRRK2 mutations or protects against LRRK2-mediated alterations in synaptic vesicle morphology has not yet been assessed.
Dynamins are small GTPases that participate in clathrin-mediated endocytosis and intracellular vesicle budding by promoting membrane scission [118, 119]. Mammals have 3 Dynamin genes (Dynamin 1, 2 and 3), all of which are expressed in the brain [118, 119]. Dynamin 1 is highly enriched in neurons; Dynamin 2 is ubiquitously expressed, and Dynamin 3 is expressed in the brain but at lower levels than Dynamin 1 [118, 119]. All 3 Dynamins participate in SVE, with Dynamin 1 playing the most prominent role in this process [118]. LRRK2 complexes with Dynamin 1, 2 and 3 in HEK-293 cells, and with Dynamin 1 in the mouse brain [115]. LRRK2 and Dynamin 1 also colocalize on early endosomes in SH-SY5Y cells [115]. Interestingly, G2019S LRRK2 overexpression in these cells enhances LRRK2-Dynamin 1 colocalization and reduces levels of Dynamin 1 at early endosomes [115]. Additionally, in vitro kinase assays show that Dynamin 1 is a modest LRRK2 kinase substrate [115]. These LRRK2-Dynamin interactions may contribute to LRRK2-mediated disruptions in SVE or intracellular trafficking defects. Further experiments examining whether LRRK2-Dynamin complexes or Dynamin 1 phosphorylation by LRRK2 contributes to SVE abnormalities caused by pathogenic LRRK2 variants will help further elucidate this pathway.
Synaptojanin-1 (SYNJ1) is an EndoA-interacting lipid phosphatase that plays a critical role in clathrin uncoating of vesicles during SVE [120]. An unbiased phospho-proteome screen in brains of a Drosophila PD model expressing human R1441C LRRK2 identified increased phosphorylation of synaptojanin-1 at two distinct sites (pThr1131 and pSer1142) [116]. This finding could also be extended to human synaptojanin-1 with direct phosphorylation of Thr1173 by LRRK2 in vitro identified by mass spectrometry [116]. As noted earlier, EndoA is also a putative kinase substrate of LRRK2 and decreases in EndoA phosphorylation appear to contribute to disruptions in SVE in LRRK2 KO Drosophila [100]. Together, these findings identify multiple LRRK2-interacting partners and LRRK2 kinase substrates that directly regulate SVE and suggest that LRRK2-mediated disruption in SVE can lead to downstream events that participate in PD pathogenesis. This idea is further supported by the identification of SYNJ1 and DNAJC6 (auxilin) mutations as causes of early-onset, recessive familial PD [121–124]. Accordingly, targeting SVE pathway deficits could potentially ameliorate at least some of the pathogenic actions of LRRK2 (or SYNJ1 or DNAJC6) mutations in PD subjects, and this approach should be further explored in cellular and rodent models as a strategy to inhibit mutant LRRK2-induced neuronal damage.
LRRK2 AND THE GOLGI NETWORK
LRRK2 has a robust effect on Golgi integrity and vesicle sorting via the Golgi. Overexpression of familial LRRK2 mutants induces Golgi fragmentation in cell lines, primary neurons, and striatal neurons [78, 82, 125, 126]. Surprisingly, LRRK2 depletion has a similar effect, leading to Golgi fragmentation and defective Golgi sorting of lysosomal hydrolases [127].
When transiently overexpressed in cultured cells, Rab29 recruits LRRK2 to the TGN, where it activates LRRK2, enhancing LRRK2 autophosphorylation and substrate phosphorylation [79, 81–84]. Rab29-mediated recruitment of LRRK2 to the TGN promotes LRRK2-mediated phosphorylation of Rab29 and recruitment and phosphorylation of 2 additional LRRK2 substrates, Rab10 and Rab8a to the TGN [79, 81–83]. This process requires Rab29 prenylation and membrane association but does not rely on other proteins that are specifically enriched on TGN membranes, as artificially targeting Rab29 to mitochondrial membranes initiates this cascade at the mitochondria [84]. Guanine nucleotide-binding of Rab29 and LRRK2 is required for this process, but Rab29 recruitment of LRRK2 does not require LRRK2 kinase activity [82, 83]. This pathway is likely important for maintaining Golgi integrity, as Rab29 depletion or expression of Rab29 dominant-negative or phospho-mimetic mutants also causes Golgi fragmentation [78, 128]. In addition to complexing with Rab29, LRRK2 also binds BAG5 and GAK proteins in cultured cells and mouse brain [126]. Overexpression of any of these individual proteins causes Golgi fragmentation which can be blocked by concurrent knockdown of any of the other proteins individually [126]. These results implicate all four complex components in Rab29/LRRK2-mediated Golgi maintenance [126].
The Rab29-LRRK2 pathway is also important for retrograde trafficking as knockout or dominant-negative Rab29 expression disrupts retrograde trafficking of M6PR, Sortilin and Furin from endosomes and lysosomes to the TGN [79, 128]. Beilina et al. has recently reported a novel interaction between LRRK2, Rab29 and the Golgi-associated retrograde protein (GARP) complex [94]. Specifically, LRRK2 and Rab29 interact with the GARP complex component VPS52 at the TGN. Overexpression of Rab29 recruits VPS52 to the TGN, and this effect is enhanced by co-expression of LRRK2 [94]. Conversely, knockdown of VPS52, or other GARP complex components (VPS53 or VPS51), inhibits Rab29-mediated recruitment of LRRK2 to the TGN, indicating a cooperative stabilization of the Rab29:LRRK2:GARP complex that requires each component [94]. LRRK2 and VPS52 both complex with VAMP4 and syntaxin-6, SNARE proteins that localize to the TGN, suggesting that LRRK2 may stabilize GARP complex interactions with SNARE proteins at the TGN to promote membrane fusion and vesicle trafficking [94]. This hypothesis is supported by the observation that LRRK2 or Rab29 knockdown destabilizes the VPS52-syntaxin-6 interaction [94]. Knockdown of Rab29, LRRK2 or VPS52 decreases retrograde trafficking of M6PR or Cholera toxin B from endosomes to the TGN and impairs anterograde trafficking of GPI proteins from the TGN to the plasma membrane [94]. Conversely, primary astrocytes from R1441C LRRK2 knockin mice exhibit a kinase-dependent increase in retrograde and anterograde vesicular trafficking [94].
This study provides valuable mechanistic insight into the role of LRRK2 in vesicle trafficking to and from the TGN. Because the GARP complex has not been shown to directly regulate anterograde trafficking, the precise mechanisms through which LRRK2 modulates this pathway are currently unclear. The effects of LRRK2 on anterograde trafficking from the TGN may be an indirect result of modulating retrograde trafficking through the GARP complex, or could be caused by direct interactions between LRRK2 and the anterograde trafficking pathway. Further examination of anterograde trafficking components and LRRK2 will be required to distinguish between these two possibilities. How chronic LRRK2-mediated enhancement of retrograde trafficking affects the endolysosomal system in vivo, and how this contributes to PD neuropathology will be an interesting area of investigation. Specifically, whether modulating the LRRK2-GARP complex interaction can ameliorate LRRK2-induced neurotoxicity will be important. Surprisingly, Beilina et al. found that neuronal silencing of the GARP complex-specific component VPS54 (but not VPS50) enhanced age-dependent dopaminergic neuronal damage induced by G2019S LRRK2 in a C. elegans model, suggesting that the GARP complex operates downstream of LRRK2 and is normally required to maintain neuronal health in vivo [94].
ArfGAP1 is a GAP for Arf1, a cis-Golgi protein that functions in the secretory pathway and regulates Golgi organization and lipid homeostasis in this organelle [129, 130]. ArfGAP1 was initially identified as a LRRK2 modifier in yeast where deletion of the ArfGAP1 ortholog, GCS1, suppressed growth deficits and endolysosomal abnormalities induced by human LRRK2 [131]. Subsequent experiments have shown that LRRK2 complexes with ArfGAP1 in mammalian cells and mouse brain, and colocalizes with ArfGAP1 in compact perinuclear structures that resemble the Golgi in primary cortical neurons [125, 132]. Additionally, both ArfGAP1 and LRRK2 are present in Golgi subcellular fractions, indicating that these proteins likely complex together at this organelle [125, 132]. In vitro, ArfGAP1 acts as a LRRK2 GAP, enhances LRRK2 kinase activity and is a robust LRRK2 kinase substrate [125, 132].
