Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models
- Departments of Biological Sciences, Cell and Developmental Biology, Kennedy Center for Research on Human Development, Vanderbilt University, Nashville, TN, USA
Proper brain function requires stringent balance of excitatory and inhibitory synapse formation during neural circuit assembly. Mutation of genes that normally sculpt and maintain this balance results in severe dysfunction, causing neurodevelopmental disorders including autism, epilepsy and Rett syndrome. Such mutations may result in defective architectural structuring of synaptic connections, molecular assembly of synapses and/or functional synaptogenesis. The affected genes often encode synaptic components directly, but also include regulators that secondarily mediate the synthesis or assembly of synaptic proteins. The prime example is Fragile X syndrome (FXS), the leading heritable cause of both intellectual disability and autism spectrum disorders. FXS results from loss of mRNA-binding FMRP, which regulates synaptic transcript trafficking, stability and translation in activity-dependent synaptogenesis and plasticity mechanisms. Genetic models of FXS exhibit striking excitatory and inhibitory synapse imbalance, associated with impaired cognitive and social interaction behaviors. Downstream of translation control, a number of specific synaptic proteins regulate excitatory versus inhibitory synaptogenesis, independently or combinatorially, and loss of these proteins is also linked to disrupted neurodevelopment. The current effort is to define the cascade of events linking transcription, translation and the role of specific synaptic proteins in the maintenance of excitatory versus inhibitory synapses during neural circuit formation. This focus includes mechanisms that fine-tune excitation and inhibition during the refinement of functional synaptic circuits, and later modulate this balance throughout life. The use of powerful new genetic models has begun to shed light on the mechanistic bases of excitation/inhibition imbalance for a range of neurodevelopmental disease states.
Introduction
The integration of excitatory and inhibitory inputs at the level of the individual neuron, and in the organization of functional units constituting neural circuits, is fundamental to the information processing that mediates brain function. Establishing and maintaining the appropriate ratio of excitatory versus inhibitory synapses (E/I ratio) is a critical factor that enables circuit threshold definition and balances measured output responsiveness. Disruption of the E/I set point beyond acceptable tolerances leads to aberrant hyper/hypo-transmissive states, which when chronically unresolved cause severe dysfunction. A growing body of evidence suggests that disrupted E/I ratios within the central nervous system may be implicated in a range of neurodevelopmental disorders. Contributing factors may include the selective loss of either excitatory or inhibitory synapses, without compensatory changes, or genetic conditions that actively favor the formation or maintenance of one class of synapse relative to the other. Such imbalances may arise during initial neural circuit formation, as a failure of ‘refinement’ mechanisms that address balance during preliminary circuit use periods, or as a selective inability to maintain the E/I ratio into maturity, perhaps owing to defective plasticity properties. This review will discuss these alternate possibilities in the context of genetic models of neurodevelopmental disorders.
We examine E/I balance based on the two primary, opposing central neurotransmitters, glutamate and γ-amino-butyric acid (GABA). Their respective excitatory and inhibitory synapses are characteristically divergent in a range of features (Boeckers, 2006 ; Craig et al., 2006 ; Okabe, 2007 ; Tretter and Moss, 2008 ; Tyagarajan and Fritschy, 2010 ). First, the cellular position of the synapses differs dramatically. Glutamatergic synapses form almost exclusively on dendritic spines after preferential association with, and stabilization of, an incoming, exploratory filopodial scout (Lohmann and Bonhoeffer, 2008 ). In contrast, GABAergic synapses do not form on spines, but rather reside on dendritic shafts, nerve cell somata and axon initial segments. These inhibitory synapses arise from predecessor axon-dendrite contacts without any apparent protrusive activity from either axon or dendrite (Wierenga et al., 2008 ). Second, ultrastructural properties of pre- versus post-synaptic specializations are readily distinguishable between synaptic classes (Gray, 1959 ; Colonnier, 1968 ; Peters and Palay, 1996 ). Excitatory contacts maintain discrete asymmetry with an electron-dense postsynaptic density opposing a presynaptic active zone. Inhibitory contacts, however, appear relatively symmetric with presynaptic vesicles clustered opposite a synaptic cleft without a robust postsynaptic density. Third, the molecular constituents of the synapses differ considerably. Beyond the ionotropic and metabotropic glutamate and GABA receptors that confer transmitter specificity, presynaptic transporters responsible for vesicle loading and organizing postsynaptic scaffolding molecules often serve as reporters for immunohistochemical synapse discrimination (Boeckers, 2006 ; Craig et al., 2006 ; Okabe, 2007 ; Tretter and Moss, 2008 ; Tyagarajan and Fritschy, 2010 ). Excitatory synapses maintain vesicular glutamate transporters (VGLUTs) and the postsynaptic density protein-95 kDa (PSD-95), whereas inhibitory synapses are marked by the presence of the vesicular GABA (VGAT) and vesicular inhibitory amino acid (VIAAT) transporters and the postsynaptic adaptor protein gephryin.
In considering the overall ratio of excitatory to inhibitory neurons, in the mammalian cortex roughly 80% of neurons are excitatory and 20% inhibitory (Rubenstein and Merzenich, 2003 ). However, this compositional ratio may vary dramatically with brain region, development or aging, and it does not necessarily reflect the E/I synaptic balance per individual neuron. The E/I ratio influencing a given neuron is often functionally studied at an electrophysiological level by examining the contribution of glutamatergic and GABAergic synaptic inputs. The ratio can be determined by variably clamping the membrane potential to differentially record excitatory and inhibitory postsynaptic currents (EPSCs or IPSCs) (Liu, 2004 ). In addition, anatomical methods can be employed to structurally characterize the E/I ratio. Postembedding immunostaining or assays of symmetrical versus asymmetrical synapses via electron microscopy can assess the prevalence of excitatory and inhibitory synapses amongst convergent contacts (Megias et al., 2001 ). Serial electron microscope reconstructions have shown that in vivo rat hippocampal CA1 pyramidal cell dendrites receive approximately 30,000 excitatory inputs and 1,700 inhibitory inputs, yielding an E/I ratio of ∼18:1 (Megias et al., 2001 ). Notably, in vitro hippocampal cultures display an immature dendritic compartment ratio of ∼3:2 at 16-days shifting to ∼4:1 by 19 days (Liu, 2004 ), which nevertheless remains strikingly different from the in vivo condition. Examination of the local inhibitory interneurons in the hippocampus reveals salient differences: Parvalbumin-containing neurons receive 16,000 inputs of which 6% are inhibitory (E/I = ∼14:1); calbindin D (28 k) neurons receive 4,000 inputs of which 30% are inhibitory (E/I = ∼2:1); and calretinin-positive neurons maintain 2,000 inputs of which 20% are inhibitory (E/I = ∼3:1) (Gulyas et al., 1999 ). Clearly then, considerable differences in E/I ratios are discernable between, and even amongst, neurons of particular classes, and ratios may be highly dynamic as a function of developmental stage.
As studies have shown the relative excitatory drive upon each neuronal subtype is likely quite distinct, the E/I ratio therefore must be carefully regulated. Establishing this balance is further complicated by the developmental and activity-dependent shifts influencing ratio modulation. For example, Xenopus tectal neurons can bidirectionally modify GABAergic inputs as a function of the prevalent E/I ratio, with the level of convergent glutamatergic input appearing to be the determining factor (Liu et al., 2007 ). Indeed, GABA can serve as a depolarizing, excitatory transmitter in immature neurons (Ben-Ari, 2002 ; Akerman and Cline, 2007 ), and, when coupled with the relatively limited glutamateric inputs at this stage, repetitive stimulation can lead to attenuating long-term depression (LTD) in GABAergic inputs (Liu et al., 2007 ). However, with the developmental elevation of glutamatergic strength and transition of GABAergic inputs to their canonical inhibitory role, similar stimulation at maturity yields GABAergic long-term potentiation (LTP) (Liu et al., 2007 ). An excitatory role for GABA is also prevalent in the developing murine neocortex. A combination of the resting membrane potential (Em) and reversal potential for GABA (EGABA) in immature animals (postnatal days 2–10) leads to pyramidal neuron and interneuron depolarization that mediates action potential generation within cortical layers 5/6 (Rheims et al., 2008 ). Moreover, excitatory and inhibitory neurotransmitters can be employed coincidentally with their co-release critical to proper circuit refinement (Noh et al., 2010 ). In the mammalian auditory system, VGLUT3 and glutamate are found at developing inhibitory GABA/glycinergic synapses in the sound localization pathway of the lateral superior olive, prior to the onset of hearing (Gillespie et al., 2005 ). Genetic disruption of the glutamatergic component in a Vglut3−/−mouse impairs the coordinate, requisite synaptic strengthening and silencing necessary for normal tonotopic map organization (Noh et al., 2010 ). Thus, successfully implementing a strategy to influence the E/I ratio depends upon not only synapse enumeration but also how specific neurotransmitters function as a consequence of developmental progression.
