Mechanisms of Aβ Clearance and Degradation by Glial Cells
 Miriam Ries
Miriam Ries Magdalena Sastre
Magdalena Sastre- Division of Brain Sciences, Imperial College London, Hammersmith Hospital, London, UK
Glial cells have a variety of functions in the brain, ranging from immune defense against external and endogenous hazardous stimuli, regulation of synaptic formation, calcium homeostasis, and metabolic support for neurons. Their dysregulation can contribute to the development of neurodegenerative disorders, including Alzheimer’s disease (AD). One of the most important functions of glial cells in AD is the regulation of Amyloid-β (Aβ) levels in the brain. Microglia and astrocytes have been reported to play a central role as moderators of Aβ clearance and degradation. The mechanisms of Aβ degradation by glial cells include the production of proteases, including neprilysin, the insulin degrading enzyme, and the endothelin-converting enzymes, able to hydrolyse Aβ at different cleavage sites. Besides these enzymes, other proteases have been described to have some role in Aβ elimination, such as plasminogen activators, angiotensin-converting enzyme, and matrix metalloproteinases. Other relevant mediators that are released by glial cells are extracellular chaperones, involved in the clearance of Aβ alone or in association with receptors/transporters that facilitate their exit to the blood circulation. These include apolipoproteins, α2macroglobulin, and α1-antichymotrypsin. Finally, astrocytes and microglia have an essential role in phagocytosing Aβ, in many cases via a number of receptors that are expressed on their surface. In this review, we examine all of these mechanisms, providing an update on the latest research in this field.
Introduction
The pathogenesis of Alzheimer’s disease (AD) has been associated with the presence of extracellular amyloid-β peptide (Aβ) aggregates, forming neuritic plaques, as well as intra-neuronal Aβ (Gouras et al., 2005; Blair et al., 2014), probably due to alterations in the mechanisms of generation and/or clearance of amyloid in the brain during aging. There is evidence of an increase in the expression of β-APP cleaving enzyme-1 (BACE1), the enzyme responsible for the cleavage of the amyloid precursor protein (APP) in the amyloidogenic pathway, in sporadic AD cases (Holsinger et al., 2002; Yang et al., 2003). In addition, the dysregulation of the systems involved in the clearance and degradation of Aβ has generated a lot of interest in the last decade, particularly their effect on the accumulation of Aβ in the blood vessel walls, leading to cerebral amyloid angiopathy (Love, 2004).
A great number of reviews have examined the main mechanisms of Aβ clearance and degradation (Bates et al., 2009; Saido and Leissring, 2012; Yoon and Jo, 2012; Tarasoff-Conway et al., 2015). These include Aβ proteases, which are enzymes that degrade or cleave Aβ into smaller fragments. Other proteins with a crucial role in Aβ clearance are apolipoprotein E (ApoE) and α2-macroglobulin (α2-M); they interact with transporters including low-density lipoprotein receptor-related protein 1 (LRP1) receptors, very low-density lipoprotein receptor (VLDLR), and P-glycoprotein, localized in astrocytes and on the abluminal side of the cerebral endothelium, where they facilitate the transport of Aβ across the blood brain barrier (BBB) into the blood circulation (Deane et al., 2009). Genetic mutations resulting in loss of function of those proteins have demonstrated their importance in disease progression, particularly in late onset AD, although there are indications of alterations of some of the Aβ proteases by environmental factors (Table 1). Experimental data have suggested that Aβ and solutes can normally also be cleared along the lymphatic drainage pathways within the basement membranes of capillaries and arteries (Carare et al., 2008; Tarasoff-Conway et al., 2015). Lastly, another way to eliminate Aβ from the brain is by the uptake and phagocytosis of Aβ by cells such as microglia, astrocytes, and macrophages.

TABLE 1. Genetic vs environmental factors affecting Aβ clearance.
Many of the mechanisms mentioned above are mediated by glial cells. Here, we review how glial cells are mediators of Aβ removal from the brain (schematic Figure 1).
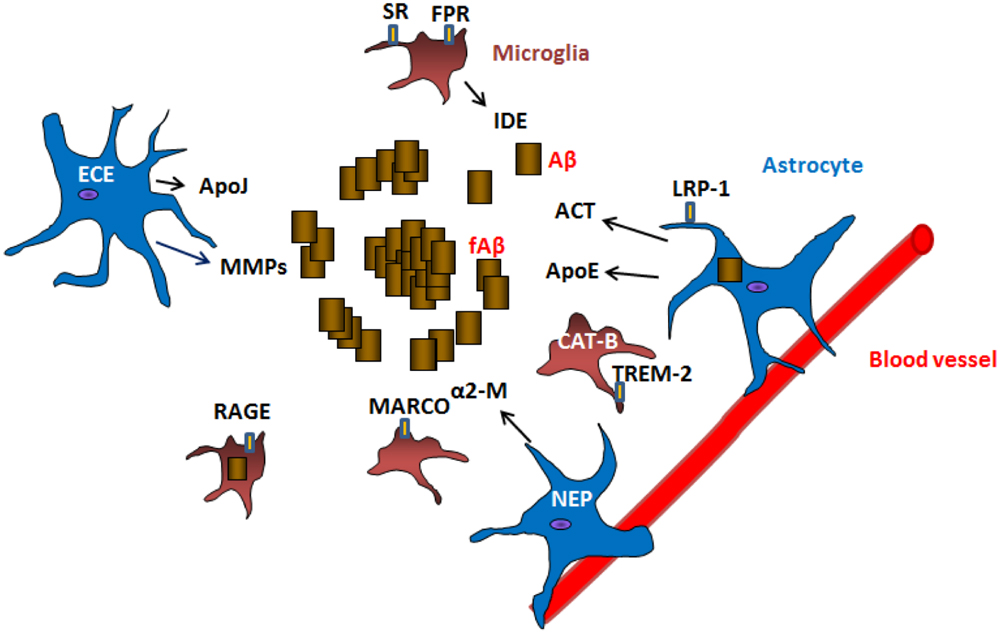
FIGURE 1. Mechanisms of Aβ clearance by glial cells. Astrocytes and microglia can produce Aβ degrading proteases neprilysin (NEP), endothelin-converting enzyme (ECE), insulin degrading enzyme (IDE), matrix metalloproteases (MMPs), cathepsin B (CAT-B) as well as chaperones apolipoprotein E (ApoE), apolipoprotein J (ApoJ), α-2 macroglobulin (α2-M), α1-antichymotrypsin (ACT) involved in the clearance of Aβ. Receptors located in the surface of glial cells such as lipoprotein receptor-related protein 1 (LRP-1), scavenger receptors (SR), formyl peptide receptors (FPR), macrophage receptor with collagenous structure (MARCO), receptor for advanced glycation end products (RAGE) and triggering receptor expressed on myeloid cells 2 (TREM-2) are involved in the uptake and clearance of Aβ (receptor mediated endocytosis). Astrocytes are also connected to blood vessels, where they are implicated in the draining of Aβ and other products out of the brain.