Consistent with the observation that GCS1 deletion suppresses LRRK2-induced toxicity in yeast, gene silencing of ArfGAP1 in primary cortical neurons suppresses G2019S LRRK2-mediated neurite outgrowth deficits [125]. Interestingly, wild-type ArfGAP1 overexpression also causes neurite outgrowth deficits in primary cortical neurons, which is suppressed by silencing LRRK2 [125]. These findings indicate a cooperative relationship where enhanced LRRK2 or ArfGAP1 expression leads to neurite outgrowth deficits and both proteins are required for this pathological effect [125]. Additionally, overexpression of either ArfGAP1 or familial LRRK2 mutants causes Golgi complex fragmentation in primary cortical neurons [125]. Whether mutant LRRK2-mediated Golgi fragmentation is modulated by ArfGAP1 expression, or vice versa, has not yet been assessed. However, given the direct interaction between ArfGAP1 and LRRK2, and the functional interactions observed in yeast and primary neurite outgrowth experiments, it is likely that ArfGAP1-LRRK2 interactions are important for maintaining Golgi complex integrity and function.
LRRK2-mediated effects on Golgi integrity may also involve the N-ethylmaleimide-sensitive fusion (NSF) protein. NSF primarily functions as a SNARE complex chaperone, facilitating vesicle fusion between intracellular membranes and SNARE complex disassembly for recycling [133]. It was originally identified as an essential factor for vesicle trafficking in the Golgi [134]. NSF has also been shown to facilitate vesicle trafficking from the endoplasmic reticulum to the Golgi, and Golgi reassembly following mitosis [135, 136]. LRRK2 complexes with NSF in mammalian cells and phosphorylates NSF in vitro [127, 137]. Interestingly, NSF knockdown phenocopies the effects of LRRK2 knockdown, in mammalian cells, with Golgi fragmentation and the formation of enlarged late endosomal structures [127]. LRRK2 or NSF knockdown also disrupts plasma membrane receptor recycling pathways and trafficking of lysosomal hydrolases from the TGN to lysosomes, resulting in impaired lysosomal degradation capacity [127]. Whether LRRK2-NSF interaction or LRRK2-mediated phosphorylation of NSF is required for Golgi maintenance and function has not yet been assessed. Similarly, whether NSF expression or phosphorylation of NSF by LRRK2 contributes to Golgi fragmentation in response to overexpression of familial LRRK2 mutants has not been tested.
In addition to maintaining Golgi integrity, LRRK2 has also been implicated in the maintenance of Endoplasmic Reticulum Exit Sites (ERESs) through interaction with the ERES protein Sec16a [138]. Sec16a recruits and stabilizes protein complexes to ERESs that facilitate the formation of COPII-coated vesicles [139]. Knockdown or overexpression of Sec16a results in secretory pathway disruption and disorganization of ERESs [139]. LRRK2 complexes with Sec16a in mammalian cells and in the mouse brain, and colocalizes with Sec16a at ERESs in primary hippocampal neurons [138]. LRRK2 knockout in primary hippocampal neurons or in MEFs leads to dispersed Sec16a and impaired ER to Golgi transport [138]. Interestingly, the familial R1441C LRRK2 mutation has impaired interaction with Sec16a compared to wild-type LRRK2. Additionally, R1441C LRRK2 knockin MEFs also have impaired ER to Golgi transport and show Sec16a dispersion [138]. These findings implicate LRRK2 in organization and maintenance of ERESs and highlight an additional role for LRRK2 in regulation of transport through the secretory pathway.
LRRK2 interacts with multiple proteins resident at the Golgi and endoplasmic reticulum to regulate Golgi integrity, and Golgi network anterograde or retrograde trafficking. Additionally, modulating the expression of individual LRRK2-interacting proteins in these pathways can prevent LRRK2-induced Golgi fragmentation (Rab29, BAG5, GAK) or LRRK2-induced toxicity (ArfGAP1) [125, 126]. Most studies examining the effects of LRRK2 on the Golgi network have been performed in cell culture models with overexpression or depletion of LRRK2. Examining Golgi integrity and vesicle trafficking in knockin rodent models where familial LRRK2 mutants are expressed at endogenous levels will be important for evaluating 1) whether the Golgi network is disrupted by familial LRRK2 mutations in vivo and 2) whether Golgi disruption contributes to LRRK2-PD and sporadic PD.
LRRK2 AND THE RETROMER
The retromer complex regulates retrograde trafficking and retrieval of transmembrane protein cargo from early endosomes to the TGN or cell surface membrane for recycling [140–142]. The retromer complex has two components, the cargo-selective complex (CSC) and a sorting nexin (SNX) dimer. VPS35 is the largest component of the CSC and is essential for retromer-mediated cargo sorting, particularly for cargo recognition, which is critical for cellular function. Recently, VPS35 has been identified as a familial PD gene, with a missense mutation (D620N) causing late-onset, autosomal dominant familial PD [143, 144]. The precise effects of the D620N mutation on VPS35 function, and the molecular mechanisms that underlie the pathogenicity of this mutation, are currently unclear [145]. The dominant inheritance pattern of PD associated with D620N VPS35 suggests that this mutation most likely manifests disease via 1) a partial dominant-negative effect on VPS35 function or 2) a toxic or normal gain-of-function effect.
Recent studies of D620N VPS35 in rodent models and PD subjects indicate that this mutation does not manifest an obvious loss-of-function effect, but potentially induces PD pathology by activating LRRK2 [146–148]. Notably, although homozygous VPS35 knockout mice die early in embryonic development, homozygous D620N VPS35 knockin mice are viable with a normal lifespan, indicating that this mutation does not result in a full loss-of-function [146]. MEF cultures carrying the D620N VPS35 mutation exhibit markedly increased LRRK2-mediated phosphorylation of Rab10 and increased LRRK2 autophosphorylation at Ser1292, both of which are decreased by treatment with the selective LRRK2 kinase inhibitor MLi-2 [147]. Increased Rab10 phosphorylation also occurs in peripheral organs and brain of D620N VPS35 knockin mice, and in neutrophils and monocytes from PD subjects carrying a heterozygous D620N mutation [147, 148]. Importantly, LRRK2 hyperactivation initially appears to be equivalently induced by heterozygous or homozygous D620N mutations in these mice supporting a threshold effect and most likely a gain-of-function mechanism [147]. Intriguingly, the D620N VPS35 mutation enhances phosphorylation of LRRK2 kinase substrates (by ∼6-fold) to a greater extent than familial LRRK2 mutations in knockin mice (2–4-fold), and endogenous VPS35 is required for the increased LRRK2 kinase activity in cells derived from mutant LRRK2 knockin mice [147]. These compelling data potentially support the concept of VPS35 as a master regulator of LRRK2 activity presumably at endosomes, similar to the effects of Rab29 at the TGN, although the underlying activation mechanism and subcellular location remain obscure.
LRRK2 may also regulate retrograde trafficking through interactions with Rab32 or Rab38. Rab32 and Rab38 both complex with LRRK2 and with sorting nexin 6 (SNX6), a SNX dimer subunit of the retromer [149, 150]. Expression of constitutively-active or inactive Rab32 impairs retrograde trafficking of the retromer cargo M6PR [149]. Whether LRRK2 modulates Rab32-mediated disruption of retrograde trafficking has not yet been tested, and it is currently not known whether Rab32 and Rab38 exist together in a complex with LRRK2 and SNX6, or independently. This putative pathway for LRRK2-retromer interaction via Rab proteins is of interest but has not yet been fully characterized. Further evaluation of LRRK2 and retromer subunit interactions will be beneficial in determining how these proteins functionally intersect and modulate each other. Evaluating whether D620N VPS35-dependent neurodegeneration can be ameliorated by LRRK2 knockout or kinase inhibition in rodent models will be important for validating the significance of their functional interaction in the context of PD-like pathology.
LRRK2 AND AUTOPHAGY
Autophagy is a critical process for maintaining protein homeostasis and preventing protein aggregation. Perturbations in autophagy have been observed in brain tissue from PD subjects and in animal models of PD. The relationship between LRRK2 and autophagy has been an area of growing interest and has been studied in many model systems. As this topic has recently been comprehensively reviewed [151], we will provide a limited discussion of LRRK2 and autophagy.