How then do neural circuits establish and maintain balanced excitation and inhibition? For the sake of stable, long-term effectiveness and integrity, network activity is proposed to be maintained within a given dynamic range by compensatory alterations preventing runaway signaling, a concept termed ‘synaptic homeostasis’ (Turrigiano and Nelson, 2004 ; Davis, 2006 ; Maffei and Fontanini, 2009 ). The initial characterization of this phenomenon was done in cultured cortical neurons (Turrigiano et al., 1998 ). Pharmacological activity blockade with tetrodotoxin resulted in an upregulation of miniature excitatory postsynaptic current (mEPSC) amplitudes to 192% of control; whereas, disruption of GABAergic inhibition with bicuculline resulted in a 2.5-fold increase in neuronal firing and decreased mEPSC amplitudes to 70% of control (Turrigiano et al., 1998 ). Thus, in response to influences forcibly altering activity, compensatory mechanisms are enlisted in an apparent attempt to restore the initial circuit set point. In addition to the modulation of synaptic strength, balance may be achieved through adaptation of synaptic efficacy, membrane excitability and/or synapse number. For example, in independent studies, activity deprivation also reduced miniature inhibitory postsynaptic current (mIPSC) amplitudes with a concomitant 50% reduction in synapses reactive for the ionotropic GABAA receptor (GABAAR) (Kilman et al., 2002 ), and diminished activity in hippocampal cultures has been shown to increase the prevalence of synaptic pairing in efforts to enhance connectivity (Nakayama et al., 2005 ). Nevertheless, many conditions exist that permanently alter the E/I balance, indicating regular failure of the apparent homeostatic mechanism. This fact suggests either that homeostasis is far from a universal property of circuits, or that many, disparate defects can prevent appropriate compensation mechanisms from being engaged or maintained, compromising neurological function.
A host of devastating neurological diseases have been proposed to result from underlying E/I imbalance, ranging from neuropsychiatric conditions such as schizophrenia (Kehrer et al., 2008 ) and Tourette’s syndrome (Wassef et al., 2003 ), to neurodegenerative conditions like Parkinson’s and Huntington’s diseases (Choonara et al., 2009 ; Cummings et al., 2009 ). In particular, a large number of neurodevelopmental diseases are proposed to result from an E/I shift. This review will specifically highlight a subset of such disorders that harbor identifiable genetic components and have been successfully modeled to enable rigorous preclinical investigations. Neurodevelopmental disorders include a myriad of conditions, which share the common theme that symptomatic presentation and disease onset is identifiable in early postnatal years, prior to full brain maturation, when critical periods of activity-dependent refinement and circuit remodeling are vulnerable. Such disorders include the autism spectrum, epilepsy, Rett syndrome (RTT), and Fragile X syndrome (FXS) (Figure 1 ). For each case, we will survey indicative E/I imbalance conditions and then provide a more thorough discussion of FXS as our prime example. We will then discuss a range of causative genetic alterations that skew E/I balance by interfering with various aspects of normal gene expression programs and synaptic formation, organization and maintenance.
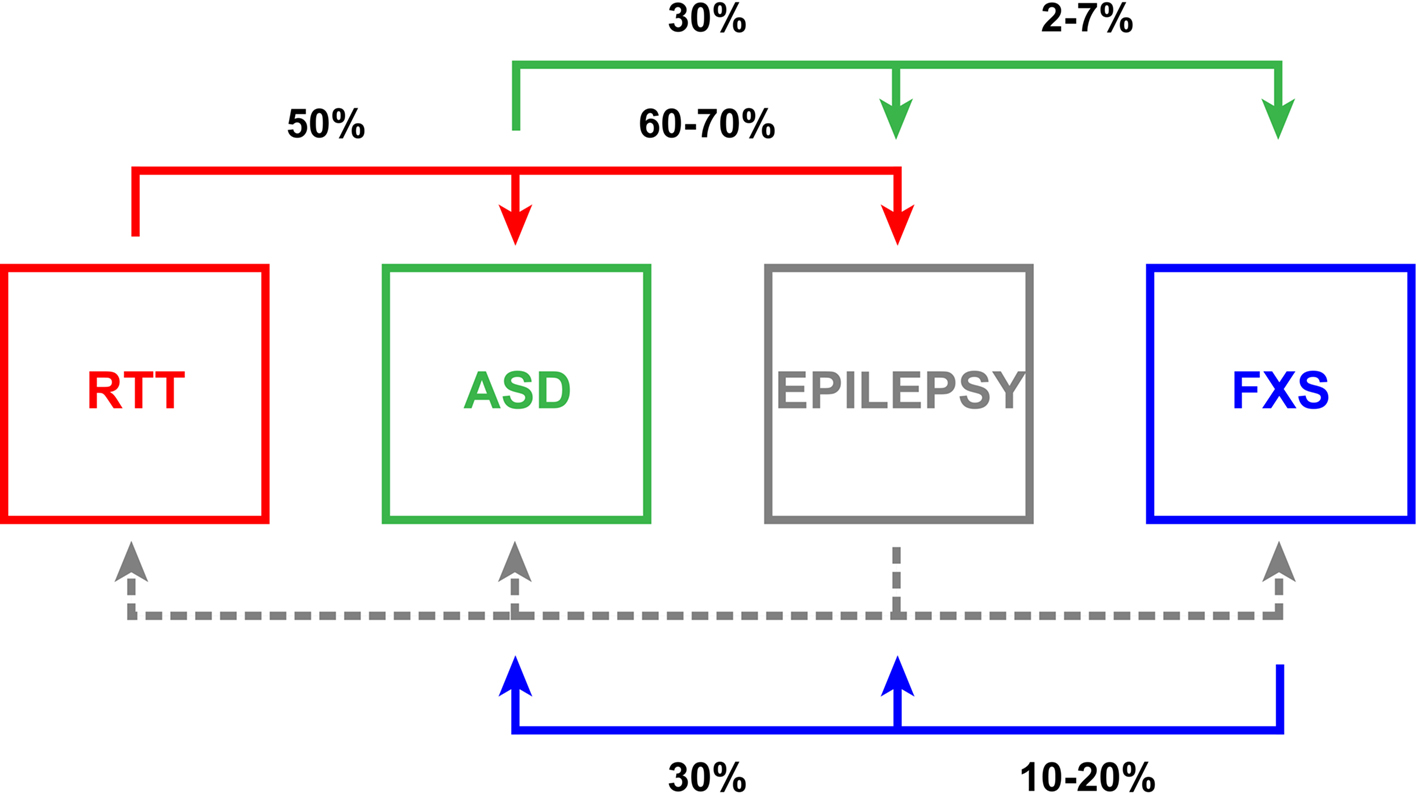
Figure 1.Relationships of neurodevelopmental diseases with E/I ratio synaptic imbalance. Rett syndrome (RTT, red), autism spectrum disorder (ASD, green), epilepsy (gray) and Fragile X syndrome (FXS, blue) manifest pathological E/I ratio imbalance with comorbidities indicated pair-wise in black. Epilepsy often presents with the other three conditions, but comparable associative statistics (dashed gray line) are not well documented in the literature. The interconnectivity amongst these disorders emphasizes the clinical significance of E/I ratio disruption and suggests that this imbalance likely renders a higher susceptibility to further dysfunction.
Neurodevelopmental Diseases with E/I Imbalance
Autism
With an incidence that some recent estimates place as high as 1:110 (Mulvihill et al., 2009 ), autism spectrum disorders (ASDs) are diseases of neurodevelopment with pervasive impact. The spectrum is characterized by compromised communication, social skills and cognition, often coupled with stereotypic repetitive behaviors and perception anomalies including hypersensitivity to tactile and auditory stimulation (APA, 2000 ; Levy et al., 2009 ; O’Hare, 2009 ). Autism is clinically obvious typically by 3 years of age; however, abnormalities in socialization and play can be ascertained as early as 12 months (Landa et al., 2007 ; Barbaro and Dissanayake, 2009 ; Zwaigenbaum, 2010 ). The early onset of ASD implicates defects in either fetal brain development or the earliest phases of postnatal maturation. Thus for effective intervention, it is critical that we push downward the age of effective diagnosis as early as possible. In addition, autism often presents with other neurological disorders (Figure 1 ). Approximately 5% (2–7%) of ASD children have the common mutation causing FXS (Rogers et al., 2001 ; Hagerman, 2008b ; Harris et al., 2008 ). Thirty percent of ASD patients also struggle with epilepsy (Tuchman and Rapin, 2002 ; Tuchman et al., 2009 ), and though current classification guidelines prevent comorbid diagnoses of autism and RTT, a recent study indicated that approximately 50% of RTT patients have been shown to meet ASD criterion employing the Developmental Behavior Checklist and the Diagnostic Interview for Social and Communication Disorders (Wulffaert et al., 2009 ).
Defects in synaptogenesis, synaptic refinement and connectivity have long been postulated to be a leading cause of the autism spectrum (Rubenstein and Merzenich, 2003 ; Rippon et al., 2007 ). Specifically, autism has been suggested to result from ‘an increased ratio of excitation/inhibition in sensory, mnemonic, social and emotional systems’ (Rubenstein and Merzenich, 2003 ). This putative imbalance is thought to contribute to the prevalence of poor signal-to-noise ratios, whereby hyperexcitable, non-tunable cortical circuits lead to critical developmental periods passing without appropriate differentiation being achieved, resulting in systemic instability (Rubenstein and Merzenich, 2003 ; Rippon et al., 2007 ). In support of this hypothesis, cytological abnormalities in the organization of mini-columns have been observed in neocortical post-mortem tissue. These vertical arrays of functionally related glutamatergic and GABAergic neurons process thalamic input and, in the autistic cortex, are smaller, more numerous and display disordered peripheral neuropil space (Casanova et al., 2002 , 2003 ; Casanova, 2006 ). Also potentially contributing to unchecked hyperexcitability, post-mortem studies have revealed that neurons in autistic patients likely display depressed GABA production as the level of glutamic acid decarboxylase (GAD), the rate limiting enzyme in GABA synthesis, is reduced by half (Fatemi et al., 2002 ). Additionally, GABA responsiveness may itself be compromised as GABAAR subunit depression is also observed with mRNA and protein studies showing altered α1–5 and β1 subunits (Fatemi et al., 2009 , 2010 ). These studies suggest a shift in the E/I ratio favoring an elevated preponderance of glutamatergic connections with inhibitory insufficiency.