Aβ Degrading Proteases
Many of the proteins involved in the enzymatic degradation of Aβ are produced by glial cells. These proteases cleave at different sites within the Aβ sequence, resulting in different enzymatic products (Nalivaeva et al., 2012). These include:
Metalloendopeptidases
Neprilysin (NEP), the insulin degrading enzyme (IDE) and endothelin-converting enzymes-1 and -2 (ECE1 and ECE2) (Eckman et al., 2001, 2003) are metalloendopeptidases particularly involved in the degradation of monomeric Aβ species (although neprilysin can also hydrolyse oligomeric forms). ECEs are expressed in neurons, endothelial cells (Naidoo et al., 2004), and astrocytes (Nakagomi et al., 2000) and primarily degrade Aβ intracellularly (Eckman et al., 2003). NEP is mainly expressed in pre-synaptic terminals of neurons (Fukami et al., 2002) but can also be found in activated astrocytes (Fisk et al., 2007; Yamamoto et al., 2014) and microglia (Hickman et al., 2008). In the brain, IDE is synthesized and secreted by neurons, oligodendrocytes, and microglia (Bernstein et al., 2008), where it is released via exosomes (Tamboli et al., 2010) especially acting on extracellular Aβ deposits. IDE is unequally expressed in human brain, majorly found in hypothalamic neurons and in hippocampus, cerebellum, and brain stem (Bernstein et al., 2008), coinciding with the location of insulin receptors in the brain.
The relevance of NEP and IDE has been proven in knockout models, whereby mice lacking those enzymes crossed with APP transgenic models show increased Aβ deposition (Iwata et al., 2001; Farris et al., 2003) in the brain. Conversely, overexpression of these proteases has been shown to reduce Aβ load (Leissring et al., 2003).
The expression of NEP and IDE in glial cells has been found to change depending on the stage of the disease. In vitro and in vivo studies have shown that IDE and ECE are up-regulated in response to increasing levels of brain Aβ (Zhao et al., 2007), while NEP has been found reduced in AD brains (Wang et al., 2010).
Plasminogen Activators and ACE
Other enzymes also involved in Aβ degradation, but whose relevance depends on the degree of Aβ pathology, are plasminogen activators and angiotensin-converting enzyme (ACE) (Hu et al., 2001), which are more effective for aggregated Aβ rather than regulating steady-state Aβ levels (Saido and Leissring, 2012). While ACE is mainly neuronal, tissue plasminogen activator (tPA) is synthesized by neurons and microglial cells (Melchor and Strickland, 2005).
Matrix Metalloproteinases (MMPs)
Matrix metalloproteinases, known to be expressed and secreted by astrocytes, play a role in the extracellular degradation of both monomeric and fibrillar forms of Aβ (Yan et al., 2006). Astrocytes surrounding amyloid plaques show enhanced expression of MMP-2 and MMP-9 in aged APP/presenilin 1 mice. Moreover, astrocyte-conditioned medium (ACM) degraded Aβ, lowering its levels and producing several fragments after incubation with synthetic human Aβ(1–40) and Aβ(1–42). This activity was attenuated with specific inhibitors of MMP-2 and -9, as well as in ACM derived from mmp-2 or -9 knockout (KO) mice (Yin et al., 2006).
Lysosomal Peptidases
It has been described that after being phagocytosed by microglia, Aβ can be degraded by cathepsin B (CAT-B) (Nakanishi, 2003; Halle et al., 2008). This enzyme is able to reduce longer forms of Aβ into shorter less toxic species, such as Aβ38. In addition, ECE-2 can be found expressed in endosomes/lysosomes where it can mediate the degradation of Aβ (Pacheco-Quinto and Eckman, 2013).
Proteasomal Degradation
The ubiquitin proteasome system (UPS) is a mechanism of protein degradation which selectively targets individual proteins, including short-lived, damaged, or defectively folded proteins (Rock et al., 1994; Lilienbaum, 2013). Before a protein is degraded by the proteasome, the ubiquitin (Ub) system selects the protein target by the conjugation of Ub carried out by the serial activity of several enzymes (E1–E3) along the pathway. It has been shown that the proteasomal degradation machinery is capable of cleaving Aβ(1–42) peptides in a dose-dependent manner, without significantly affecting the overall catalytic function of the proteasome (Zhao and Yang, 2010). In addition, it was shown that E2 conjugating enzymes, E3 ligases, and de-ubiquitinating enzymes play a pivotal role in the proteasomal degradation of Aβ (Hong et al., 2014). Interestingly, it seems that the UPS system in glial cells is more efficient at degrading aggregated proteins compared with neurons (Jansen et al., 2014), and this could explain why they do not contain protein aggregates. However, in neurodegenerative diseases the UPS system in glia could become dysfunctional and contribute to the progression of the disease.
Autophagic Degradation
The autophagy pathway is critical for the turnover of cell organelles and degradation of aggregated proteins in cells under stress. It was hypothesized that a defective clearance of Aβ-generating autophagic vacuoles creates conditions favorable for Aβ accumulation in AD and this is supported by data indicating that increasing autophagy by rapamycin reduces amyloid burden in vivo (Nixon, 2007). The role of autophagy in the degradation of Aβ has not been investigated in glial cells until recently. Cho and colleagues reported the importance of autophagy in the clearance of extracellular Aβ fibrils by microglia and in the regulation of the Aβ-induced NLRP3 (NLR Family, Pyrin Domain Containing 3) inflammasome using microglia from atg7 knockout mice and in vitro cultures (Cho et al., 2014). Interestingly, microglia isolated from human AD brains show significantly reduced beclin 1 and retromer protein levels (Lucin et al., 2013). In addition, astrocytes from transgenic AD models showed strong expression of microtubule-associated protein light chain 3 (LC3), and autophagy seems to be involved in Aβ internalization by those cells (Pomilio et al., 2016), providing a link between autophagy and phagocytosis.