Expression of familial LRRK2 mutants in rodents, or in LRRK2-PD brains, leads to alterations in macroautophagy, chaperone-mediated autophagy (CMA) and mitophagy in the brain [107, 152–157]. G2019S LRRK2 inhibits CMA in isolated lysosomes, HEK-293 cells and in cultured primary midbrain neurons [158]. Additionally, brain tissue from G2019S-PD subjects displays increased levels of the CMA protein LAMP2a, confirming alterations in this pathway [158]. R1441G LRRK2 knockin mice also show signs of impaired CMA with accumulation of amyloid-like α-synuclein oligomers and increased membrane-bound LAMP2a and HSPA8 in striatal tissue [106].
Familial LRRK2 mutations can alter mitophagy and macroautophagy in diverse model systems, however, the impact of LRRK2 on these pathways varies in both magnitude and direction. These variations in effect size likely result from differences in experimental systems including cell type, cellular context, and timing and dosage of LRRK2 expression. For example, human fibroblasts derived from PD subjects harboring G2019S or R1441C LRRK2 mutations exhibit decreased mitophagy that results from enhanced phosphorylation of Rab10 [157]. Rab10 phosphorylation impairs the Rab10-optineurin interaction and prevents Rab10 from recruiting optineurin to depolarized mitochondria to initiate mitophagy [157]. Alternatively, transient overexpression of G2019S or R1441C LRRK2 in primary cortical neurons enhances calcium uptake by mitochondria, leading to mitochondrial membrane depolarization and increased mitophagy specifically in dendrites, where transient calcium influxes are highest [152, 156].
LRRK2 knockout mice exhibit altered expression of autophagy markers and formation of large autophagic vacuoles in kidneys, but lack autophagic pathology in the brain [88, 90, 92, 93]. This is likely due to LRRK2 being highly expressed in kidneys relative to the brain, and LRRK2 knockout in the brain may potentially be compensated for by LRRK1 expression. Interestingly, LRRK1/2 double knockout in mice causes age-dependent impairment of the autophagy pathway in the brain that results in the formation of p62-positive puncta and the accumulation of autophagic vacuoles in the substantia nigra [95]. Familial LRRK2 mutations appear to have subtle effects on macroautophagy in vivo with modest variations between model systems. An early study detected the abnormal accumulation of autophagic vacuoles, often containing damaged or condensed mitochondria, by transmission electron microscopy in the striatum and cortex of aged human G2019S or R1441C LRRK2 transgenic mice [159]. Aged R1441G LRRK2 knockin mice have no apparent alterations in autophagy markers or protein aggregation in the midbrain [160]. Likewise, mice that overexpress human R1441C LRRK2 selectively in dopaminergic neurons do not develop protein aggregation or changes in autophagy markers in the nigra with age [161]. G2019S LRRK2 knockin mice have slightly elevated levels of the autophagy marker, LC3-II, in hemi-brain extracts and decreased LC3-I protein levels in the cortex, but no change in p62 levels in hemi-brain or cortical extracts [107, 162]. Aged transgenic rats overexpressing human R1441C or G2019S LRRK2 have increased LC3-positive puncta in midbrain dopaminergic neurons compared to transgenic rats expressing wild-type LRRK2 or non-transgenic controls, suggesting that mutant LRRK2 induces autophagy or impairs autophagic flux [97]. Corresponding evidence for altered autophagy in G2019S LRRK2-PD brains is also modest, with milder effects than in sporadic PD overall, yet with a clear increase in p62-positive inclusions and staining relative to control subjects [154]. Therefore, evidence for altered autophagy in brain tissue of mutant LRRK2 models and PD subjects is rather modest and depends to a large degree on gene dosage, cell type or brain region in each rodent model or PD subject.
CONCLUSIONS
The effects of LRRK2 knockout or PD-linked familial LRRK2 mutations are pervasive throughout the endolysosomal system and occur in multiple intersecting pathways, including endocytosis, lysosomal function, Golgi sorting, retrograde endosomal trafficking, and autophagy. These findings suggest that LRRK2 functions at many points in the endolysosomal system, with increases or decreases in LRRK2 expression levels and/or kinase activity negatively impacting this system. Specific tissues and cell types appear to have distinct requirements for LRRK2 kinase activity. For example, the organs most affected by LRRK2 KO are the lungs and kidneys where LRRK2 expression levels are highest [88–92]. However, similar endolysosomal abnormalities have not been reported in the brain of LRRK2 KO rodents, and aged LRRK2 KO rodents do not exhibit dopaminergic neurodegeneration [88, 90–92]. Together, this suggests that the brain may be more sensitive to increases in LRRK2 kinase activity due to PD-linked mutations while peripheral organs with comparatively higher levels of LRRK2 expression are more sensitive to the loss of LRRK2 expression. In addition, it is plausible that unique compensatory mechanisms exist in the mammalian brain that mask the detrimental effects of LRRK2 loss-of-function in KO rodent models.
Lysosomes appear to be particularly sensitive to changes in LRRK2 expression and kinase activity. Impaired lysosomal degradation capacity and im-paired autophagy are emerging as recurring themes in distinct PD models. Identifying the mechanisms through which LRRK2 activity modulates lysosome homeostasis and protein degradation will be important for understanding how LRRK2 mutations contribute to PD. Lysosomes are promising therapeutic targets for treating PD, and recent findings support the idea of utilizing LRRK2 kinase inhibitors to attenuate endolysosomal perturbations in subjects with sporadic PD or GBA-linked PD [67, 68, 111, 112]. Evaluating the neuroprotective effects of LRRK2 kinase inhibition in additional genetic models of PD will provide an indication of how effective this approach might be for treating a broader population of familial or sporadic PD subjects. The recent discovery that D620N VPS35 induces LRRK2 hyperactivation might suggest that kinase inhibition could also be an effective strategy for treating VPS35-linked PD [147]. This raises the broader question of whether LRRK2 activation represents a common outcome of different genetic forms of PD, with compelling evidence for the PD risk factor, Rab29, and VPS35 as master regulators of LRRK2 activity within distinct vesicular compartments [81, 82, 126, 147]. Likewise, LRRK2 kinase activity may be functionally linked to GBA deficiency [111–113], whereas non-mutated LRRK2 might be activated in sporadic PD brains or by environmental neurotoxins [67, 68]. Many gene products linked to PD can be categorized as either activators of LRRK2 or targets of LRRK2 activity either directly or potentially indirectly via phosphorylation of a subset of Rabs that decorate the endolysosomal system and mediate membrane-targeting events [163, 164]. A detailed mechanistic understanding of how other PD-linked gene products functionally cooperate with LRRK2 in the endolysosomal pathway in addition to elucidating the discrete effects mediated by LRRK2 and/or Rab phosphorylation within this pathway will form pivotal next steps in further uncovering the pathophysiological mechanisms underlying PD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors work is supported in part by grants from the National Institutes of Health (R01 NS091719, R01 NS105432, R01 NS117137, to D.J.M.) and the Parkinson’s Foundation (PF-FBS-1894, to M.E.).