In addition to FXS and RTT, a number of genetic susceptibilities have been linked to elevated autism risk including maternal 15q11–13 chromosomal duplications, anomalies in the tumor suppressor genes NF1, TSC1/TSC2, and PTEN that activate mammalian target of rapamycin (mTOR)/phosphatidylinositol 3-kinase (PI3K) signaling pathways, and mutations in a range of synaptic genes such as the neurexins, neuroligins, SHANK3 and CNTNAP2 (Betancur et al., 2009 ; Bourgeron, 2009 ; Kumar and Christian, 2009 ). For example, mutations in the postsynaptic cell adhesion molecule neuroligin-3 (NLGN3), particularly a single amino acid substitution at the Arg451 to Cys451 (R451C), have been well-characterized (Jamain et al., 2003 ). This genetic variant has been introduced in mice and behaviorally produces impaired social interaction, albeit with a surprisingly enhanced capacity for spatial learning (Tabuchi et al., 2007 ). At the E/I level, the R451C–NLGN3 knock-in shows increased VGAT and gephyrin levels. Although no change in inhibitory synapse number was observed, a 50% increased mIPSC frequency and increased amplitude of evoked IPSCs in whole-cell recordings of layer 2/3 somatosensory barrel cortex slices was observed in the absence of any accompanying changes in excitatory transmission (Tabuchi et al., 2007 ). Interestingly, these changes did not occur in the complete NLGN3 knockout (KO), indicating R451C–NLGN3 presumably represents a gain-of-function variation. In vitro connectivity studies employing dissociated rat hippocampal cultures indicated that NLGN3 introduction enhances optically observable spontaneous synchrony, whereas R471C-NLGN3 reduces this capacity (Gutierrez et al., 2009 ). Further, studies of axonal topography revealed R471C-NLGN3 expression results in shorter, less interconnected neurons and the preferential degeneration of inhibitory neurons. Thus, the overall network influence trends toward a reduction of inhibitory activity (Gutierrez et al., 2009 ). Collectively, these ASD model studies indicate that E/I imbalance can present differentially in various circuits; and, importantly, structural and functional changes do not always coincide.
Epilepsy
Epilepsy refers to a family of chronic neurological disorders with a frequency of approximately 7:1,000 individuals (Hirtz et al., 2007 ), characterized by a tendency toward recurrent non-provoked, spontaneous seizures (Scharfman, 2007 ; Jacobs et al., 2009 ; Reid et al., 2009 ). Epilepsy is often discussed as resulting from disruption in the balance of neuronal excitation and inhibition coupled with asynchrony generating episodic periods of abnormal activity (Fritschy, 2008 ) and can be caused by neurological channelopathies with mutations in voltage-gated (N+, K+, Ca2+ and Cl−) and ligand-gated (nicotinic acetylcholine and GABAAR) channels (Kang and Macdonald, 2009 ; Reid et al., 2009 ; Kullmann, 2010 ). Furthermore, epilepsy is often strikingly associated with other neurodevelopmental disorders (Figure 1 ). Epilepsy develops in 60–70% of RTT patients (Huppke et al., 2007 ; Moser et al., 2007 ), and abnormal electroencephalographs (EEGs) are observed in 60% of autistic children with 30% having recurrent seizures (Tuchman and Rapin, 2002 ; Tuchman et al., 2009 ; Coury, 2010 ). Likewise, altered EEGs are observed commonly in FXS children, and 10–20% display benign focal epilepsy with childhood seizures, typically remitting in late adolescence (Musumeci et al., 1999 ; Sabaratnam et al., 2001 ; Berry-Kravis, 2002 ; Qiu et al., 2008 ; Hagerman and Stafstrom, 2009 ). This remission prevalence and the incidence of other childhood epilepsies that wane with age are indicative of higher epileptogenic susceptibility to E/I imbalance in the immature brain (Rakhade and Jensen, 2009 ). During development, homeostatic mechanisms may not be effectively engaged, or may require a substantial temporal window to effect the functional compensation necessary to offset alterations in activity. However, resolution is not a consistent feature of epileptic disease progressions, indicating an insufficiency and/or failure of long-range homeostatic influence under certain circumstances.
The clear propensity of E/I imbalance involvement in epilepsy obviates the need to explore and define this in effective epilepsy model systems (Fritschy, 2008 ). In rodents, experimental cortical dysplasia induced via in utero irradiation mirrors altered cortical abnormalities seen in intractable epilepsy (Roper, 1998 ; Porter et al., 2003 ). Initial findings showed reduced inhibitory synaptic currents in pyramidal cells (Zhu and Roper, 2000 ) and reduced density of inhibitory interneurons (Roper et al., 1999 ) with depressed excitatory drive (Xiang et al., 2006 ). A more recent study specifically assessed the inhibitory synaptic activity in fast-spiking (FS) interneurons in these animals, showing a reduction in frequency, though not amplitude, of spontaneous IPSCs (sIPSCs) and aberrant short-term facilitation upon evocation (Zhou et al., 2009 ). Moreover, simultaneously monitoring excitatory and inhibitory postsynaptic currents revealed an E/I shift favoring inhibition in these interneurons, which typically serve to temper activity, thus leading to a net circuit effect of unchecked excitation. Altered E/I parameters are also seen upon modeling temporal lobe epilepsy, which is characterized by neuron loss in the hippocampal dentate gyrus (Dudek and Sutula, 2007 ). A recent study of the pilocarpine-elicited status epilepticus model revealed a preliminary loss of GABAergic neurons and decrement of inhibitory synapses to minimally 70% of control levels with concomitant excitatory synaptic loss onto the granule cells (Thind et al., 2010 ). Excitatory synapses were reported to later reestablish normally, whereas the surviving interneurons initiate robust synaptogenesis and supersede control levels by 120–150%. However, it was previously shown that disrupted chloride regulation, via loss of Cl− extrusive function in the K+/Cl− KCC2 co-transporter, leads to a reduction in inhibition efficacy and enhanced granule cell excitability (Pathak et al., 2007 ). These results suggest that the E/I quantitative ratio is not always solely indicative of the exerted circuit influence, and coordinate functional determinations remain imperative.
Rett Syndrome
Rett syndrome (RTT) is a relatively rare (1:10,000) but insidious, progressive X-linked neurodevelopmental disease that is a leading cause of intellectual disability in females (Glaze, 2002 ; Percy and Lane, 2005 ; Segawa and Nomura, 2005 ; Chahrour and Zoghbi, 2007 ; Monteggia and Kavalali, 2009 ). Development appears to progress normally in RTT children until 6–18 months of age, when symptomatic presentation begins. The disorder is then associated with developmental reversion involving loss of acquired proficiencies including expressivity and motor skills, continuing cognitive impairment, autistic behaviors and seizures (Figure 1 ). The most common etiology is de novo mutation in the transcriptional regulator, methyl-CpG-binding protein 2 (MECP2) (Amir et al., 1999 ). MECP2 disruptions account for up to 90% of RTT cases, although MECP2 duplication events (Shi et al., 2005 ; Van Esch et al., 2005 ; del Gaudio et al., 2006 ; Bunyan and Robinson, 2008 ; Smyk et al., 2008 ) and mutations in cyclin-dependent kinase-like 5 (CDKL5) (Weaving et al., 2004 ; Mari et al., 2005 ; Sprovieri et al., 2009 ) and the Forkhead family transcriptional repressor FOXG1 (Ariani et al., 2008 ; Jacob et al., 2009a ; Mencarelli et al., 2010 ) have also been implicated.
Rett syndrome has been successfully modeled in mice via deletion of all or the third exon of MECP2 (Chen et al., 2001 ; Guy et al., 2001 ; Shahbazian et al., 2002 ). Mutant animals display age-progressive motor impairment, tremors, increased anxiety and seizures. In agreement with analyses of post-mortem tissue samples (Belichenko et al., 1994 ; Chapleau et al., 2009 ), MECP2 mutants display a significant reduction in dendritic spine number (Belichenko et al., 2009 ; Chapleau et al., 2009 ), suggesting decreased excitatory synapse capacity. Electrophysiological examinations of cortical slices from the MECP2 KO mouse indicate a reduction in spontaneous pyramidal neuron activity resulting from a reduction in mEPSC amplitude as compared to maintained mIPSCs, thus favoring inhibition (Dani et al., 2005 ). Deficits in LTP upon MECP2 truncation have also been reported (Moretti et al., 2006 ); however, cortical LTP induction mechanisms appear to be retained in early-symptomatic MECP2 KO animals though with fewer and weaker connections, as revealed by a 50% reduction in connection probability and a 45% reduction in excitatory postsynaptic potential (EPSP) amplitude, respectively (Dani and Nelson, 2009 ). Recent in utero transfection of short hairpin RNA (shRNA) constructs directed against MECP2 support these findings with intracortical networks showing at least 30% reduction in excitatory synaptic input from cortical layers L3/5A (Wood et al., 2009 ). Glutamate uncaging coupled with laser scanning photostimulation likewise revealed a 40% reduced excitatory input to layer 2/3 neurons (Wood et al., 2009 ). In addition, impaired hippocampus-dependent spatial memory, contextual fear memory, social memory, and LTP arise with MECP2 truncation (Moretti et al., 2006 ). In contrast to cortical studies, hippocampal network examination of the MECP2 KO animals revealed a tendency toward hyperexcitability, as spontaneous inhibitory rhythmic activity was selectively reduced in frequency (Zhang et al., 2008 ). However, the MECP2 KO displays a 20% reduction in excitatory synapses in the stratum radiatum of hippocampal CA1 neurons in vivo, although this appears to resolve by 5 weeks of age (Chao et al., 2007 ). Thus, as in ASD, E/I imbalance in RTT may be circuit specific and temporally modulated.