Aβ Clearance by Extracellular Chaperones
These include proteins that bind Aβ in plasma and cerebrospinal fluid (CSF), such as albumin, α2M, α1-antichymotrypsin (ACT), serum amyloid P component (SAP), complement proteins, transthyretin, apoferritin, apolipoproteins, and lipoproteins (Bates et al., 2009), which are essential because they modulate the formation of Aβ fibrils and mediate the interaction of Aβ with LRP-1 receptors in astrocytes.
Apolipoprotein E (ApoE)
It is the major risk factor for late onset AD. It is produced primarily by astrocytes in the brain and has been implicated in the degradation of Aβ in these cells (Koistinaho et al., 2004), although it can be produced by microglia and neurons in response to stimuli from glial cells (Saura et al., 2003; Harris et al., 2004). It has been shown that ApoE binds Aβ, and this association is more efficient with the ApoE2 and E3 isoforms than with ApoE4; these complexes are thought to influence both seeding of fibrillar Aβ and transport of soluble Aβ (Wildsmith et al., 2013). However, some recent controversial reports indicate that ApoE does not bind Aβ but competes with Aβ for binding to LRP-1 in astrocytes, and this could impact Aβ clearance by glia and across the blood–brain barrier (BBB) (Verghese et al., 2013). However, it is clear that ApoE has a role on Aβ clearance because bexarotene, which enhances ApoE expression, clears Aβ and improves cognition in mice (Cramer et al., 2012).
Apolipoprotein J (ApoJ or Clusterin)
Apolipoprotein J has been shown to interact with Aβ and alters its ability to form fibrils as well as modifying Aβ-mediated neurotoxicity. Similarly to ApoE, ApoJ is also produced in astrocytes. ApoJ is known to facilitate the transport of Aβ and hence the clearance across the BBB through the megalin/LRP-2 receptor. Upon exposure to Aβ combined with ApoE, ApoJ, ACT and a combination of SAP and complement C1q, a clear reduction in astrocytic but not microglial oligomeric Aβ uptake was observed (Mulder et al., 2014).
α1-Antichymotrypsin (ACT)
α1-Antichymotrypsin is a serine protease inhibitor that has been reported to bind Aβ and is overexpressed in the brain of AD patients (Abraham, 2001). ACT has been shown to be produced in astrocytes (Abraham et al., 1989) and its expression is regulated by proinflammatory cytokines including interleukin (IL)-1, oncostatin M (OSM), and complexes of IL-6, soluble IL-6 receptors and transcriptional regulators such as nuclear factor 1-X and activator protein 1 (AP-1; Gopalan et al., 2006a,b).
α2-Macroglobulin (α2-M)
α2-Macroglobulin is a matrix metalloproteinase inhibitor that is also released by glial cells, in particular perivascular astrocytes (Cucullo et al., 2003). Microinjection of clusterin and α2-M into the hippocampus of rat brains were found to prevent Aβ42-induced learning and memory impairments and reduce Aβ42-induced glial inflammation and neuronal degeneration (Cascella et al., 2013), suppressing oligomer cytotoxicity. α2-M can act as a ligand for LRP-1 (Kanekiyo et al., 2014), promoting increased neurite outgrowth in primary sensory neurons (Yamauchi et al., 2013).
Aβ Internalisation
An important mechanism of Aβ clearance from the brain is its uptake by glial cells. Aβ can be internalized by microglia, astrocytes, and other immune cells such as macrophages.
Pinocytosis
Soluble Aβ can be cleared by microglia through fluid phase pinocytosis (Mandrekar et al., 2009), with spontaneous formation and internalization of pinosomes from membrane ruffles. Furthermore, soluble Aβ(1–42) is able to induce its pinocytic self-uptake by stimulating the P2Y4 receptor and the PI3kinase/Akt cascade through autocrine ATP signaling on microglia (Li et al., 2013).
Phagocytosis
It is widely accepted that microglia phagocytose fibrillar Aβ, particularly more vigorously when bound by the C3b complement system (Lee and Landreth, 2010). Interestingly, the induction of microglial phagocytosis by fibrillar Aβ is attenuated by oligomeric Aβ (Pan et al., 2011). Astrocytes can also endocytose monomeric and oligomeric Aβ through actin regulation (Lee et al., 2015). In addition, there is evidence that astrocytes are able to phagocytose neurons containing Aβ (Nagele et al., 2003).
Receptor-Mediated Endocytosis
Oligomeric and fibrillar Aβ are primarily internalized though receptor-mediated endocytosis, via a number of receptors that are expressed on the surface of microglia and astrocytes:
Scavenger Receptors (SR)
Scavenger receptor type-A (SR-A), type B1 (SR-B1), CD36 and CD40 are able to bind and mediate the endocytosis of oligomeric and fibrillar Aβ (Paresce et al., 1996; Coraci et al., 2002; Husemann et al., 2002; El Khoury et al., 2003; Yang et al., 2011). SR-As can act in conjunction with other receptors, such as complement receptor 3 (also known as Mac-1/CD11b) to promote the uptake of fibrillar Aβ in microglia (Fu et al., 2012). On the other hand, the class B scavenger receptors CD36/SR-BII are not involved in oligomeric Aβ clearance, but can affect the recruitment and activation of microglia in response to fibrillar Aβ (Coraci et al., 2002; El Khoury et al., 2003). Fibrillar Aβ also acts as a scaffold for the assembly of a receptor complex consisting of CD36, alpha6beta1-integrin, and CD47 (Bamberger et al., 2003). Besides microglia, some types of SRs are expressed by astrocytes, including SR-BI and SR-MARCO (macrophage receptor with collagenous structure; Alarcon et al., 2005).