REFERENCES
[1] | Lang AE , Lozano AM ((1998) ) Parkinson’s disease. First of two parts. N Engl J Med 339: , 1044–1053. |
[2] | Lang AE , Lozano AM ((1998) ) Parkinson’s disease. Second of two parts. N Engl J Med 339: , 1130–1143. |
[3] | Hernandez DG , Reed X , Singleton AB ((2016) ) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139: , 59–74. |
[4] | Trinh J , Farrer M ((2013) ) Advances in the genetics of Parkinson disease. Nat Rev Neurol 9: , 445–454. |
[5] | Blauwendraat C , Nalls MA , Singleton AB ((2020) ) The genetic architecture of Parkinson’s disease. Lancet Neurol 19: , 170–178. |
[6] | Ferreira M , Massano J ((2017) ) An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol Scand 135: , 273–284. |
[7] | Aharon-Peretz J , Rosenbaum H , Gershoni-Baruch R ((2004) ) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351: , 1972–1977. |
[8] | Chang D , Nalls MA , Hallgrímsdóttir IB , Hunkapiller J , van der Brug M , Cai F , Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[9] | Di Fonzo A , Wu-Chou YH , Lu CS , van Doeselaar M , Simons EJ , Rohé CF , Chang HC , Chen RS , Weng YH , Vanacore N , Breedveld GJ , Oostra BA , Bonifati V ((2006) ) A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics 7: , 133–138. |
[10] | Goker-Alpan O , Schiffmann R , LaMarca ME , Nussbaum RL , McInerney-Leo A , Sidransky E ((2004) ) Parkinsonism among Gaucher disease carriers. J Med Genet 41: , 937–940. |
[11] | Hamza TH , Zabetian CP , Tenesa A , Laederach A , Montimurro J , Yearout D , Kay DM , Doheny KF , Paschall J , Pugh E , Kusel VI , Collura R , Roberts J , Griffith A , Samii A , Scott WK , Nutt J , Factor SA , Payami H ((2010) ) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42: , 781–785. |
[12] | Krohn L , Öztürk TN , Vanderperre B , Ouled Amar Bencheikh B , Ruskey JA , Laurent SB , Spiegelman D , Postuma RB , Arnulf I , Hu MTM , Dauvilliers Y , Högl B , Stefani A , Monaca CC , Plazzi G , Antelmi E , Ferini-Strambi L , Heidbreder A , Rudakou U , Cochen De Cock V , Young P , Wolf P , Oliva P , Zhang XK , Greenbaum L , Liong C , Gagnon JF , Desautels A , Hassin-Baer S , Montplaisir JY , Dupré N , Rouleau GA , Fon EA , Trempe JF , Lamoureux G , Alcalay RN , Gan-Or Z ((2020) ) Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann Neurol 87: , 139–153. |
[13] | Lwin A , Orvisky E , Goker-Alpan O , LaMarca ME , Sidransky E ((2004) ) Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 81: , 70–73. |
[14] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simon-Sanchez J , Schulte C , Sharma M , Krohn L , Pihlstrom L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB , 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium ((2019) ) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol 18: , 1091–1102. |
[15] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R ; International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI); 23andMe; GenePD; NeuroGenetics Research Consortium (NGRC); Hussman Institute of Human Genomics (HIHG); Ashkenazi Jewish Dataset Investigator; Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE); North American Brain Expression Consortium (NABEC); United Kingdom Brain Expression Consortium (UKBEC); Greek Parkinson’s Disease Consortium; Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46: , 989–993. |
[16] | Satake W , Nakabayashi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A , Tomiyama H , Nakashima K , Hasegawa K , Obata F , Yoshikawa T , Kawakami H , Sakoda S , Yamamoto M , Hattori N , Murata M , Nakamura Y , Toda T ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41: , 1303–1307. |
[17] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A , Chen CM , Clark LN , Condroyer C , De Marco EV , Dürr A , Eblan MJ , Fahn S , Farrer MJ , Fung HC , Gan-Or Z , Gasser T , Gershoni-Baruch R , Giladi N , Griffith A , Gurevich T , Januario C , Kropp P , Lang AE , Lee-Chen GJ , Lesage S , Marder K , Mata IF , Mirelman A , Mitsui J , Mizuta I , Nicoletti G , Oliveira C , Ottman R , Orr-Urtreger A , Pereira LV , Quattrone A , Rogaeva E , Rolfs A , Rosenbaum H , Rozenberg R , Samii A , Samaddar T , Schulte C , Sharma M , Singleton A , Spitz M , Tan EK , Tayebi N , Toda T , Troiano AR , Tsuji S , Wittstock M , Wolfsberg TG , Wu YR , Zabetian CP , Zhao Y , Ziegler SG ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361: , 1651–1661. |
[18] | Simón-Sánchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Krüger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[19] | Arranz AM , Delbroek L , Van Kolen K , Guimaraes MR , Mandemakers W , Daneels G , Matta S , Calafate S , Shaban H , Baatsen P , De Bock PJ , Gevaert K , Vanden Berghe P , Verstreken P , De Strooper B , Moechars D ((2015) ) LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J Cell Sci 128: , 541–552. |
[20] | Cao M , Wu Y , Ashrafi G , McCartney AJ , Wheeler H , Bushong EA , Boassa D , Ellisman MH , Ryan TA , De Camilli P ((2017) ) Parkinson sac domain mutation in Synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron 93: , 882–896.e885. |
[21] | Cremona O , Di Paolo G , Wenk MR , Lüthi A , Kim WT , Takei K , Daniell L , Nemoto Y , Shears SB , Flavell RA , McCormick DA , De Camilli P ((1999) ) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99: , 179–188. |
[22] | Vargas KJ , Makani S , Davis T , Westphal CH , Castillo PE , Chandra SS ((2014) ) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34: , 9364–9376. |
[23] | Ungewickell E , Ungewickell H , Holstein SE , Lindner R , Prasad K , Barouch W , Martin B , Greene LE , Eisenberg E ((1995) ) Role of auxilin in uncoating clathrin-coated vesicles. Nature 378: , 632–635. |
[24] | Lee DW , Zhao X , Zhang F , Eisenberg E , Greene LE ((2005) ) Depletion of GAK/auxilin 2 inhibits receptor-mediated endocytosis and recruitment of both clathrin and clathrin adaptors. J Cell Sci 118: , 4311–4321. |
[25] | Schuske KR , Richmond JE , Matthies DS , Davis WS , Runz S , Rube DA , van der Bliek AM , Jorgensen EM ((2003) ) Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron 40: , 749–762. |
[26] | Verstreken P , Kjaerulff O , Lloyd TE , Atkinson R , Zhou Y , Meinertzhagen IA , Bellen HJ ((2002) ) Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell 109: , 101–112. |
[27] | Migheli R , Del Giudice MG , Spissu Y , Sanna G , Xiong Y , Dawson TM , Dawson VL , Galioto M , Rocchitta G , Biosa A , Serra PA , Carri MT , Crosio C , Iaccarino C ((2013) ) LRRK2 affects vesicle trafficking, neurotransmitter extracellular level and membrane receptor localization. PLoS One 8: , e77198. |
[28] | Paravicini G , Horazdovsky BF , Emr SD ((1992) ) Alternative pathways for the sorting of soluble vacuolar proteins in yeast: A vps35 null mutant missorts and secretes only a subset of vacuolar hydrolases. Mol Biol Cell 3: , 415–427. |
[29] | Mignogna ML , Giannandrea M , Gurgone A , Fanelli F , Raimondi F , Mapelli L , Bassani S , Fang H , Van Anken E , Alessio M , Passafaro M , Gatti S , Esteban JA , Huganir R , D’Adamo P ((2015) ) The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat Commun 6: , 6504. |
[30] | Matsuda N , Sato S , Shiba K , Okatsu K , Saisho K , Gautier CA , Sou YS , Saiki S , Kawajiri S , Sato F , Kimura M , Komatsu M , Hattori N , Tanaka K ((2010) ) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: , 211–221. |
[31] | Narendra D , Tanaka A , Suen DF , Youle RJ ((2008) ) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: , 795–803. |
[32] | Narendra DP , Jin SM , Tanaka A , Suen DF , Gautier CA , Shen J , Cookson MR , Youle RJ ((2010) ) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: , e1000298. |