Fragile X Syndrome
Fragile X syndrome (FXS) is the leading monogenic heritable cause of intellectual disability and ASD, conservatively affecting 1:4,000 males and 1:8,000 females (O’Donnell and Warren, 2002 ; Penagarikano et al., 2007 ; Cornish et al., 2008 ; Garber et al., 2008 ), although recent estimates place the full mutation frequency as high as 1:2,500 in the general population (Hagerman, 2008a ). Clinical FXS symptoms include delayed and depressed developmental trajectories (Bailey et al., 2001a ,b ; Hall et al., 2008 ), short-term working memory deficits (Munir et al., 2000 ; Cornish et al., 2001 ; Kwon et al., 2001 ; Lanfranchi et al., 2009 ), disordered sleep (Gould et al., 2000 ; Miano et al., 2008 ), seizures (Kluger et al., 1996 ; Musumeci et al., 1999 ; Singh et al., 1999 ; Sabaratnam et al., 2001 ; Berry-Kravis, 2002 ; Di Bonaventura et al., 2006 ; Qiu et al., 2008 ), elevated anxiety (Tsiouris and Brown, 2004 ), and attention-deficit hyperactivity disorder (Hatton et al., 2002 ; Sullivan et al., 2006 ). Significantly, 30% of FXS patients are also diagnosed with autism (Figure 1 ) (Kau et al., 2004 ; Hagerman, 2008b ; Harris et al., 2008 ).
FXS is caused by the loss of fragile X mental retardation 1 (FMR1) gene function (Verkerk et al., 1991 ), most commonly due to unstable CGG-trinucleotide repeat expansion in the 5′ regulatory region that leads to hypermethylation and transcriptional silencing (Sutcliffe et al., 1992 ). The FMR1 product (FMRP) is an mRNA-binding, polysome-associated protein that typically acts as a negative regulator of protein translation (Laggerbauer et al., 2001 ; Li et al., 2001 ; Sung et al., 2003 ; Zalfa et al., 2003 ; Lu et al., 2004 ; Qin et al., 2005 ; Tessier and Broadie, 2008 ), facilitates mRNA trafficking (Dictenberg et al., 2008 ; Estes et al., 2008 ), and influences mRNA stability (Zalfa et al., 2005 ; Zhang et al., 2007 ). Murine and Drosophila FXS models allow mechanistic examinations into the disease (Bakker et al., 1994 ; Zhang et al., 2001 ; Mientjes et al., 2006 ), and both models recapitulate numerous clinical FXS features, displaying neuronal overgrowth, altered synaptogenesis and synaptic plasticity coupled with defective circadian rhythms, learning and memory (Bassell and Warren, 2008 ; Gatto and Broadie, 2009b ; Mercaldo et al., 2009 ; Pfeiffer and Huber, 2009 ).
In light of E/I balance, it is important to note that FMRP is activity-regulated. Downstream of glutamatergic neurotransmission, metabotropic glutamate receptor (mGluR)-mediated translational induction elevates FMRP levels (Weiler and Greenough, 1993 ; Weiler et al., 1997 ). Moreover, peak endogenous FMRP levels in vivo correlate with postnatal periods of significant use-dependent synaptic refinement and later plasticity (Singh et al., 2007 ; Tessier and Broadie, 2008 ). As we have discussed previously (Gatto and Broadie, 2009b ; Tessier and Broadie, 2009 ), considerable work has shown that sensory stimulation also serves as an effective means to elicit activity-responsive FMRP enhancement and can be achieved via exposure to complex, stimulating environments and motor skill training (Irwin et al., 2000 , 2005 ). Levels of sensory stimulation directly impact FMRP levels and function. In rodents, whisker stimulation increases FMRP expression in synapse- and polysome-containing fractions (Todd and Mack, 2000 ) as does visual experience in dark-reared/light-exposed animals (Gabel et al., 2004 ). Consistently, in Drosophila, sensory deprivation yields depressed dFMRP levels, as does genetic disruption of visual or olfactory sensory modalities (Tessier and Broadie, 2008 ). Thus, activity positively regulates FMRP, and FMRP functions in activity-dependent synaptic mechanisms.
E/I Balance in FXS
The above studies suggest that conditions favoring an excitation-dominant E/I ratio elevate FMRP levels and function. Conversely, FMRP itself may impinge upon the balance of excitatory and inhibitory synapse formation during the genesis and maintenance of neural circuits (Figure 2 ). Post-mortem examinations in FXS patients reveal aberrant, supernumerary cortical dendritic spines, suggesting an architectural framework for elevated glutamatergic synaptogenesis (Rudelli et al., 1985 ; Hinton et al., 1991 ). However, as with the potential E/I ratio variability, FMRP function may be brain region specific. GluR1-containing α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPAR) and LTP are depressed in the mouse FXS model cortex (Li et al., 2002 ; Zhao et al., 2005 ; Desai et al., 2006 ; Wilson and Cox, 2007 ), whereas mGluR-mediated LTD is enhanced in the hippocampus (Huber et al., 2002 ; Nosyreva and Huber, 2006 ). Moreover, in conditioned hippocampal slices from FMR1 KO mice, synaptically released glutamate induced prolonged group I mGluR-mediated epileptic discharges, whereas induction of this type of activity in wild-type animals normally requires mGluR activation with (RS)-3,5-dihydroxyphenylglycine (DHPG). The induced epileptiform activity in the KO mouse could be blocked with translational inhibitors and suppressed with mGluR antagonists (Chuang et al., 2005 ). In Drosophila fmr1 (dfmr1) mutants, electrophysiological studies have indicated increased glutamatergic vesicle fusion event frequency and enhanced exocytic vesicle cycling (Zhang et al., 2001 ; Gatto and Broadie, 2008 ). High frequency stimulation paradigms revealed multiple synaptic transmission events in response to discrete stimuli and periodic amplitude cycling during the high frequency challenge. These defects are partially rescued in dfmr1; DmGluRA double null mutants, which lack all mGluR-mediated signaling (Repicky and Broadie, 2008 ). Taken together, these studies indicate exaggerated mGluR signaling function in FXS models, establishing a hyperexcitable state (Figure 2 ).
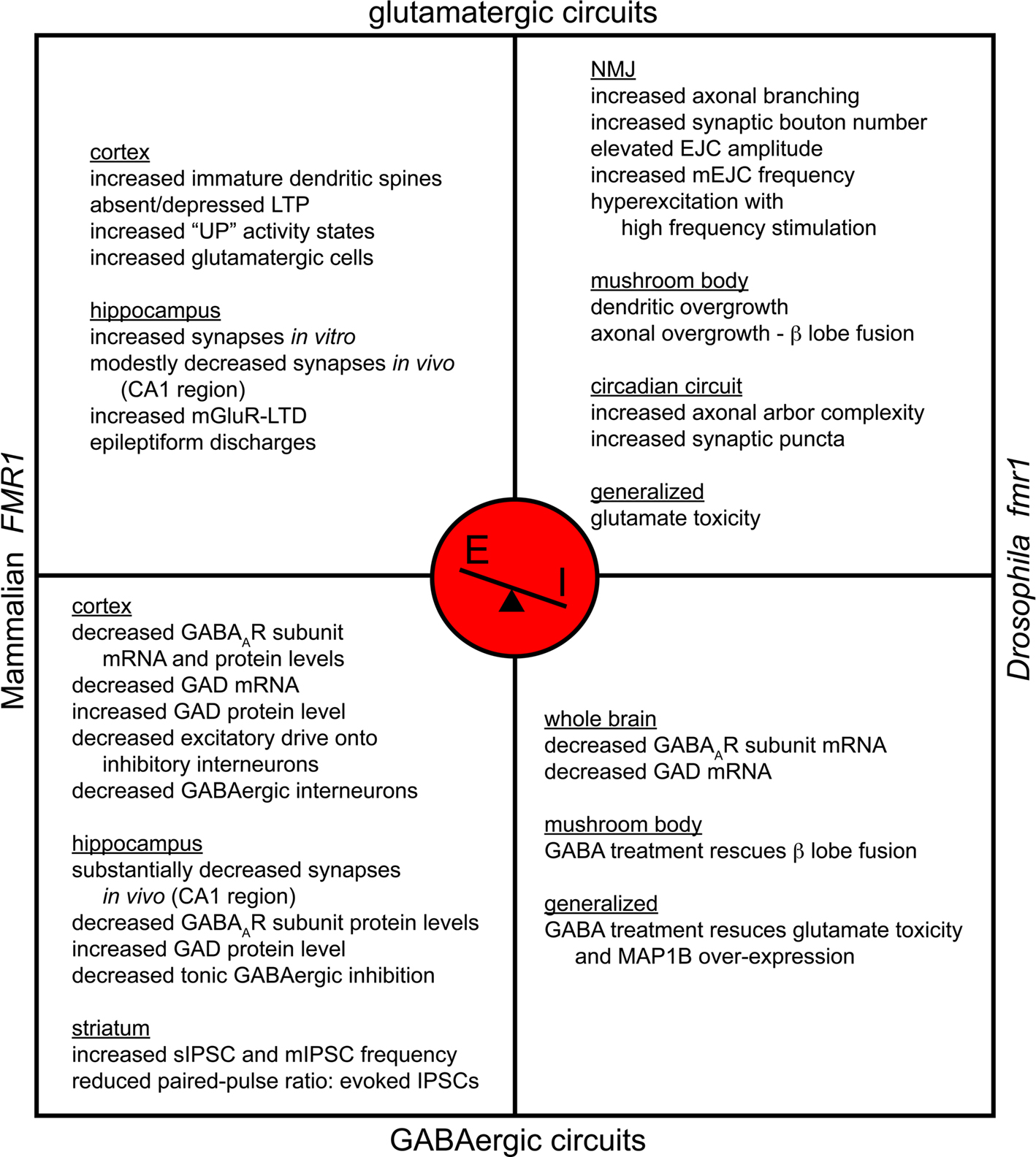
Figure 2. Fragile X syndrome is a disease of excitation-dominance. FXS models have been established via mutation of mouse and Drosophila FMR1. Both systems manifest a shift in synaptic E/I ratio with hyperexcitation favored. Model-specific alterations are diagrammed for glutamatergic and GABAergic circuits. Both models share the prevalence of increased numbers of excitatory synaptic boutons, elevated glutamatergic transmission, susceptibility to hyperexcitability and decreased GABAAR expression. FXS is thus characterized by glutamatergic elevation as well as GABAergic depression.