Toll-Like Receptors (TLR)
Toll-like receptors are involved in the microglial clearance of monomeric, oligomeric, and fibrillar Aβ (Tahara et al., 2006; Reed-Geaghan et al., 2009). TLR2 and TLR4 are directly involved in the microglial phagocytic response to Aβ, or indirectly with other receptors, as in the case of TLR9 activation by Aβ through the upregulation of formyl peptide receptor-2 (FPR2; Iribarren et al., 2005). The phagocytosis of fibrillar Aβ(1–42) by microglia can also be mediated through the LPS receptor CD14, a co-receptor of the functional TLR complex (Liu et al., 2005). Studies performed in TLR2 and TLR4 knockout mice have confirmed the importance of these receptors in Aβ clearance, showing increased Aβ deposition (Tahara et al., 2006; Richard et al., 2008; Birch et al., 2014). Some TLRs are also expressed in astrocytes and respond to TLR activators by secreting pro-inflammatory molecules (Gorina et al., 2009; van Noort and Bsibsi, 2009; Trudler et al., 2010).
Receptor for Advanced Glycation End Products (RAGE)
Although RAGE receptors have been mainly involved in mediating the inflammatory cascade in activated microglia (Solito and Sastre, 2012), their role in Aβ phagocytosis in astrocytes has been recently demonstrated when blocking these receptors with specific antibodies (Jones et al., 2013).
Formyl Peptide Receptors (FPR)
Formyl peptide receptors are a group of seven-transmembrane G protein coupled receptors (Le et al., 2002) and are expressed in neurons, microglia and astrocytes (Panaro et al., 2007; Braun et al., 2011). There is evidence that the FPRL1/FPR2 subtype binds to Aβ(1–42) and activates microglial internalization of the Aβ/FPRL1 complex in a PLD dependent-manner in microglia (Le et al., 2001; Brandenburg et al., 2008) and astrocytes (Brandenburg et al., 2008). In addition, both RAGE and SR-MARCO are known to form complexes with FPRL1/FPR2 in the presence of Aβ, initiating microglial signaling cascades in response to Aβ (Brandenburg et al., 2010; Slowik et al., 2012).
Fc Receptors (FcRs)
Fc receptors are expressed in microglia, astrocytes, oligodendrocytes, and neurons (Okun et al., 2010). Peress and colleagues first reported FcγRI, FcγRII, and FcγRIII immunoreactivity in senile plaques and on ramified microglia throughout the cortex and white matter of healthy controls and AD patients (Peress et al., 1993). The FcRs have been shown to mediate Aβ clearance in the presence of anti-Aβ antibodies (Bard et al., 2000; Wilcock et al., 2003), as observed in Aβ immunization therapies (Bacskai et al., 2002). The degree of involvement of Fc receptors in the clearance of Aβ bound by endogenous antibodies such as IgGs is currently not well elucidated (Doens and Fernández, 2014).
Triggering Receptor Expressed on Myeloid Cells 2 (TREM2)
Genetic variants of TREM2 receptors were recently identified as causing increased susceptibility to late onset AD. TREM2 can activate phagocytosis in microglia and reduce TLR-mediated signaling in macrophages (Klesney-Tait et al., 2006). Missense mutations associated with FTD and FTD-like syndrome have been shown to reduce TREM2 maturation and impair the phagocytic activity of TREM2-expressing cells (Kleinberger et al., 2014). More recently, it has been demonstrated that TREM2 is able to specifically sense fibrillar Aβ, activating microglial clustering around plaques, thereby limiting Aβ diffusion and subsequent toxicity (Wang et al., 2015, 2016).
Lipoprotein Receptor-Related Proteins (LRPs)
Lipoprotein receptor-related protein 1 (LRP1) is a large endocytic receptor for more than 40 ligands, including ApoE, α2-M and Aβ, and it is expressed by neurons, vascular cells and glial cells in the brain. Astrocytes take up Aβ through LRP1 either directly or indirectly in the presence of amyloid-associated protein ApoE, with perivascular astrocytes in AD brains found to contain both Aβ and ApoE (Utter et al., 2008; Kanekiyo et al., 2014). ApoE deficient astrocytes do not respond as well as wild type astrocytes to amyloid deposits, suggesting that ApoE is needed for astrocyte clearance of Aβ (Koistinaho et al., 2004). However, as indicated above, a recent paper has suggested that in fact ApoE may compete for the binding of Aβ to LRP-1. LRP-1 can also mediate Aβ phagocytosis in microglia, confirmed in vitro using LRP1 deficient cells (N’Songo et al., 2013). Furthermore, Aβ can be taken up when bound to LRP2 together with ApoJ and the megalin receptor (Zlokovic et al., 1996).
LGI3
The transmembrane protein leucine-rich glioma inactivated protein 3 co-localizes with Aβ at the astrocytic cell membrane (Kimura et al., 2007), and its downregulation reduces Aβ internalization by astrocytes (Okabayashi and Kimura, 2008).
Astrocytes and the “Glymphatic” System
It has been recently shown that astrocytes may contribute to the clearance of debris from the brain thanks to their projections around blood vessels, creating a sort of network that drains Aβ and other products out of the brain. In vessels, astrocyte end feet appear to connect to the smooth muscle layer (Morris et al., 2014). High expression of the channel aquaporin 4 (AQP-4) at the astrocyte end feet is thought to help solute clearance due to its role in water transport (Igarashi et al., 2014). In fact, AQP-4 knock-out mice show hindered solute clearance including that of Aβ (Iliff and Nedergaard, 2013).
Conclusion
Glial cells represent around 50% of the cells in the human brain (Azevedo et al., 2009). It is well established that in AD there is an up-regulation in the number and/or activation of microglia and astrocytes, associated with the deposition of Aβ. Although many studies have supported the notion that the activation of these glial cells may have detrimental effects due to the release of pro-inflammatory mediators such as certain cytokines and reactive oxygen species, there is evidence that supports their “protective” role by promoting the removal of Aβ. This function seems to be associated with a special and particular phenotype in microglia (formally known as M2 or alternatively activated) in contrast with the pro-inflammatory M1 state (Tang and Le, 2016). Therefore, many of the proteins that have been described in this review may not be expressed throughout life by glial cells, but their presence may depend on the activation status of those cells. This may also change during aging, when there is a dysregulation of glial function and these systems may become defective, contributing to the accumulation of Aβ in the brain.
A number of the studies reported here have outlined the difficulties of dissecting out each of these specific mechanisms of Aβ clearance only by using animal models with a specific deletion for one of those proteins, because many mechanisms of clearance are interconnected. Besides, some of these proteins have additional roles in the brain that are not directly related to the clearance of Aβ and may interfere with the interpretation of the results.