[33] | Weinreb NJ , Brady RO , Tappel AL ((1968) ) The lysosomal localization of sphingolipid hydrolases. Biochim Biophys Acta 159: , 141–146. |
[34] | Brady RO , Kanfer JN , Bradley RM , Shapiro D ((1966) ) Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J Clin Invest 45: , 1112–1115. |
[35] | Dehay B , Ramirez A , Martinez-Vicente M , Perier C , Canron MH , Doudnikoff E , Vital A , Vila M , Klein C , Bezard E ((2012) ) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109: , 9611–9616. |
[36] | Usenovic M , Tresse E , Mazzulli JR , Taylor JP , Krainc D ((2012) ) Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci 32: , 4240–4246. |
[37] | van Veen S , Martin S , Van den Haute C , Benoy V , Lyons J , Vanhoutte R , Kahler JP , Decuypere J-P , Gelders G , Lambie E , Zielich J , Swinnen JV , Annaert W , Agostinis P , Ghesquière B , Verhelst S , Baekelandt V , Eggermont J , Vangheluwe P ((2020) ) ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578: , 419–424. |
[38] | Reczek D , Schwake M , Schröder J , Hughes H , Blanz J , Jin X , Brondyk W , Van Patten S , Edmunds T , Saftig P ((2007) ) LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131: , 770–783. |
[39] | Jinn S , Drolet RE , Cramer PE , Wong AH-K , Toolan DM , Gretzula CA , Voleti B , Vassileva G , Disa J , Tadin-Strapps M , Stone DJ ((2017) ) TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A 114: , 2389–2394. |
[40] | Hsin IL , Sheu GT , Jan MS , Sun HL , Wu TC , Chiu LY , Lue KH , Ko JL ((2012) ) Inhibition of lysosome degradation on autophagosome formation and responses to GMI, an immunomodulatory protein from Ganoderma microsporum. Br J Pharmacol 167: , 1287–1300. |
[41] | McGlinchey RP , Lee JC ((2015) ) Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci U S A 112: , 9322–9327. |
[42] | Qi X , Man SM , Malireddi RK , Karki R , Lupfer C , Gurung P , Neale G , Guy CS , Lamkanfi M , Kanneganti TD ((2016) ) Cathepsin B modulates lysosomal biogenesis and host defense against Francisella novicida infection. J Exp Med 213: , 2081–2097. |
[43] | Di Fonzo A , Rohé CF , Ferreira J , Chien HF , Vacca L , Stocchi F , Guedes L , Fabrizio E , Manfredi M , Vanacore N , Goldwurm S , Breedveld G , Sampaio C , Meco G , Barbosa E , Oostra BA , Bonifati V ((2005) ) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365: , 412–415. |
[44] | Zimprich A , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Muller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , Gasser T ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |
[45] | Paisan-Ruiz C , Jain S , Evans EW , Gilks WP , Simon J , van der Brug M , Lopez de Munain A , Aparicio S , Gil AM , Khan N , Johnson J , Martinez JR , Nicholl D , Marti Carrera I , Pena AS , de Silva R , Lees A , Marti-Masso JF , Perez-Tur J , Wood NW , Singleton AB ((2004) ) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44: , 595–600. |
[46] | Ross OA , Toft M , Whittle AJ , Johnson JL , Papapetropoulos S , Mash DC , Litvan I , Gordon MF , Wszolek ZK , Farrer MJ , Dickson DW ((2006) ) Lrrk2 and Lewy body disease. Ann Neurol 59: , 388–393. |
[47] | Henderson MX , Sengupta M , Trojanowski JQ , Lee VMY ((2019) ) Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun 7: , 183. |
[48] | Rajput A , Dickson DW , Robinson CA , Ross OA , Dächsel JC , Lincoln SJ , Cobb SA , Rajput ML , Farrer MJ ((2006) ) Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67: , 1506–1508. |
[49] | Gilks WP , Abou-Sleiman PM , Gandhi S , Jain S , Singleton A , Lees AJ , Shaw K , Bhatia KP , Bonifati V , Quinn NP , Lynch J , Healy DG , Holton JL , Revesz T , Wood NW ((2005) ) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365: , 415–416. |
[50] | Lesage S , Janin S , Lohmann E , Leutenegger A-L , Leclere L , Viallet Fo , Pollak P , Durif F , Thobois S , Layet V , Vidailhet M , Agid Y , Durr A , Brice A ((2007) ) LRRK2 exon 41 mutations in sporadic Parkinson disease in Europeans. Arch Neurol 64: , 425–430. |
[51] | Shu L , Zhang Y , Sun Q , Pan H , Tang B ((2019) ) A comprehensive analysis of population differences in LRRK2 variant distribution in Parkinson’s disease. Front Aging Neurosci 11: , 13. |
[52] | Sen S , Webber PJ , West AB ((2009) ) Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem 284: , 36346–36356. |
[53] | Berger Z , Smith KA , Lavoie MJ ((2010) ) Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49: , 5511–5523. |
[54] | Greggio E , Zambrano I , Kaganovich A , Beilina A , Taymans JM , Daniels V , Lewis P , Jain S , Ding J , Syed A , Thomas KJ , Baekelandt V , Cookson MR ((2008) ) The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem 283: , 16906–16914. |
[55] | Aasly JO , Vilariño-Güell C , Dachsel JC , Webber PJ , West AB , Haugarvoll K , Johansen KK , Toft M , Nutt JG , Payami H , Kachergus JM , Lincoln SJ , Felic A , Wider C , Soto-Ortolaza AI , Cobb SA , White LR , Ross OA , Farrer MJ ((2010) ) Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson’s disease. Mov Disord 25: , 2156–2163. |
[56] | Rudenko IN , Cookson MR ((2014) ) Heterogeneity of leucine-rich repeat kinase 2 mutations: Genetics, mechanisms and therapeutic implications. Neurotherapeutics 11: , 738–750. |
[57] | Haugarvoll K , Rademakers R , Kachergus JM , Nuytemans K , Ross OA , Gibson JM , Tan EK , Gaig C , Tolosa E , Goldwurm S , Guidi M , Riboldazzi G , Brown L , Walter U , Benecke R , Berg D , Gasser T , Theuns J , Pals P , Cras P , De Deyn PP , Engelborghs S , Pickut B , Uitti RJ , Foroud T , Nichols WC , Hagenah J , Klein C , Samii A , Zabetian CP , Bonifati V , Van Broeckhoven C , Farrer MJ , Wszolek ZK ((2008) ) Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 70: , 1456–1460. |
[58] | Lin CH , Tzen KY , Yu CY , Tai CH , Farrer MJ , Wu RM ((2008) ) LRRK2 mutation in familial Parkinson’s disease in a Taiwanese population: Clinical, PET, and functional studies. J Biomed Sci 15: , 661–667. |
[59] | Mata IF , Kachergus JM , Taylor JP , Lincoln S , Aasly J , Lynch T , Hulihan MM , Cobb SA , Wu RM , Lu CS , Lahoz C , Wszolek ZK , Farrer MJ ((2005) ) Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 6: , 171–177. |
[60] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AHV , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[61] | West AB , Moore DJ , Biskup S , Bugayenko A , Smith WW , Ross CA , Dawson VL , Dawson TM ((2005) ) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102: , 16842–16847. |
[62] | Greggio E , Jain S , Kingsbury A , Bandopadhyay R , Lewis P , Kaganovich A , van der Brug MP , Beilina A , Blackinton J , Thomas KJ , Ahmad R , Miller DW , Kesavapany S , Singleton A , Lees A , Harvey RJ , Harvey K , Cookson MR ((2006) ) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23: , 329–341. |
[63] | West AB , Moore DJ , Choi C , Andrabi SA , Li X , Dikeman D , Biskup S , Zhang Z , Lim KL , Dawson VL , Dawson TM ((2007) ) Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 16: , 223–232. |
[64] | Steger M , Tonelli F , Ito G , Davies P , Trost M , Vetter M , Wachter S , Lorentzen E , Duddy G , Wilson S , Baptista MA , Fiske BK , Fell MJ , Morrow JA , Reith AD , Alessi DR , Mann M ((2016) ) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5: , e12813. |
[65] | Sheng Z , Zhang S , Bustos D , Kleinheinz T , Le Pichon CE , Dominguez SL , Solanoy HO , Drummond J , Zhang X , Ding X , Cai F , Song Q , Li X , Yue Z , van der Brug MP , Burdick DJ , Gunzner-Toste J , Chen H , Liu X , Estrada AA , Sweeney ZK , Scearce-Levie K , Moffat JG , Kirkpatrick DS , Zhu H ((2012) ) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 4: , 164ra161. |
[66] | Steger M , Diez F , Dhekne HS , Lis P , Nirujogi RS , Karayel O , Tonelli F , Martinez TN , Lorentzen E , Pfeffer SR , Alessi DR , Mann M ((2017) ) Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6: , e31012. |