The FXS hyperexcitable state resulting from an E/I imbalance was recently demonstrated in acute neocortical slices isolated from the FMR1 KO mouse (Gibson et al., 2008 ). In layer 4 recordings, mutants showed ∼50% reduction in excitatory drive onto FS inhibitory neurons, with EPSC frequency and amplitude diminished due to compromised connectivity. In addition, there was a ∼20% decrease in excitatory drive onto excitatory neurons (Gibson et al., 2008 ). Together, these conditions foster an inhibition of inhibitory neurons within the circuit promoting an E/I balance in favor of elevated excitation. Moreover, the intrinsic excitability of the excitatory neurons was itself enhanced yielding a 2-fold increase in epochs of persistent activity, or ‘UP’ states, albeit with altered synchrony ascribed to the disrupted FS circuitry (Gibson et al., 2008 ). Consistently, the FMR1 KO may yield more cells that are responsive to mGluR activation (Castren et al., 2005 ) and increased neocortical glutamatergic cell differentiation (Tervonen et al., 2009 ), further predisposing the FXS state for hyperexcitation.
In addition to enhanced excitation, dysfunction in FXS neuronal circuits can also be attributed to more direct GABAergic involvement (Figure 2 ). In FMR1 KO animals, initial examination of subicular neuron function, located in the most inferior aspect of the hippocampus, revealed electrophysiological responses indicating decreased GABAergic inhibition (D’Antuono et al., 2003 ). Further functional characterization with patch-clamp recordings in FMR1 null organotypic slice cultures revealed a 91% decrease in current density for tonic, but not phasic, inhibition in subicular pyramidal cells, suggesting a defect in peri- and extra-synaptic GABA receptors (Curia et al., 2009 ). This tonic depression was associated with corollary α5 and δ GABAAR subunit under-expression, with mRNA levels depressed 26 and 35% by real-time RT-PCR and protein levels depressed 13 and 28% by Western blot, respectively (Curia et al., 2009 ). In the somatosensory neocortex, there was a 20% reduction in parvalbumin-reactive GABAergic interneurons, and GABAergic neurons were also decreased in Layers II/III/IV (Selby et al., 2007 ), suggesting aberrant local inhibitory contributions. These changes are clearly brain region specific and may be absent, or even opposite, in other brain regions. In fact, in light of frontostriatal circuit disruption reported in clinical FXS evaluations (Menon et al., 2004b ), striatal slices from the FMR1 KO reveal abnormal GABA-mediated transmission in spiny neurons, although within this region inhibitory transmission was enhanced with an increased frequency of sIPSCs and mIPSCs (Centonze et al., 2008 ). Interestingly, this change was not due to increased GABAergic synapse number, as VGAT-reactive puncta were actually decreased, but rather an enhanced release probability as evidenced by the reduced paired-pulse ratio of evoked IPSCs (Centonze et al., 2008 ).
Mechanistic Theories Behind E/I imbalance in FXS
The ‘mGluR Theory of FXS’ places FMRP function downstream of mGluR activation at the synapse (Bear et al., 2004 ; Bear, 2005 ), based on observations that Group I mGluR1/5 signaling via Gq-mediated transduction stimulates synaptic translation via FMRP (Weiler and Greenough, 1993 ; Weiler et al., 1997 , 2004 ; Greenough et al., 2001 ; Job and Eberwine, 2001 ; Muddashetty et al., 2007 ; Westmark and Malter, 2007 ). In wild-type mice, mGluR-dependent LTD requires stimulus-responsive protein synthesis (Massey and Bashir, 2007 ), but FMR1 KO mice do not display mGluR-triggered polysome assembly or activity induced protein synthesis (Todd et al., 2003 ; Weiler et al., 2004 ; Muddashetty et al., 2007 ; Westmark and Malter, 2007 ; Park et al., 2008 ). The result is elevated mGluR-triggered LTD (Huber et al., 2002 ; Nosyreva and Huber, 2006 ), suggesting that FMRP usually contributes feedback by repressing LTD-enhancing elements. mGluR5 antagonists rescue many FMR1 KO defects, including audiogenic seizure susceptibility, open-field exploratory hyperactivity and prepulse startle inhibition (Yan et al., 2005 ; de Vrij et al., 2008 ). Similarly, genetic reduction of mGluR5 signaling (mGluR5/+ heterozygotes) in the FMR1 null background alleviates most defined neurological dysfunctions, including altered ocular dominance plasticity, cortical neuron dendritic spine density, increased basal protein synthesis, inhibitory avoidance extinction and audiogenic seizure sensitivity (Dolen et al., 2007 ).
Despite the clear importance of the mGluR signaling mechanism, it alone does not account for the full range of FXS abnormalities. More recent work has established links to fast cholinergic transmission and slow modulatory transmission mediated by biogenic amines (e.g. dopamine) and neuropeptides (Zhang et al., 2005 ; Wang et al., 2008 ). Examination of M1 muscarinic acetylcholine receptor (mAChR) activated-LTD revealed a protein synthesis-dependent mechanism and AMPAR internalization associated with elevated FMRP levels (Volk et al., 2007 ). mAChR-mediated LTD was enhanced in FMR1 KO mice, associated with misregulation of FMRP targets, i.e. elongation factor 1α (EF1α) and calcium/calmodulin-dependent kinase II (CaMKII), uncoupling LTD induction from requisite protein synthesis. Thus, Gq-coupled ACh and glutamate receptors both appear to mediate protein synthesis-dependent synaptic changes mechanistically involving FMRP. Moreover, female FMR1 KO mice display increased dopamine turnover in cortical regions, striatum and hippocampus (Gruss and Braun, 2004 ), although this is reported not to occur in KO males (Gruss and Braun, 2001 ). A proteomic approach employing 2D electrophoresis in dfmr1 mutants indicated elevations in phenylalanine hydroxylase and GTP cyclohydrolase, driving the upregulated biosynthesis of dopamine and serotonin (Zhang et al., 2005 ). Critically, as dopamine D1 receptors contribute to ionotropic glutamate AMPAR trafficking, D1 stimulation in FMR1 KO cultured prefrontal cortex neurons caused a 37% decrease in GluR1-containing AMPAR surface expression and compromised phosphorylation (Wang et al., 2008 ). Moreover, D1 receptor signaling was itself impaired, and the redistribution of its G protein-coupled receptor kinase 2 (GRK2) to the membrane was also observed, likely yielding D1 receptor hyperphosphorylation and disrupted function. The in vivo relevance of these studies was highlighted by finding that amphetamine introduction in the FMR1 KO mouse elevated dopamine release in the prefrontal cortex and improved object recognition (Ventura et al., 2004 ), and D1 agonist treatment reduced open-field hyperactivity (Wang et al., 2008 ). These results indicate that FMRP’s mode of action is more complex than being solely mGluR-reactionary and, in fact, suggest that FMRP serves more generally as part of a feedback loop linking neurotransmitter signaling via Gq-coupled receptors for a range of neurotransmitters and neuromodulators. This model is described as the ‘Gq Theory of FXS’.
Disruption of inhibitory GABAergic signaling is also a vitally important component of the disease state (Figure 2 ), as expressed by the ‘GABAAR Theory of FXS’ (D’Hulst and Kooy, 2007 , 2009 ). In addition to the functional defects in inhibitory signaling discussed above, GABAAR subunit mRNA and protein levels are depressed in both murine and Drosophila FXS models, suggesting that FMRP must contribute to the stability and/or translation of GABAAR transcripts (El Idrissi et al., 2005 ; D’Hulst et al., 2006 , 2009b ; Gantois et al., 2006 ). The Drosophila model further manifests reduced mRNA levels for the GABA synthesizing enzyme GAD (D’Hulst et al., 2009b ), and likewise, the FMR1 KO mouse has been reported to show decreased GAD mRNA in the cortex (D’Hulst et al., 2009b ). However, in sharp contrast, the FMR1 KO displays increased GAD protein expression where decreased GABAAR β subunits are detected (i.e. in cortex, hippocampus, diencephalon and brainstem), perhaps indicating a partial compensatory mechanism (El Idrissi et al., 2005 ). Most notably, GABA administration in the Drosophila FXS model reportedly blocks glutamate toxicity and rescues microtubule-associated protein 1B (MAP1B)/Futsch over-expression, neuronal overgrowth defects and courtship memory impairment (Chang et al., 2008 ). Thus, FXS is likely a disease of neuronal hyperexcitation, not only due to overactive Gq-signaling downstream of glutamate and other transmitters, but also due to hypoinhibition, due to reduced expression and function of GABAARs.
Genetic Mechanisms Regulating E/I Balance
Although homeostatic mechanisms and corrective programs may be in play to address synaptic stability, E/I imbalance remains as a prevalent underpinning in many types of neurological dysfunction. It is therefore critical to understand at what level the genetic and molecular regulation goes awry to compromise circuit integrity. In recent years, a growing number of genes have been identified as participating in sculpting and maintaining the delicate balance of excitatory and inhibitory synaptogenesis, in different animal models and in different classes of circuit. These factors range from transcriptional and translational regulators, to specific resident synaptic proteins; moreover, mutations in these same genes are often clinically linked to the manifestation of the neurodevelopmental diseases discussed above (Figure 3 ).