The therapeutic approaches targeting these clearance mechanisms have provided promising results, including the design of vectors carrying genes for NEP, for instance, or the discovery of drugs that enhance the synthesis of ApoE, showing reductions in Aβ deposition and improving cognitive impairments. Therefore, research in this field holds great potential for the development of new treatments to cure/stop the progression of AD.
Author Contributions
MR wrote the mechanisms of Aβ internalization and organized the reference list and MS wrote the rest of the manuscript.
Funding
Studentship to MR was funded by the Imperial College Medical Research Council Doctoral Training Centre.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Prof. Steve Gentleman (Imperial College London) for critical reading of the manuscript.
References
Abraham, C., Selkoe, D., Potter, H., Price, D., and Cork, L. (1989). α1-Antichymotrypsin is present together with the β-protein in monkey brain amyloid deposits. Neuroscience 32, 715–720. doi: 10.1016/0306-4522(89)90292-3
Abraham, C. R. (2001). Reactive astrocytes and α1-antichymotrypsin in Alzheimer’s disease. Neurobiol. Aging 22, 931–936. doi: 10.1016/S0197-4580(01)00302-5
Alarcon, R., Fuenzalida, C., Santibanez, M., and von Bernhardi, R. (2005). Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J. Biol. Chem. 280, 30406–30415. doi: 10.1074/jbc.M414686200
Azevedo, F. A., Carvalho, L. R., Grinberg, L. T., Farfel, J. M., Ferretti, R. E., Leite, R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541. doi: 10.1002/cne.21974
Bacskai, B. J., Kajdasz, S. T., McLellan, M. E., Games, D., Seubert, P., Schenk, D., et al. (2002). Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J. Neurosci. 22, 7873–7878.
Bamberger, M. E., Harris, M. E., McDonald, D. R., Husemann, J., and Landreth, G. E. (2003). A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 23, 2665–2674.
Bard, F., Cannon, C., Barbour, R., Burke, R., Games, D., Grajeda, H., et al. (2000). Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919. doi: 10.1038/78682
Bates, K., Verdile, G., Li, Q., Ames, D., Hudson, P., Masters, C., et al. (2009). Clearance mechanisms of Alzheimer’s amyloid-β peptide: implications for therapeutic design and diagnostic tests. Mol. Psychiatry 14, 469–486. doi: 10.1038/mp.2008.96
Bernstein, H., Lendeckel, U., Bukowska, A., Ansorge, S., Ernst, T., Stauch, R., et al. (2008). Regional and cellular distribution patterns of insulin-degrading enzyme in the adult human brain and pituitary. J. Chem. Neuroanat. 35, 216–224. doi: 10.1016/j.jchemneu.2007.12.001
Birch, A. M., Katsouri, L., and Sastre, M. (2014). Modulation of inflammation in transgenic models of Alzheimer’s disease. J. Neuroinflammation 11:25. doi: 10.1186/1742-2094-11-25
Blair, J. A., Siedlak, S. L., Wolfram, J. A., Nunomura, A., Castellani, R. J., Ferreira, S. T., et al. (2014). Accumulation of intraneuronal Amyloid-β is common in normal brain. Curr. Alzheimer Res. 11, 317–324. doi: 10.2174/1567205011666140302200902
Brandenburg, L., Konrad, M., Wruck, C., Koch, T., Pufe, T., and Lucius, R. (2008). Involvement of formyl-peptide-receptor-like-1 and phospholipase D in the internalization, and signal transduction of amyloid beta 1-42 in glial cells. Neuroscience 156, 266–276. doi: 10.1016/j.neuroscience.2008.07.042
Brandenburg, L., Konrad, M., Wruck, C. J., Koch, T., Lucius, R., and Pufe, T. (2010). Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1–42-induced signal transduction in glial cells. J. Neurochem. 113, 749–760. doi: 10.1111/j.1471-4159.2010.06637.x
Braun, B. J., Slowik, A., Leib, S. L., Lucius, R., Varoga, D., Wruck, C. J., et al. (2011). The formyl peptide receptor like-1 and scavenger receptor MARCO are involved in glial cell activation in bacterial meningitis. J. Neuroinflammation 8:11. doi: 10.1186/1742-2094-8-11
Carare, R., Bernardes-Silva, M., Newman, T., Page, A., Nicoll, J., Perry, V., et al. (2008). Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 34, 131–144. doi: 10.1111/j.1365-2990.2007.00926.x
Cascella, R., Conti, S., Tatini, F., Evangelisti, E., Scartabelli, T., Casamenti, F., et al. (2013). Extracellular chaperones prevent Aβ 42-induced toxicity in rat brains. Biochim. Biophys. Acta 1832, 1217–1226. doi: 10.1016/j.bbadis.2013.04.012
Cho, M., Cho, K., Kang, H., Jeon, E., Kim, H., Kwon, H., et al. (2014). Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10, 1761–1775. doi: 10.4161/auto.29647
Coraci, I. S., Husemann, J., Berman, J. W., Hulette, C., Dufour, J. H., Campanella, G. K., et al. (2002). CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to β-amyloid fibrils. Am. J. Pathol. 160, 101–112. doi: 10.1016/S0002-9440(10)64354-4
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Cucullo, L., Marchi, N., Marroni, M., Fazio, V., Namura, S., and Janigro, D. (2003). Blood-brain barrier damage induces release of alpha2-macroglobulin. Mol. Cell. Proteomics 2, 234–241. doi: 10.1074/mcp.M200077-MCP200
Deane, R., Bell, R. D., Sagare, A., and Zlokovic, B. V. (2009). Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 8, 16–30. doi: 10.2174/187152709787601867
Doens, D., and Fernández, P. L. (2014). Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflammation 11:48. doi: 10.1186/1742-2094-11-48
Eckman, E. A., Reed, D. K., and Eckman, C. B. (2001). Degradation of the Alzheimer’s amyloid beta peptide by endothelin-converting enzyme. J. Biol. Chem. 276, 24540–24548. doi: 10.1074/jbc.M007579200
Eckman, E. A., Watson, M., Marlow, L., Sambamurti, K., and Eckman, C. B. (2003). Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 278, 2081–2084. doi: 10.1074/jbc.C200642200