[67] | Maio RD , Hoffman EK , Rocha EM , Keeney MT , Sanders LH , Miranda BRD , Zharikov A , Laar AV , Stepan AF , Lanz TA , Kofler JK , Burton EA , Alessi DR , Hastings TG , Greenamyre JT ((2018) ) LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 10: , eaar5429. |
[68] | Rocha EM , Miranda BRD , Castro S , Drolet R , Hatcher NG , Yao L , Smith SM , Keeney MT , Maio RD , Kofler J , Hastings TG , Greenamyre JT ((2020) ) LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol Dis 134: , 104626. |
[69] | Abeliovich A , Gitler AD ((2016) ) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539: , 207–216. |
[70] | Wallings RL , Humble SW , Ward ME , Wade-Martins R ((2019) ) Lysosomal dysfunction at the centre of Parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci 42: , 899–912. |
[71] | Hu YB , Dammer EB , Ren RJ , Wang G ((2015) ) The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl Neurodegener 4: , 18. |
[72] | Winckler B , Faundez V , Maday S , Cai Q , Guimas Almeida C , Zhang H ((2018) ) The endolysosomal system and proteostasis: From development to degeneration. J Neurosci 38: , 9364–9374. |
[73] | Inpanathan S , Botelho RJ ((2019) ) The lysosome signaling platform: Adapting with the times. Front Cell Dev Biol 7: , 113. |
[74] | Higashi S , Moore DJ , Yamamoto R , Minegishi M , Sato K , Togo T , Katsuse O , Uchikado H , Furukawa Y , Hino H , Kosaka K , Emson PC , Wada K , Dawson VL , Dawson TM , Arai H , Iseki E ((2009) ) Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in Lewy body disease. J Neuropathol Exp Neurol 68: , 994–1005. |
[75] | Jeong GR , Jang EH , Bae JR , Jun S , Kang HC , Park CH , Shin JH , Yamamoto Y , Tanaka-Yamamoto K , Dawson VL , Dawson TM , Hur EM , Lee BD ((2018) ) Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol Neurodegener 13: , 8. |
[76] | Bonet-Ponce L , Cookson MR ((2019) ) The role of Rab GTPases in the pathobiology of Parkinson’ disease. Curr Opin Cell Biol 59: , 73–80. |
[77] | Zhen Y , Stenmark H ((2015) ) Cellular functions of Rab GTPases at a glance. J Cell Sci 128: , 3171–3176. |
[78] | Fujimoto T , Kuwahara T , Eguchi T , Sakurai M , Komori T , Iwatsubo T ((2018) ) Parkinson’s disease-associated mutant LRRK2 phosphorylates Rab7L1 and modifies trans-Golgi morphology. Biochem Biophys Res Commun 495: , 1708–1715. |
[79] | MacLeod DA , Rhinn H , Kuwahara T , Zolin A , Di Paolo G , McCabe BD , Marder KS , Honig LS , Clark LN , Small SA , Abeliovich A ((2013) ) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77: , 425–439. |
[80] | Eguchi T , Kuwahara T , Sakurai M , Komori T , Fujimoto T , Ito G , Yoshimura SI , Harada A , Fukuda M , Koike M , Iwatsubo T ((2018) ) LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A 115: , E9115–E9124. |
[81] | Liu Z , Bryant N , Kumaran R , Beilina A , Abeliovich A , Cookson MR , West AB ((2018) ) LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum Mol Genet 27: , 385–395. |
[82] | Purlyte E , Dhekne HS , Sarhan AR , Gomez R , Lis P , Wightman M , Martinez TN , Tonelli F , Pfeffer SR , Alessi DR ((2018) ) Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J 37: , 1–18. |
[83] | Madero-Perez J , Fernandez B , Lara Ordonez AJ , Fdez E , Lobbestael E , Baekelandt V , Hilfiker S ((2018) ) RAB7L1-mediated relocalization of LRRK2 to the Golgi complex causes centrosomal deficits via RAB8A. Front Mol Neurosci 11: , 417. |
[84] | Gomez RC , Wawro P , Lis P , Alessi DR , Pfeffer SR ((2019) ) Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J Cell Biol 218: , 4157–4170. |
[85] | Dhekne HS , Yanatori I , Gomez RC , Tonelli F , Diez F , Schule B , Steger M , Alessi DR , Pfeffer SR ((2018) ) A pathway for Parkinson’s disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. Elife 7: , e40202. |
[86] | Lara Ordonez AJ , Fernandez B , Fdez E , Romo-Lozano M , Madero-Perez J , Lobbestael E , Baekelandt V , Aiastui A , Lopez de Munain A , Melrose HL , Civiero L , Hilfiker S ((2019) ) RAB8, RAB10 and RILPL1 contribute to both LRRK2 kinase-mediated centrosomal cohesion and ciliogenesis deficits. Hum Mol Genet 28: , 3552–3568. |
[87] | Madero-Perez J , Fdez E , Fernandez B , Lara Ordonez AJ , Blanca Ramirez M , Gomez-Suaga P , Waschbusch D , Lobbestael E , Baekelandt V , Nairn AC , Ruiz-Martinez J , Aiastui A , Lopez de Munain A , Lis P , Comptdaer T , Taymans JM , Chartier-Harlin MC , Beilina A , Gonnelli A , Cookson MR , Greggio E , Hilfiker S ((2018) ) Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol Neurodegener 13: , 3. |
[88] | Tong Y , Yamaguchi H , Giaime E , Boyle S , Kopan R , Kelleher RJ 3rd , Shen J ((2010) ) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A 107: , 9879–9884. |
[89] | Baptista MA , Dave KD , Frasier MA , Sherer TB , Greeley M , Beck MJ , Varsho JS , Parker GA , Moore C , Churchill MJ , Meshul CK , Fiske BK ((2013) ) Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One 8: , e80705. |
[90] | Herzig MC , Kolly C , Persohn E , Theil D , Schweizer T , Hafner T , Stemmelen C , Troxler TJ , Schmid P , Danner S , Schnell CR , Mueller M , Kinzel B , Grevot A , Bolognani F , Stirn M , Kuhn RR , Kaupmann K , van der Putten PH , Rovelli G , Shimshek DR ((2011) ) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet 20: , 4209–4223. |
[91] | Kuwahara T , Inoue K , D’Agati VD , Fujimoto T , Eguchi T , Saha S , Wolozin B , Iwatsubo T , Abeliovich A ((2016) ) LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci Rep 6: , 29945. |
[92] | Hinkle KM , Yue M , Behrouz B , Dächsel JC , Lincoln SJ , Bowles EE , Beevers JE , Dugger B , Winner B , Prots I , Kent CB , Nishioka K , Lin W-L , Dickson DW , Janus CJ , Farrer MJ , Melrose HL ((2012) ) LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol Neurodegener 7: , 25. |
[93] | Tong Y , Giaime E , Yamaguchi H , Ichimura T , Liu Y , Si H , Cai H , Bonventre JV , Shen J ((2012) ) Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener 7: , 2. |
[94] | Beilina A , Bonet-Ponce L , Kumaran R , Kordich JJ , Ishida M , Mamais A , Kaganovich A , Saez-Atienzar S , Gershlick DC , Roosen DA , Pellegrini L , Malkov V , Fell MJ , Harvey K , Bonifacino JS , Moore DJ , Cookson MR ((2020) ) The Parkinson’s disease protein LRRK2 interacts with the GARP complex to promote retrograde transport to the trans-Golgi network. Cell Rep 31: , 107614. |
[95] | Giaime E , Tong Y , Wagner LK , Yuan Y , Huang G , Shen J ((2017) ) Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron 96: , 796–807. |
[96] | Henry AG , Aghamohammadzadeh S , Samaroo H , Chen Y , Mou K , Needle E , Hirst WD ((2015) ) Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet 24: , 6013–6028. |
[97] | Wallings R , Connor-Robson N , Wade-Martins R ((2019) ) LRRK2 interacts with the vacuolar-type}+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet 28: , 2696–2710. |
[98] | Cheung G , Cousin MA ((2012) ) Adaptor protein complexes 1 and 3 are essential for generation of synaptic vesicles from activity-dependent bulk endosomes. J Neurosci 32: , 6014–6023. |
[99] | Park SY , Guo X ((2014) ) Adaptor protein complexes and intracellular transport. Biosci Rep 34: , e00123. |
[100] | Matta S , Van Kolen K , da Cunha R , van den Bogaart G , Mandemakers W , Miskiewicz K , De Bock PJ , Morais VA , Vilain S , Haddad D , Delbroek L , Swerts J , Chavez-Gutierrez L , Esposito G , Daneels G , Karran E , Holt M , Gevaert K , Moechars DW , De Strooper B , Verstreken P ((2012) ) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75: , 1008–1021. |