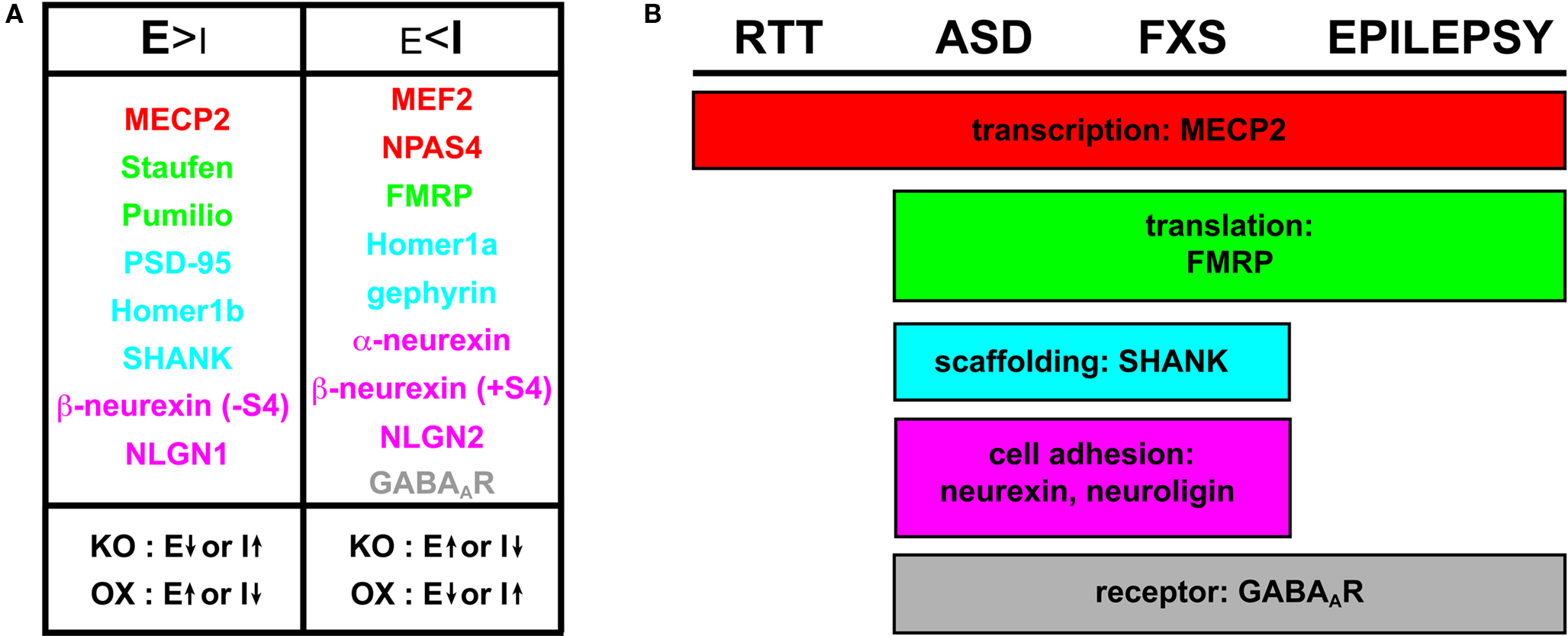
Figure 3. Molecular players influencing E/I ratio and commonalities in neurodevelopmental disorders. (A) Molecular regulators of E/I synaptic balance include transcriptional regulators (red), translational regulators (green), scaffolding proteins (blue), cell adhesion molecules (magenta) and receptors (gray). Components that promote excitatory synapse formation (E > I) are evident when knockout (KO) or knockdown yields depressed E or elevated I; likewise, over-expression (OX) of an excitation-favoring element yields elevated E or depressed I. With the converse class of regulators, inhibitory synapse formation is favored (E < I) when KO yields elevated E or depressed I, and OX yields depressed E or elevated I. (B) Regulators implicated in neurodevelopmental disease states illustrating the molecular overlap in RTT, ASD, FXS and epilepsy. The list includes players based on clinically implicated genetic influence and/or identification in neurodevelopmental model systems. As indicated in Figure 1 , robust inter-relationships suggest that several of these players are likely more widespread in their involvement, especially pertaining to epilepsy.
Transcriptional Regulators
To keep the synaptic E/I ratio balanced, transcriptional regulators likely provide a first line of defense (Figure 3 ). For example, MECP2 binds methylated DNA to mediate recruitment of a repressor complex including Sin3A and histone deacetylases; its mutation is the leading cause of RTT (Amir et al., 1999 ). Exemplifying the interconnectedness of neurodevelopmental dysfunction, MECP2 mRNA levels are also dramatically misregulated in FMR1 KO mice; decreased in hippocampus, cortex, diencephalon and brain stem, and conversely elevated in the cerebellum (Zhang et al., 2009 ). Reduced MECP2 expression is also noted in the frontal cortex in non-FXS autism (Nagarajan et al., 2006 ). To control E/I balance, MECP2 serves to promote excitatory synapse formation. MECP2 null neurons maintain a 40% reduction in VGLUT/PSD-95 puncta and a 46% reduction in EPSC amplitude, whereas transgenic 2-fold over-expression of MECP2 yields a 60% increase in synapse number with a 116% increase in EPSC amplitude (Chao et al., 2007 ). As a range of neurodevelopmental disorders display compromised MECP2 levels, and thus potentially favor depressed excitatory synapse formation and function, how these findings align with the network-specific hyperexcitability characteristic of RTT, FXS and autism remains a confounding issue.
The myocyte enhancer factor 2 (MEF2) family of transcription factors, initially implicated in muscle development (Buskin and Hauschka, 1989 ; Gossett et al., 1989 ), also regulates excitatory synapse number (Flavell et al., 2006 ; Barbosa et al., 2008 ). In contrast to MECP2, however, the MEF2 family serves a suppressive role. In cultured hippocampal neurons, RNAi-mediated knockdown of MEF2A and D caused a significant increase in the number of synapsin/PSD-95 reactive synapses and increased mEPSC frequency (Flavell et al., 2006 ). These effects are attributed to MEF2 promoting the transcription of targets that negatively regulate excitatory synapse development, including activity-regulated cytoskeletal-associated protein (Arc) and synaptic Ras guanosine triphosphate activating protein (synGAP), as indicated by real-time quantitative PCR and Western blotting (Flavell et al., 2006 ). MEF2-dependent negative regulation of excitatory synapse number and function was confirmed in vivo using a brain-specific deletion of MEF2C (Barbosa et al., 2008 ), although surprisingly, neurons formed normal synapses in culture. Behaviorally, MEF2C deletion significantly impaired hippocampal-dependent learning and memory (Barbosa et al., 2008 ). Thus, there is a correlation in this case between increased excitatory synapse number, potentiated synaptic transmission, and impaired behavioral output.
Inhibitory synapse development appears to be independently driven by a different dedicated set of transcription factors. The basic helix-loop-helix Per-ARNT-Sim (bHLH-PAS) family member Npas4 was identified in a screen for neuronal genes induced upon membrane depolarization via calcium influx (Lin et al., 2008 ). Target genes were selected based on excitatory activity being temporally coordinated with inhibitory synapse development. RNAi against Npas4 in vitro significantly reduced inhibitory synapse number, to less than 50% of control, whereas Npas-minigene over-expression doubled inhibitory synapse density (Lin et al., 2008 ). These studies showed no impact on excitatory synapse number, suggesting Npas4 activity is specifically driving inhibitory synapse formation. Functionally, deletion of Npas4 in organotypic slice cultures decreased inter-event intervals (increased frequency) of spontaneous mEPSCs, while over-expression increased their inter-event interval (decreased frequency) and decreased their amplitude (Lin et al., 2008 ). Thus, Npas4 selectively and positively regulates inhibitory synaptogenesis. Taken together, the above studies show that different transcription factors control excitatory and inhibitory synapse formation independently and, apparently, without a detectable level of E/I balance compensation.
Translational Regulators
Downstream of transcription, activity-dependent regulators of protein expression are prime candidates to modulate the E/I ratio. The importance of spatially restricted, local translation modulating functional synapse changes is well documented, particularly in mediating plasticity (Steward and Schuman, 2001 ; Sutton and Schuman, 2006 ; Bassell and Warren, 2008 ; Waung and Huber, 2009 ). The FMRP translational regulator perhaps represents the archetypal example. FMRP was initially proposed to bind ∼4% of human fetal brain mRNAs in vitro (Ashley et al., 1993 ), but thus far only a handful of in vivo targets have been validated. The best-supported FMRP targets include Arc, PSD-95, AMPAR subunits GluR1/2, GABAAR subunit δ, actin-binding profilin, MAPIB and CaMKII (Bassell and Warren, 2008 ; Gatto and Broadie, 2009b ). While much attention had been paid to the morphological phenotypes associated with FXS model neurons, including the excessive formation of dendritic filopodia/immature spines and supernumerary bouton formation (Comery et al., 1997 ; Nimchinsky et al., 2001 ; Zhang et al., 2001 ; Morales et al., 2002 ; Galvez et al., 2003 ; Michel et al., 2004 ; Pan et al., 2004 ; Gatto and Broadie, 2008 , 2009a ; Tessier and Broadie, 2008 ), relatively little information is available regarding any corollary alteration in E/I ratio at the level of synapse number. One recent study indicated FMR1-deficient neurons manifest 10% fewer excitatory synapses and 50% more inhibitory synapses in the hippocampus CA1 region (Dahlhaus et al., 2009 ). Importantly, FMRP re-introduction in a range of FXS model systems, from mammalian dissociated FMR1 null hippocampal cultures to the Drosophila dfmr1 mutant NMJ and circadian circuits (Pfeiffer and Huber, 2007 ; Gatto and Broadie, 2008 , 2009a ), has been shown to rescue synapse over elaboration by inducing synapse loss. These findings highlight the normal function of FMRP in restricting synapse number.