El Khoury, J. B., Moore, K. J., Means, T. K., Leung, J., Terada, K., Toft, M., et al. (2003). CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 197, 1657–1666. doi: 10.1084/jem.20021546
Farfara, D., Trudler, D., Segev-Amzaleg, N., Galron, R., Stein, R., and Frenkel, D. (2011). γ-Secretase component presenilin is important for microglia β-amyloid clearance. Ann. Neurol. 69, 170–180. doi: 10.1002/ana.22191
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Fisk, L., Nalivaeva, N. N., Boyle, J. P., Peers, C. S., and Turner, A. J. (2007). Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurones and astrocytes. Neurochem. Res. 32, 1741–1748. doi: 10.1007/s11064-007-9349-2
Fu, H., Liu, B., Frost, J. L., Hong, S., Jin, M., Ostaszewski, B., et al. (2012). Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 60, 993–1003. doi: 10.1002/glia.22331
Fukami, S., Watanabe, K., Iwata, N., Haraoka, J., Lu, B., Gerard, N. P., et al. (2002). Aβ-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Aβ pathology. Neurosci. Res. 43, 39–56. doi: 10.1016/S0168-0102(02)00015-9
Gopalan, S. M., Wilczynska, K. M., Konik, B. S., Bryan, L., and Kordula, T. (2006a). Astrocyte-specific expression of the alpha1-antichymotrypsin, and glial fibrillary acidic protein genes requires activator protein-1. J. Biol. Chem. 281, 1956–1963. doi: 10.1074/jbc.M510935200
Gopalan, S. M., Wilczynska, K. M., Konik, B. S., Bryan, L., and Kordula, T. (2006b). Nuclear factor-1-X regulates astrocyte-specific expression of the alpha1-antichymotrypsin, and glial fibrillary acidic protein genes. J. Biol. Chem. 281, 13126–13133. doi: 10.1074/jbc.M601194200
Gorina, R., Santalucia, T., Petegnief, V., Ejarque-Ortiz, A., Saura, J., and Planas, A. M. (2009). Astrocytes are very sensitive to develop innate immune responses to lipid-carried short interfering RNA. Glia 57, 93–107. doi: 10.1002/glia.20738
Gouras, G. K., Almeida, C. G., and Takahashi, R. H. (2005). Intraneuronal Aβ accumulation, and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 26, 1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU, and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
Harris, F. M., Tesseur, I., Brecht, W. J., Xu, Q., Mullendorff, K., Chang, S., et al. (2004). Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer’s disease. J. Biol. Chem. 279, 3862–3868. doi: 10.1074/jbc.M309475200
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction, and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Holsinger, R., McLean, C. A., Beyreuther, K., Masters, C. L., and Evin, G. (2002). Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann. Neurol. 51, 783–786. doi: 10.1002/ana.10208
Hong, L., Huang, H., and Jiang, Z. (2014). Relationship between amyloid-beta, and the ubiquitin–proteasome system in Alzheimer’s disease. Neurol. Res. 36, 276–282. doi: 10.1179/1743132813Y.0000000288
Hu, J., Igarashi, A., Kamata, M., and Nakagawa, H. (2001). Angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide (Aβ); retards. (Aβ) aggregation, deposition, fibril formation; and inhibits cytotoxicity. J. Biol. Chem. 276, 47863–47868. doi: 10.1074/jbc.M104068200
Husemann, J., Loike, J. D., Anankov, R., Febbraio, M., and Silverstein, S. C. (2002). Scavenger receptors in neurobiology, and neuropathology: their role on microglia, and other cells of the nervous system. Glia 40, 195–205. doi: 10.1002/glia.10148
Igarashi, H., Suzuki, Y., Kwee, I. L., and Nakada, T. (2014). Water influx into cerebrospinal fluid is significantly reduced in senile plaque bearing transgenic mice, supporting beta-amyloid clearance hypothesis of Alzheimer’s disease. Neurol. Res. 36, 1094–1098. doi: 10.1179/1743132814Y.0000000434
Iliff, J. J., and Nedergaard, M. (2013). Is there a cerebral lymphatic system? Stroke 44, S93–S95. doi: 10.1161/STROKEAHA.112.678698
Iribarren, P., Chen, K., Hu, J., Gong, W., Cho, E. H., Lockett, S., et al. (2005). CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1-42 peptide by up-regulating the expression of the G-protein- coupled receptor mFPR2. FASEB J. 19, 2032–2034. doi: 10.1096/fj.05-4578fje
Iwata, H., Tomita, T., Maruyama, K., and Iwatsubo, T. (2001). Subcellular compartment, and molecular subdomain of beta-amyloid precursor protein relevant to the Abeta 42-promoting effects of Alzheimer mutant presenilin 2. J. Biol. Chem. 276, 21678–21685. doi: 10.1074/jbc.M007989200
Jansen, A. H., Reits, E. A., and Hol, E. M. (2014). The ubiquitin proteasome system in glia, and its role in neurodegenerative diseases. Front. Mol. Neurosci. 7:73. doi: 10.3389/fnmol.2014.00073
Jones, R. S., Minogue, A. M., Connor, T. J., and Lynch, M. A. (2013). Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47, and RAGE. J. Neuroimmune Pharmacol. 8, 301–311. doi: 10.1007/s11481-012-9427-3
Kanekiyo, T., Xu, H., and Bu, G. (2014). ApoE, and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754. doi: 10.1016/j.neuron.2014.01.045
Kimura, N., Ishii, Y., Suzaki, S., Negishi, T., Kyuwa, S., and Yoshikawa, Y. (2007). Aβ upregulates, and colocalizes with LGI3 in cultured rat astrocytes. Cell Mol. Neurobiol. 27, 335–350. doi: 10.1007/s10571-006-9127-8
Kleinberger, G., Yamanishi, Y., Suarez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6:243ra86. doi: 10.1126/scitranslmed.3009093
Klesney-Tait, J., Turnbull, I. R., and Colonna, M. (2006). The TREM receptor family, and signal integration. Nat. Immunol. 7:1266–1273. doi: 10.1038/ni1411
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726. doi: 10.1038/nm1058
Le, Y., Gong, W., Tiffany, H. L., Tumanov, A., Nedospasov, S., Shen, W., et al. (2001). Amyloid ß42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 21.