[101] | Pan PY , Li X , Wang J , Powell J , Wang Q , Zhang Y , Chen Z , Wicinski B , Hof P , Ryan TA , Yue Z ((2017) ) Parkinson’s disease-associated LRRK2 hyperactive kinase mutant disrupts synaptic vesicle trafficking in ventral midbrain neurons. J Neurosci 37: , 11366–11376. |
[102] | Shin N , Jeong H , Kwon J , Heo HY , Kwon JJ , Yun HJ , Kim CH , Han BS , Tong Y , Shen J , Hatano T , Hattori N , Kim KS , Chang S , Seol W ((2008) ) LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res 314: , 2055–2065. |
[103] | Maas JW , Yang J , Edwards RH ((2017) ) Endogenous leucine-rich repeat kinase 2 slows synaptic vesicle recycling in striatal neurons. Front Synaptic Neurosci 9: , 5. |
[104] | Piccoli G , Condliffe SB , Bauer M , Giesert F , Boldt K , De Astis S , Meixner A , Sarioglu H , Vogt-Weisenhorn DM , Wurst W , Gloeckner CJ , Matteoli M , Sala C , Ueffing M ((2011) ) LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci 31: , 2225–2237. |
[105] | Hockey LN , Kilpatrick BS , Eden ER , Lin-Moshier Y , Brailoiu GC , Brailoiu E , Futter CE , Schapira AH , Marchant JS , Patel S ((2015) ) Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci 128: , 232–238. |
[106] | Ho PW , Leung CT , Liu H , Pang SY , Lam CS , Xian J , Li L , Kung MH , Ramsden DB , Ho SL ((2020) ) Age-dependent accumulation of oligomeric SNCA/alpha-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 16: , 347–370. |
[107] | Schapansky J , Khasnavis S , DeAndrade MP , Nardozzi JD , Falkson SR , Boyd JD , Sanderson JB , Bartels T , Melrose HL , LaVoie MJ ((2018) ) Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol Dis 111: , 26–35. |
[108] | Herbst S , Campbell P , Harvey J , Bernard EM , Papayannopoulos V , Wood NW , Morris HR , Gutierrez MG ((2020) ) LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J, doi: 10.15252/embj.2020104494 |
[109] | Lee H , Flynn R , Sharma I , Haberman E , Carling PJ , Nicholls FJ , Stegmann M , Vowles J , Haenseler W , Wade-Martins R , James WS , Cowley SA ((2020) ) LRRK2 is recruited to phagosomes and co-recruits RAB8 and RAB10 in human pluripotent stem cell-derived macrophages. Stem Cell Rep 14: , 940–955. |
[110] | Nguyen M , Krainc D ((2018) ) LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A 115: , 5576–5581. |
[111] | Ysselstein D , Nguyen M , Young TJ , Severino A , Schwake M , Merchant K , Krainc D ((2019) ) LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun 10: , 5570. |
[112] | Sanyal A , DeAndrade MP , Novis HS , Lin S , Chang J , Lengacher N , Tomlinson JJ , Tansey MG , LaVoie MJ ((2020) ) Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord 35: , 760–773. |
[113] | Sanyal A , Novis HS , Gasser E , Lin S , LaVoie MJ ((2020) ) LRRK2 kinase inhibition rescues deficits in lysosome function due to heterozygous GBA1 expression in human iPSC-derived neurons. Front Neurosci 14: , 442. |
[114] | Yim YI , Sun T , Wu LG , Raimondi A , De Camilli P , Eisenberg E , Greene LE ((2010) ) Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc Natl Acad Sci U S A 107: , 4412–4417. |
[115] | Stafa K , Tsika E , Moser R , Musso A , Glauser L , Jones A , Biskup S , Xiong Y , Bandopadhyay R , Dawson VL , Dawson TM , Moore DJ ((2014) ) Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet 23: , 2055–2077. |
[116] | Islam MS , Nolte H , Jacob W , Ziegler AB , Putz S , Grosjean Y , Szczepanowska K , Trifunovic A , Braun T , Heumann H , Heumann R , Hovemann B , Moore DJ , Kruger M ((2016) ) Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet 25: , 5365–5382. |
[117] | Soukup SF , Kuenen S , Vanhauwaert R , Manetsberger J , Hernandez-Diaz S , Swerts J , Schoovaerts N , Vilain S , Gounko NV , Vints K , Geens A , De Strooper B , Verstreken P ((2016) ) A LRRK2-Dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92: , 829–844. |
[118] | Ferguson SM , De Camilli P ((2012) ) Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 13: , 75–88. |
[119] | Praefcke GJ , McMahon HT ((2004) ) The dynamin superfamily: Universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 5: , 133–147. |
[120] | Gad H , Ringstad N , Low P , Kjaerulff O , Gustafsson J , Wenk M , Paolo GD , Nemoto Y , Crum J , Ellisman MH , Camilli PD , Shupliakov O , Brodin L ((2000) ) Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of Endophilin. Neuron 27: , 301–312. |
[121] | Quadri M , Fang M , Picillo M , Olgiati S , Breedveld GJ , Graafland J , Wu B , Xu F , Erro R , Amboni M , Pappatà S , Quarantelli M , Annesi G , Quattrone A , Chien HF , Barbosa ER , Oostra BA , Barone P , Wang J , Bonifati V ((2013) ) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34: , 1208–1215. |
[122] | Olgiati S , Quadri M , Fang M , Rood JP , Saute JA , Chien HF , Bouwkamp CG , Graafland J , Minneboo M , Breedveld GJ , Zhang J , Verheijen FW , Boon AJ , Kievit AJ , Jardim LB , Mandemakers W , Barbosa ER , Rieder CR , Leenders KL , Wang J , Bonifati V ((2016) ) DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann Neurol 79: , 244–256. |
[123] | Edvardson S , Cinnamon Y , Ta-Shma A , Shaag A , Yim YI , Zenvirt S , Jalas C , Lesage S , Brice A , Taraboulos A , Kaestner KH , Greene LE , Elpeleg O ((2012) ) A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7: , e36458. |
[124] | Krebs CE , Karkheiran S , Powell JC , Cao M , Makarov V , Darvish H , Di Paolo G , Walker RH , Shahidi GA , Buxbaum JD , De Camilli P , Yue Z , Paisán-Ruiz C ((2013) ) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34: , 1200–1207. |
[125] | Stafa K , Trancikova A , Webber PJ , Glauser L , West AB , Moore DJ ((2012) ) GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet 8: , e1002526. |
[126] | Beilina A , Rudenko IN , Kaganovich A , Civiero L , Chau H , Kalia SK , Kalia LV , Lobbestael E , Chia R , Ndukwe K , Ding J , Nalls MA , Consortium IPDG , Consortium NABE , Olszewski M , Hausera DN , Kumaran R , Lozano AM , Baekelandt V , Greene LE , Taymans J-M , Greggio E , Cookson MR ((2014) ) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A 111: , 2626–2631. |
[127] | Lanning NJ , VanOpstall C , Goodall ML , MacKeigan JP , Looyenga BD ((2018) ) LRRK2 deficiency impairs trans-Golgi to lysosome trafficking and endocytic cargo degradation in human renal proximal tubule epithelial cells. Am J Physiol Renal Physiol 315: , F1465–F1477. |
[128] | Wang S , Ma Z , Xu X , Wang Z , Sun L , Zhou Y , Lin X , Hong W , Wang T ((2014) ) A role of Rab29 in the integrity of the trans-Golgi network and retrograde trafficking of mannose-6-phosphate receptor. PLoS One 9: , e96242. |
[129] | D’Souza-Schorey C , Chavrier P ((2006) ) ARF proteins: Roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7: , 347–358. |
[130] | Donaldson JG , Jackson CL ((2011) ) ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat Rev Mol Cell Biol 12: , 362–375. |
[131] | Xiong Y , Coombes CE , Kilaru A , Li X , Gitler AD , Bowers WJ , Dawson VL , Dawson TM , Moore DJ ((2010) ) GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet 6: , e1000902. |
[132] | Xiong Y , Yuan C , Chen R , Dawson TM , Dawson VL ((2012) ) ArfGAP1 is a GTPase activating protein for LRRK2: Reciprocal regulation of ArfGAP1 by LRRK2. J Neurosci 32: , 3877–3886. |
[133] | Zhao C , Slevin JT , Whiteheart SW ((2007) ) Cellular functions of NSF: Not just SNAPs and SNAREs. FEBS Lett 581: , 2140–2149. |
[134] | Block MR , Glick BS , Wilcox CA , Wieland FT , Rothman JE ((1988) ) Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A 85: , 7852–7856. |
[135] | Rabouille C , Kondo H , Newman R , Hui N , Freemont P , Warren G ((1998) ) Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92: , 603–610. |