Numerous other translational regulators, many with direct or indirect interactions with FMRP, similarly regulate E/I balance in developing neuronal circuits, including Staufen and Pumilio (Figure 3 ). The double-strand RNA-binding protein Staufen has been implicated in mRNA localization, translational control and decay in dendrites important for long-term memory formation (Tang et al., 2001 ; Dubnau et al., 2003 ; Dugre-Brisson et al., 2005 ; Kim et al., 2005 ). Staufen co-purifies with FMRP in ribonucleoprotein complexes (Villace et al., 2004 ), which are dendritically recruited in an activity-dependent mechanism (Kim and Kim, 2006 ). More significantly, in Drosophila, dfmr1 and staufen (stau) interact to mediate long-term memory consolidation (Bolduc et al., 2008 ). Coincident reduction in double heterozygotes (stauD3/+; Fmr13/+) showed defective 1-day memory after spaced training in an olfactory conditioning paradigm. With respect to E/I balance, examination in cultured hippocampal neurons demonstrated that RNAi-mediated Staufen2 knockdown decreased the number of PSD-95-reactive synapses by ∼70% (Goetze et al., 2006 ). This postsynaptic effect was associated with a less marked ∼25% decrease in presynaptic synapsin puncta, indicating compromised excitatory connectivity. In addition to synapse loss, there was a significant reduction in the amplitude of recorded mEPSCs (Goetze et al., 2006 ). Similarly, downregulation of Staufen1 in hippocampal slice cultures decreased mEPSC frequency and amplitude (Lebeau et al., 2008 ). Interestingly, Staufen1 disruption caused structural changes resulting in elongated dendritic spines without altered density (Lebeau et al., 2008 ). Notably, as previously indicated, similar immature-type dendritic spines are a clinical hallmark in the cytological presentation of FXS (Rudelli et al., 1985 ; Hinton et al., 1991 ).
The mRNA-binding protein Pumilio has been shown to serve as a sequence-specific translational repressor and, like Staufen and FMRP, facilitates long-term memory formation (Dubnau et al., 2003 ; Bolduc et al., 2008 ). In fact, murine microarray screens have identified pumilio as an FMRP target (Brown et al., 2001 ). In Drosophila, Pumilio positively regulates synapse number while negatively regulating the GluRIIA glutamate receptor subunit (Menon et al., 2004a , 2009 ), and Pumilio regulates neuronal excitability by repressing the voltage-gated sodium channel, paralytic (Mee et al., 2004 ). Efforts to elucidate the synaptic components regulated by Drosophila Pumilio revealed a host of key partners including Discs-Large (DLG; PSD-95 homolog), Cysteine String Protein (CSP) and GAD (Chen et al., 2008 ). Most recently, RNAi-mediated knockdown of mammalian pumilio2 in cultured hippocampal neurons caused a reduction in mature dendritic spine number coupled with an elevation in elongated dendritic filopodia (Vessey et al., 2010 ). Despite this alteration in spine morphology, and contrary to results in the Drosophila studies, increased numbers of excitatory synapses presented on the dendritic shafts and were functionally associated with an increased mEJC frequency (Vessey et al., 2010 ). Despite some differences, it is clear that FMRP, Staufen, and Pumilio coordinately regulate synaptic mechanisms in both Drosophila and mouse systems. These three translational regulators possess the capacity to modulate relative expression of a range of synaptic constituents and provide an effective means of altering E/I ratios within neural circuits underlying memory formation and likely other behavioral outputs.
Scaffolding Proteins
Beneath the synaptic membrane, numerous scaffold/adaptor proteins provide positional anchoring, coupling membrane proteins to underlying cytoskeletal elements and signaling machinery. The composition of this scaffold network, and its preferential retention of specific membrane proteins, directly promotes the differential formation of excitatory versus inhibitory synapses (Figure 3 ). A prime example of such a key scaffolding element is the membrane-associated guanylate kinase (MAGUK) family member PSD-95 found exclusively at glutamatergic excitatory synapses (Kim and Sheng, 2004 ). Despite this apparent specificity, however, PSD-95 seems to exert influence upon both excitatory and inhibitory synapse classes (Levinson and El-Husseini, 2005b ; Han and Kim, 2008 ; Keith and El-Husseini, 2008 ). PSD-95 over-expression in primary hippocampal neuron cultures was shown to enhance excitatory synapse size and spontaneous firing frequency, while reducing the number of VGAT-reactive inhibitory contacts by 50% (Prange et al., 2004 ). In agreement, PSD-95 siRNA induced a 30% decrease in excitatory synaptic contacts and 1.5-fold increase in inhibitory synapses (Prange et al., 2004 ). With such dual capacity, it is important to reiterate that PSD-95 has been linked to FXS. FMRP binds the 3′-UTR of PSD-95 conferring enhanced message stability (Zalfa et al., 2007 ), which could perhaps contribute to elevated glutamatergic synapse formation. PSD-95 is also rapidly translated upon mGluR stimulation with DHPG, and this transient response is lost in FMRP deficient neurons (Todd et al., 2003 ; Muddashetty et al., 2007 ).
Other important scaffolding components in excitatory synapses include Homer and SH3 and ankyrin repeat containing-protein (SHANK) (Boeckers, 2006 ; Okabe, 2007 ). The neuronal immediate early gene Homer, with its Ena/VASP homology 1 (EVH1) and coiled-coil domains, dimerizes and serves to link mGluR, inositol 1,4,5-triphosphate receptor (IP3R) and N-methyl-D-aspartate (NMDA) receptor complexes (Brakeman et al., 1997 ; Kato et al., 1998 ; Xiao et al., 2000 ; Duncan et al., 2005 ). As a putative E/I modulator, Homer functions bidirectionally depending on its prevailing isoform. Homer1b has been shown to induce synaptic growth and maturation cooperatively with SHANK (Sala et al., 2001 ), whereas Homer1a, lacking the coiled-coil domain and potentially serving as an endogenous dominant negative incapable of dimerization, induces a 50% reduction in excitatory spine density (Sala et al., 2003 ). Homer1a over-expression also results in the redistribution of SHANK away from its synaptic clusters and a reduction in the size of PSD-95 and GKAP (a PSD-95/SHANK linker) clusters by 30% (Sala et al., 2003 ).
SHANK proteins are multi-modal adaptors that harbor ankyrin repeats, SH3, PDZ and proline rich motifs (Sheng and Kim, 2000 ). These scaffolds multimerize and couple to actin-binding proteins and mGluR through interaction with Homer (Naisbitt et al., 1999 ; Tu et al., 1999 ). Mutations in SHANK3 are linked to autism (Durand et al., 2007 ; Moessner et al., 2007 ). Importantly, SHANK1 is elevated in FMR1 KO mice at 2 weeks of age in the neocortex and in the hippocampus by 2 months, and SHANK3 is elevated in the FMR1-deficient neocortex at 2 months (Schutt et al., 2009 ). In vitro, SHANK is required for spine maintenance in hippocampal neurons and transgenic over-expression induces de novo functional spine formation in aspiny cerebellar granule cell cultures (Roussignol et al., 2005 ). In addition, the SHANK1 KO displays altered postsynaptic density composition with reduced GKAP and Homer1b/c levels, smaller dendritic spines and weaker basal synaptic transmission (Hung et al., 2008 ). Thus, in concert, Homer and SHANK appear to mediate both structural and functional synaptic alterations in establishing E/I balance.
Different scaffolds reside at inhibitory synapses. Gephyrin is a key tubulin-binding scaffold restricted to inhibitory synapses that establishes a hexagonal lattice serving to anchor both glycine and GABAA receptors (Kirsch et al., 1991 ; Schrader et al., 2004 ; Sola et al., 2004 ; Fritschy et al., 2008 ; Tretter et al., 2008 ); although, some controversy remains as to whether gephyrin is obligatory in mediating GABAAR clustering (Kneussel et al., 1999 ; Levi et al., 2004 ). Knockdown experiments with shRNAs and titration of endogenous gephyrin with an aggregating gephyrin-EGFP fusion in cultured hippocampal neurons yield depression of GABAAR clusters, without affecting the number of glutamatergic synapses (Yu et al., 2007 ; Yu and De Blas, 2008 ). This impairment was associated with the loss of ∼50% of GAD-positive GABAergic innervating boutons and corollary reduction in the sIPSC amplitude and frequency (Yu et al., 2007 ). Importantly, there was no disruption of whole-cell GABA currents, indicating the specificity of the effect is restricted to the synapse. These studies indicate a highly specific role of gephyrin in inhibitory synapses. However, perhaps due to opportunistic compensation, gephyrin reduction has been shown to increase the size of PSD-95 clusters and their apposing glutamatergic VGLUT1-reactive presynaptic termini (Yu and De Blas, 2008 ). This interaction again reveals the likely dynamic interplay between inhibitory and excitatory synapses in establishing the E/I ratio.
Cell Adhesion Molecules
Cell adhesion molecules mediating synaptic partner recognition and persistent trans-synaptic signaling are ideally situated to modulate excitatory and inhibitory synaptic formation and stabilization. In particular, presynaptic neurexin and postsynaptic neuroligin form a heterophilic adhesion complex possessing the capacity to drive E/I balance (Figure 3 ). These proteins present isoforms differentially expressed between excitatory and inhibitory synapse classes and have therefore been proposed to specify synapse identities (Levinson and El-Husseini, 2005a ,b ; Craig and Kang, 2007 ). The influence they exert may be substantial, although their roles remain somewhat elusive in vivo.