Le, Y., Murphy, P. M., and Wang, J. M. (2002). Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548. doi: 10.1016/S1471-4906(02)02316-5
Lee, C. D., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural. Transm. 117, 949–960. doi: 10.1007/s00702-010-0433-4
Lee, S., Seo, B., and Koh, J. (2015). Metallothionein-3 modulates the amyloid β endocytosis of astrocytes through its effects on actin polymerization. Mol. Brain. 8:84. doi: 10.1186/s13041-015-0173-3
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi: 10.1016/S0896-6273(03)00787-6
Li, H. Q., Chen, C., Dou, Y., Wu, H. J., Liu, Y. J., Lou, H. F., et al. (2013). P2Y4 receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol. Cell Biol. 33, 4282–4293. doi: 10.1128/MCB.00544-13
Lilienbaum, A. (2013). Relationship between the proteasomal system, and autophagy. Int. J. Biochem. Mol. Biol. 4, 1–26.
Liu, Y., Walter, S., Stagi, M., Cherny, D., Letiembre, M., Schulz-Schaeffer, W., et al. (2005). LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 128, 1778–1789. doi: 10.1093/brain/awh531
Love, S. (2004). Contribution of cerebral amyloid angiopathy to Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 75, 1–4.
Lucin, K. M., O’Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., et al. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886. doi: 10.1016/j.neuron.2013.06.046
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009
Melchor, J. P., and Strickland, S. (2005). Tissue plasminogen activator in central nervous system physiology, and pathology. Thromb. Haemost. 93, 655–660. doi: 10.1160/TH04-12-0838
Morris, A. W., Carare, R. O., Schreiber, S., and Hawkes, C. A. (2014). The cerebrovascular basement membrane: role in the clearance of β-amyloid, and cerebral amyloid angiopathy. Front. Aging Neurosci. 6:251. doi: 10.3389/fnagi.2014.00251
Mulder, S. D., Nielsen, H. M., Blankenstein, M. A., Eikelenboom, P., and Veerhuis, R. (2014). Apolipoproteins E, and J interfere with amyloid-beta uptake by primary human astrocytes, and microglia in vitro. Glia 62, 493–503. doi: 10.1002/glia.22619
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., and Wang, H. (2003). Astrocytes accumulate Aβ42, and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209. doi: 10.1016/S0006-8993(03)02361-8
Naidoo, V., Naidoo, S., Mahabeer, R., and Raidoo, D. (2004). Cellular distribution of the endothelin system in the human brain. J. Chem. Neuroanat. 27, 87–98. doi: 10.1016/j.jchemneu.2003.12.002
Nakagomi, S., Kiryu-Seo, S., and Kiyama, H. (2000). Endothelin-converting enzymes, and endothelin receptor B messenger RNAs are expressed in different neural cell species, and these messenger RNAs are coordinately induced in neurons, and astrocytes respectively following nerve injury. Neuroscience 101, 441–449. doi: 10.1016/S0306-4522(00)00345-6
Nakanishi, H. (2003). Microglial functions, and proteases. Mol. Neurobiol. 27, 163–176. doi: 10.1385/MN:27:2:163
Nalivaeva, N. N., Beckett, C., Belyaev, N. D., and Turner, A. J. (2012). Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 120, 167–185. doi: 10.1111/j.1471-4159.2011.07510.x
Nixon, R. A. (2007). Autophagy, amyloidogenesis, and Alzheimer disease. J. Cell Sci. 120, 4081–4091. doi: 10.1242/jcs.019265
N’Songo, A., Kanekiyo, T., and Bu, G. (2013). LRP1 plays a major role in the amyloid-β clearance in microglia. Mol. Neurodegener. 8:33. doi: 10.1186/1750-1326-8-S1-P33
Okabayashi, S., and Kimura, N. (2008). Leucine-rich glioma inactivated 3 is involved in amyloid beta peptide uptake by astrocytes, and endocytosis itself. Neuroreport 19, 1175–1179. doi: 10.1097/WNR.0b013e32830a4cc4
Okun, E., Mattson, M. P., and Arumugam, T. V. (2010). Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular Med. 12, 164–178. doi: 10.1007/s12017-009-8099-5
Pacheco-Quinto, J., and Eckman, E. A. (2013). Endothelin-converting enzymes degrade intracellular beta-amyloid produced within the endosomal/lysosomal pathway, and autophagosomes. J. Biol. Chem. 288, 5606–5615. doi: 10.1074/jbc.M112.422964
Pan, X., Zhu, Y., Lin, N., Zhang, J., Ye, Q., Huang, H., et al. (2011). Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: implications for Alzheimer’s disease. Mol. Neurodegener. 6, 1–18. doi: 10.1186/1750-1326-6-45
Panaro, M., Acquafredda, A., Sisto, M., Lisi, S., Calvello, R., Mitolo, C., et al. (2007). Formyl peptide receptors on immune, and nonimmune cells: analysis of sequence conservation in FPR genes. Immunopharmacol. Immunotoxicol. 29, 243–269. doi: 10.1080/08923970701512544
Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996). Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron 17, 553–565. doi: 10.1016/S0896-6273(00)80187-7
Peress, N. S., Fleit, H. B., Perillo, E., Kuljis, R., and Pezzullo, C. (1993). Identification of FcγRI, II, and III on normal human brain ramified microglia, and on microglia in senile plaques in Alzheimer’s disease. J. Neuroimmunol. 48, 71–79. doi: 10.1016/0165-5728(93)90060-C
Pomilio, C., Pavia, P., Gorojod, R. M., Vinuesa, A., Alaimo, A., Galvan, V., et al. (2016). Glial alterations from early to late stages in a model of Alzheimer’s disease: evidence of autophagy involvement in Aβ internalization. Hippocampus 26, 194–210. doi: 10.1002/hipo.22503
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G., and Landreth, G. E. (2009). CD14, and toll-like receptors 2, and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 29, 11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009
Richard, K. L., Filali, M., Prefontaine, P., and Rivest, S. (2008). Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42, and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 28, 5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008
Rock, K. L., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L., et al. (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. doi: 10.1016/S0092-8674(94)90462-6
Saido, T., and Leissring, M. A. (2012). Proteolytic degradation of amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2:a006379. doi: 10.1101/cshperspect.a006379
Sastre, M., Ritchie, C. W., and Hajji, N. (2015). Metal ions in Alzheimer’s disease brain. JSM Alzheimer’s Dis. Relat. Dement. 2:1014.