[136] | Beckers CJ , Block MR , Glick BS , Rothman JE , Balch WE ((1989) ) Vesicular transport between the endoplasmic reticulum and the Golgi stack requires the NEM-sensitive fusion protein. Nature 339: , 397–398. |
[137] | Belluzzi E , Gonnelli A , Cirnaru MD , Marte A , Plotegher N , Russo I , Civiero L , Cogo S , Carrion MP , Franchin C , Arrigoni G , Beltramini M , Bubacco L , Onofri F , Piccoli G , Greggio E ((2016) ) LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol Neurodegener 11: , 1. |
[138] | Cho HJ , Yu J , Xie C , Rudrabhatla P , Chen X , Wu J , Parisiadou L , Liu G , Sun L , Ma B , Ding J , Liu Z , Cai H ((2014) ) Leucine-rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER-Golgi export. EMBO J 33: , 2314–2331. |
[139] | Budnik A , Stephens DJ ((2009) ) ER exit sites–localization and control of COPII vesicle formation. FEBS Lett 583: , 3796–3803. |
[140] | Zhang H , Huang T , Hong Y , Yang W , Zhang X , Luo H , Xu H , Wang X ((2018) ) The retromer complex and sorting nexins in neurodegenerative diseases. Front Aging Neurosci 10: , 79. |
[141] | Burd C , Cullen PJ ((2014) ) Retromer: A master conductor of endosome sorting. Cold Spring Harb Perspect Biol 6: , a016774. |
[142] | Seaman MNJ ((2012) ) The retromer complex –endosomal protein recycling and beyond. J Cell Sci 125: , 4693–4702. |
[143] | Zimprich A , Benet-Pages A , Struhal W , Graf E , Eck SH , Offman MN , Haubenberger D , Spielberger S , Schulte EC , Lichtner P , Rossle SC , Klopp N , Wolf E , Seppi K , Pirker W , Presslauer S , Mollenhauer B , Katzenschlager R , Foki T , Hotzy C , Reinthaler E , Harutyunyan A , Kralovics R , Peters A , Zimprich F , Brucke T , Poewe W , Auff E , Trenkwalder C , Rost B , Ransmayr G , Winkelmann J , Meitinger T , Strom TM ((2011) ) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89: , 168–175. |
[144] | Vilarino-Guell C , Wider C , Ross OA , Dachsel JC , Kachergus JM , Lincoln SJ , Soto-Ortolaza AI , Cobb SA , Wilhoite GJ , Bacon JA , Behrouz B , Melrose HL , Hentati E , Puschmann A , Evans DM , Conibear E , Wasserman WW , Aasly JO , Burkhard PR , Djaldetti R , Ghika J , Hentati F , Krygowska-Wajs A , Lynch T , Melamed E , Rajput A , Rajput AH , Solida A , Wu RM , Uitti RJ , Wszolek ZK , Vingerhoets F , Farrer MJ ((2011) ) VPS35 mutations in Parkinson disease. Am J Hum Genet 89: , 162–167. |
[145] | Williams ET , Chen X , Moore DJ ((2017) ) VPS35, the retromer complex and Parkinson’s disease. J Parkinsons Dis 7: , 219–233. |
[146] | Chen X , Kordich JK , Williams ET , Levine N , Cole-Strauss A , Marshall L , Labrie V , Ma J , Lipton JW , Moore DJ ((2019) ) Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc Natl Acad Sci U S A 116: , 5765–5774. |
[147] | Mir R , Tonelli F , Lis P , Macartney T , Polinski NK , Martinez TN , Chou MY , Howden AJM , Konig T , Hotzy C , Milenkovic I , Brucke T , Zimprich A , Sammler E , Alessi DR ((2018) ) The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J 475: , 1861–1883. |
[148] | Nguyen APT , Tsika E , Kelly K , Levine N , Chen X , West AB , Boularand S , Barneoud P , Moore DJ ((2020) ) Dopaminergic neurodegeneration induced by Parkinson’s disease-linked G2019S LRRK2 is dependent on kinase and GTPase activity. Proc Natl Acad Sci U S A 117: , 17296–17307. |
[149] | Waschbusch D , Hubel N , Ossendorf E , Lobbestael E , Baekelandt V , Lindsay AJ , McCaffrey MW , Khan AR , Barnekow A ((2019) ) Rab32 interacts with SNX6 and affects retromer-dependent Golgi trafficking. PLoS One 14: , e0208889. |
[150] | Waschbusch D , Michels H , Strassheim S , Ossendorf E , Kessler D , Gloeckner CJ , Barnekow A ((2014) ) LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS One 9: , e111632. |
[151] | Albanese F , Novello S , Morari M ((2019) ) Autophagy and LRRK2 in the aging brain. Front Neurosci 13: , 1352. |
[152] | Cherra SJ , Steer E , Gusdon AM , Kiselyov K , Chu CT ((2013) ) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182: , 474–484. |
[153] | Ho DH , Kim H , Nam D , Sim H , Kim J , Kim HG , Son I , Seol W ((2018) ) LRRK2 impairs autophagy by mediating phosphorylation of leucyl-tRNA synthetase. Cell Biochem Funct 36: , 431–442. |
[154] | Mamais A , Manzoni C , Nazish I , Arber C , Sonustun B , Wray S , Warner TT , Cookson MR , Lewis PA , Bandopadhyay R ((2018) ) Analysis of macroautophagy related proteins in G2019S LRRK2 Parkinson’s disease brains with Lewy body pathology. Brain Res 1701: , 75–84. |
[155] | Plowey ED , Cherra SJ 3rd , Liu YJ , Chu CT ((2008) ) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105: , 1048–1056. |
[156] | Verma M , Callio J , Otero PA , Sekler I , Wills ZP , Chu CT ((2017) ) Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J Neurosci 37: , 11151–11165. |
[157] | Wauters F , Cornelissen T , Imberechts D , Martin S , Koentjoro B , Sue C , Vangheluwe P , Vandenberghe W ((2020) ) LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16: , 203–222. |
[158] | Orenstein SJ , Kuo SH , Tasset I , Arias E , Koga H , Fernandez-Carasa I , Cortes E , Honig LS , Dauer W , Consiglio A , Raya A , Sulzer D , Cuervo AM ((2013) ) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16: , 394–406. |
[159] | Ramonet D , Daher JP , Lin BM , Stafa K , Kim J , Banerjee R , Westerlund M , Pletnikova O , Glauser L , Yang L , Liu Y , Swing DA , Beal MF , Troncoso JC , McCaffery JM , Jenkins NA , Copeland NG , Galter D , Thomas B , Lee MK , Dawson TM , Dawson VL , Moore DJ ((2011) ) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6: , e18568. |
[160] | Liu HF , Lu S , Ho PW , Tse HM , Pang SY , Kung MH , Ho JW , Ramsden DB , Zhou ZJ , Ho SL ((2014) ) LRRK2 R1441G mice are more liable to dopamine depletion and locomotor inactivity. Ann Clin Transl Neurol 1: , 199–208. |
[161] | Tsika E , Kannan M , Foo CS , Dikeman D , Glauser L , Gellhaar S , Galter D , Knott GW , Dawson TM , Dawson VL , Moore DJ ((2014) ) Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol Dis 71: , 345–358. |
[162] | Yue M , Hinkle KM , Davies P , Trushina E , Fiesel FC , Christenson TA , Schroeder AS , Zhang L , Bowles E , Behrouz B , Lincoln SJ , Beevers JE , Milnerwood AJ , Kurti A , McLean PJ , Fryer JD , Springer W , Dickson DW , Farrer MJ , Melrose HL ((2015) ) Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis 78: , 172–195. |
[163] | Cunningham LA , Moore DJ ((2020) ) Endosomal sorting pathways in the pathogenesis of Parkinson’s disease. Prog Brain Res 252: , 271–306. |
[164] | Taylor M , Alessi DR ((2020) ) Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr Opin Cell Biol 63: , 102–113. |
[165] | Schluter OM , Schmitz F , Jahn R , Rosenmund C , Sudhof TC ((2004) ) A complete genetic analysis of neuronal Rab3 function. J Neurosci 24: , 6629–6637. |
[166] | Dodson MW , Zhang T , Jiang C , Chen S , Guo M ((2012) ) Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet 21: , 1350–1363. |
[167] | Yun HJ , Kim H , Ga I , Oh H , Ho DH , Kim J , Seo H , Son I , Seol W ((2015) ) An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J Biochem 157: , 485–495. |
[168] | Rivero-Rios P , Romo-Lozano M , Madero-Perez J , Thomas AP , Biosa A , Greggio E , Hilfiker S ((2019) ) The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J Biol Chem 294: , 4738–4758. |
[169] | Dodson MW , Leung LK , Lone M , Lizzio MA , Guo M ((2014) ) Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis Model Mech 7: , 1351–1363. |
[170] | Chua CE , Lim YS , Tang BL ((2010) ) Rab35–a vesicular traffic-regulating small GTPase with actin modulating roles. FEBS Lett 584: , 1–6. |
[171] | Li C , Wei Z , Fan Y , Huang W , Su Y , Li H , Dong Z , Fukuda M , Khater M , Wu G ((2017) ) The GTPase Rab43 controls the anterograde ER-Golgi trafficking and sorting of GPCRs. Cell Rep 21: , 1089–1101. |