Saura, J., Petegnief, V., Wu, X., Liang, Y., and Paul, S. M. (2003). Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J. Neurochem. 85, 1455–1467. doi: 10.1046/j.1471-4159.2003.01788.x
Slowik, A., Merres, J., Elfgen, A., Jansen, S., Mohr, F., Wruck, C. J., et al. (2012). Involvement of formyl peptide receptors in receptor for advanced glycation end products(RAGE)-and amyloid beta 1-42-induced signal transduction in glial cells. Mol. Neurodegener. 7, 1–18. doi: 10.1186/1750-1326-7-55
Solito, E., and Sastre, M. (2012). Microglia function in Alzheimer’s disease. Front. Pharmacol. 3:14. doi: 10.3389/fphar.2012.00014
Steneberg, P., Bernardo, L., Edfalk, S., Lundberg, L., Backlund, F., Ostenson, C. G., et al. (2013). The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of alpha-synuclein levels in beta-cells. Diabetes 62, 2004–2014. doi: 10.2337/db12-1045
Stocker, R., and Keaney, J. F. Jr. (2004). Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84, 1381–1478. doi: 10.1152/physrev.00047.2003
Tahara, K., Kim, H. D., Jin, J. J., Maxwell, J. A., Li, L., and Fukuchi, K. (2006). Role of toll-like receptor signalling in Abeta uptake, and clearance. Brain 129, 3006–3019. doi: 10.1093/brain/awl249
Tamboli, I. Y., Barth, E., Christian, L., Siepmann, M., Kumar, S., Singh, S., et al. (2010). Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 285, 37405–37414. doi: 10.1074/jbc.M110.149468
Tang, Y., and Le, W. (2016). Differential roles of M1, and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Tarasoff-Conway, J. M., Carare, R. O., Osorio, R. S., Glodzik, L., Butler, T., Fieremans, E., et al. (2015). Clearance systems in the brain - implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470. doi: 10.1038/nrneurol.2015.119
Trudler, D., Farfara, D., and Frenkel, D. (2010). Toll-like receptors expression, and signaling in glia cells in neuro-amyloidogenic diseases: towards future therapeutic application. Mediators Inflamm. 2010:12. doi: 10.1155/2010/497987
Utter, S., Tamboli, I. Y., Walter, J., Upadhaya, A. R., Birkenmeier, G., Pietrzik, C. U., et al. (2008). Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J. Neuropathol. Exp. Neurol. 67, 842–856. doi: 10.1097/NEN.0b013e3181836a71
van Noort, J. M., and Bsibsi, M. (2009). Toll-like receptors in the CNS: implications for neurodegeneration, and repair. Prog. Brain Res. 175, 139–148. doi: 10.1016/S0079-6123(09)17509-X
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110, E1807–E1816. doi: 10.1073/pnas.1220484110
Wang, S., Wang, R., Chen, L., Bennett, D. A., Dickson, D. W., and Wang, D. (2010). Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J. Neurochem. 115, 47–57. doi: 10.1111/j.1471-4159.2010.06899.x
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wilcock, D. M., DiCarlo, G., Henderson, D., Jackson, J., Clarke, K., Ugen, K. E., et al. (2003). Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J. Neurosci. 23, 3745–3751.
Wildsmith, K. R., Holley, M., Savage, J. C., Skerrett, R., and Landreth, G. E. (2013). Evidence for impaired amyloid beta clearance in Alzheimer’s disease. Alzheimers Res. Ther. 5:33. doi: 10.1186/alzrt187
Yamamoto, N., Tanida, M., Ono, Y., Kasahara, R., Fujii, Y., Ohora, K., et al. (2014). Leptin inhibits amyloid β-protein degradation through decrease of neprilysin expression in primary cultured astrocytes. Biochem. Biophys. Res. Commun. 445, 214–217. doi: 10.1016/j.bbrc.2014.01.168
Yamauchi, K., Yamauchi, T., Mantuano, E., Murakami, K., Henry, K., Takahashi, K., et al. (2013). Low-density lipoprotein receptor related protein-1 (LRP1)-dependent cell signaling promotes neurotrophic activity in embryonic sensory neurons. PLoS ONE 8:e75497. doi: 10.1371/journal.pone.0075497
Yan, P., Hu, X., Song, H., Yin, K., Bateman, R. J., Cirrito, J. R., et al. (2006). Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem. 281, 24566–24574. doi: 10.1074/jbc.M602440200
Yang, C., Shiao, Y., Shie, F., Guo, B., Chen, P., Cho, C., et al. (2011). Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiol. Dis. 42, 221–230. doi: 10.1016/j.nbd.2011.01.005
Yang, L., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X., et al. (2003). Elevated β-secretase expression, and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4. doi: 10.1038/nm0103-3
Yin, K. J., Cirrito, J. R., Yan, P., Hu, X., Xiao, Q., Pan, X., et al. (2006). Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 26, 10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006
Yoon, S., and Jo, S. A. (2012). Mechanisms of amyloid-β peptide clearance: potential therapeutic targets for Alzheimer’s disease. Biomol. Ther. 20, 245–255. doi: 10.4062/biomolther.2012.20.3.245
Zhao, X., and Yang, J. (2010). Amyloid-β peptide is a substrate of the human 20S proteasome. ACS Chem. Neurosci. 1, 655–660. doi: 10.1021/cn100067e
Zhao, Z., Xiang, Z., Haroutunian, V., Buxbaum, J. D., Stetka, B., and Pasinetti, G. (2007). M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol. Aging 28, 824–830. doi: 10.1016/j.neurobiolaging.2006.05.001
Zlokovic, B. V., Martel, C. L., Matsubara, E., McComb, J. G., Zheng, G., McCluskey, R. T., et al. (1996). Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone, and in a complex with Alzheimer disease amyloid beta at the blood-brain, and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. U.S.A. 93, 4229–4234. doi: 10.1073/pnas.93.9.4229
Keywords: astrocytes, microglia, amyloid-β, Alzheimer’s disease, proteases, phagocytosis
Citation: Ries M and Sastre M (2016) Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
Received: 17 May 2016; Accepted: 17 June 2016;
Published: 05 July 2016.
Edited by:
Roxana Octavia Carare, University of Southampton, UKReviewed by:
Francisco G. Wandosell, Centro de Biologia Molecular “Seveo Ochoa", SpainRamesh Kandimalla, Emory University, USA
Copyright © 2016 Ries and Sastre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Sastre, m.sastre@imperial.ac.uk