In mammals, the 3-gene neurexin family provides considerable diversity based on the formation of α and β isoforms, splice variants and post-translational glycosylation (Rowen et al., 2002 ; Tabuchi and Sudhof, 2002 ; Comoletti et al., 2003 ). Neurexin1 (α and β) disruptions are associated with autism (Feng et al., 2006 ; Kim et al., 2008 ; Yan et al., 2008 ), and deletion and copy number variants are also linked to schizophrenia (Kirov et al., 2008 ; Vrijenhoek et al., 2008 ; Rujescu et al., 2009 ). Importantly, a recent proteomic analysis revealed decreased neurexin1α in the FMR1 KO mouse (Liao et al., 2008 ). To modulate E/I balance, α isoforms typically associate with inhibitory synapses, while the more promiscuous β isoforms are present at both excitatory and inhibitory synapses (Craig and Kang, 2007 ). In fibroblast-neuronal co-culture studies, α-neurexins were able to cluster gephyrin, but not PSD-95, whereas β-neurexins could cluster gephyrin or PSD-95 (Graf et al., 2004 ; Kang et al., 2008 ). However, in β-neurexins, a particular splice variant (S4) shifts its capacity, such that its inclusion (+S4) favors GABAergic synapse development and its omission (−S4) favors glutamatergic synapses. Significantly, the developmental acquisition of an elevated −S4:+S4 utilization in β-neurexins was observed both in vivo (embryonic day 18 vs. postnatal day 18) and in vitro (embryonic day 18 cultures maintained for 7–22 days in vitro) (Kang et al., 2008 ). Despite these compelling findings, the in vivo significance of α-neurexins has been questioned as double- and triple-KO animals, viability issues aside, display only marginal alterations in synapse presentation (Missler et al., 2003 ; Dudanova et al., 2007 ). However, in Drosophila the sole neurexin gene, dnrx, does influence synapse formation in the central nervous system and at the glutamatergic NMJ (Li et al., 2007 ; Zeng et al., 2007 ). In the brain, dnrx disruption depressed levels of the active zone component Bruchpilot (Brp) and resulted in 50% fewer synapses identifiable by electron microscopy (Zeng et al., 2007 ). At the NMJ, dnrx nulls display shortened axonal branches and 40–60% fewer synaptic boutons with reduced Brp (Li et al., 2007 ; Sun et al., 2009 ; Zweier et al., 2009 ). Moreover, the pre- and post-synaptic engagement appeared compromised in dnrx mutants with evident membrane detachments (Li et al., 2007 ), and neurotransmission is defective as evidenced by decreased evoked excitatory junctional potential (EJP) amplitude and increased miniature EJP amplitude and frequency (Li et al., 2007 ; Sun et al., 2009 ). Thus, the Drosophila studies provide in vivo evidence for a neurexin role, at least in excitatory synapse development.
Mammalian neuroligins (NLGN) are a 4-member family, with mutation in NLGN3 (single amino acid substitution – R451C) and NLGN4 (frameshift resulting in premature termination) presenting in autism (Jamain et al., 2003 ; Laumonnier et al., 2004 ). Importantly, NLGN1/2 mRNAs interact with FMRP, and protein analysis revealed NLGN1 levels depressed in the hippocampus and cerebellum of FMR1 KO mice (Dahlhaus and El-Husseini, 2009 ). NLGN1 primarily localizes to excitatory synapses (Song et al., 1999 ), whereas NLGN2 resides at inhibitory synapses (Varoqueaux et al., 2004 ). However, in terms of influencing E/I ratio, neuronal transfection of hemagglutinin-tagged NLGN1 (HA-NLGN1) caused a 3-fold increase in VGAT-positive and a 1.5-fold increase in VGLUT-positive presynaptic contacts (Prange et al., 2004 ); this trend was also observed with NLGN2 and NLGN3 over-expression (Chih et al., 2005 ; Levinson et al., 2005 ). These results suggest that neuroligins possess more generalized synaptogenic properties, although notably, NLGN1 over-expression increased the density of PSD-95 and Homer containing puncta, and NLGN2 was more effective at inhibitory synaptic induction than either NLGN1 or NLGN3 (Chih et al., 2005 ). The influences that neuroligins exert on E/I balance are also complicated by their interactions with PSD-95. PSD-95 over-expression altered NLGN2 distribution, delivering it to excitatory synapses (Levinson et al., 2005 ). Functionally, NLGN1 over-expression enhanced excitatory synaptic transmission but not inhibitory, and the converse occurred with NLGN2 (Chubykin et al., 2007 ). In vivo NLGN1 over-expression increased the number of asymmetric synapses assayed by electron microscopy without influencing symmetric synapses, thereby apparently increasing the E/I ratio, despite elevations in gephyrin/VGAT as well as PSD-95/VGLUT (Dahlhaus et al., 2009 ). Conversely, in vivo NLGN2 over-expression decreased the E/I ratio by increasing symmetric synapse number (Hines et al., 2008 ). RNAi directed against NLGN1, 2 or 3, expressed singly or in combination, reduced excitatory synapse formation, with the triple knockdown yielding a 70% reduction (Chih et al., 2005 ). However, confoundingly, no corresponding alteration in mEPSCs was detected. Even more surprising, although excitatory and inhibitory transmission largely fail in the brainstem of the NLGN1-3 triple-KO mouse yielding perinatal respiratory arrest, the density of synaptic contacts was largely unaltered (Varoqueaux et al., 2006 ). These complications aside, it is of particular interest that transgenic over-expression of HA-NLGN1 in the FMR1-deficient background alters the E/I ratio. This transgenic intervention overcorrects the modestly depressed excitatory synapse prevalence in FMR1 nulls, shifting it from 10% less to 50% more than is seen in wild type. Conversely, introduction of HA-NLGN1 in the FMR1 KO mediates the reciprocal effect on inhibitory synapses, taking them from 50% more to 10% less as compared to control (Dahlhaus et al., 2009 ). Behaviorally, this transgenic intervention improves socialization and reduces hyperactivity in the mouse FXS model, but provides no remediation of memory deficits.
Channels/Receptors
By directly mediating membrane ion flux, synaptic channels and receptors are clearly positioned to shift the functional E/I balance (Figure 3 ). For example, GABAAR is a heteropentameric ion channel permeant to Cl/−HCO3− ions, which is most prevalently comprised of 2α, 2β, and either δ or γ subunits (Tretter and Moss, 2008 ; D’Hulst et al., 2009a ). GABAARs have been linked to autism via a two-locus gene-gene effect involving α4 through interaction with β1 (Ma et al., 2005 ). Moreover, discrete abnormalities on chromosome 15 region q11–13, containing three GABAAR subunits (α5, β3, and γ3), are also linked to autism (Cook et al., 1998 ; Shao et al., 2003 ). In these disease conditions, significant GABAAR subunit reductions were detected in parietal cortex, cerebellum and superior frontal cortex (Fatemi et al., 2009 , 2010 ). Moreover, the maternal GABAAR β3 signal peptide variant P11S, associated with 15q11–q13 duplication, has been implicated in both childhood absence epilepsy and autism owing to reduced functionality of receptors containing the β3 subunit (Delahanty et al., 2009 ). Importantly, GABAAR abundance was also reportedly decreased in FXS, at both the mRNA and protein level (El Idrissi et al., 2005 ; D’Hulst et al., 2006 , 2009b ; Gantois et al., 2006 ), and α5 or δ GABAAR KO mice display induced epileptiform hyperexcitability due to reduced tonic inhibition (Spigelman et al., 2002 ; Glykys and Mody, 2006 ).
Several lines of evidence implicate a role for the GABAAR itself in directly influencing synaptogenesis. Studies of the GABAAR γ2 subunit KO mouse indicated that the subunit was necessary for the clustering and retention of GABAARs (Essrich et al., 1998 ; Schweizer et al., 2003 ). γ2 shRNA in vitro and in vivo demonstrate the subunit was also required to establish normal postsynaptic gephyrin accumulation, and its disruption lead to the depression of incoming presynaptic GABAergic innervation (Li et al., 2005 ). In addition, these receptors typically show significant and constitutive turnover; as such, impairing receptor subunit binding to the clathrin adaptor protein-2 elevated GABAAR retention at the membrane. By over-expressing an altered β3 subunit (β3S408/9A) in cultured hippocampal neurons, this mechanism increased the number of inhibitory synapses formed to 144% of control values and enhanced inhibitory synaptic transmission by elevating mIPSC amplitudes (Jacob et al., 2009b ). Moreover, the elevated GABAAR presentation impaired dendritic spine maturity and shifted the immature filopodia versus mature mushroom-shaped spine ratio from 1:1 in controls to 2:1 in β3S408/9A-expressing neurons and decreased the density of PSD-95 clusters by 40% (Jacob et al., 2009b ). These recent findings again highlight the interdependence of excitatory and inhibitory synaptogenesis and bolster the conclusion that synapse class-specific alterations are not mutually exclusive in their synapse class effects.
Concluding Remarks
Proper brain function requires a delicate balance of excitatory and inhibitory synaptogenesis during neural circuit establishment and employment. Examination of ASD, epilepsy, RTT, and FXS demonstrate a high incidence of non-compensated disruption of E/I synapse balance. This suggests that disease-related homeostatic insufficiency and/or failure of compensatory mechanisms impair the development, connectivity and transmissive properties of neural circuits. E/I ratio imbalance may often be attributed to mutations in genes that facilitate synaptogenesis and/or synaptic maintenance through transcriptional control, translational control, structural scaffolding, cell adhesion and signaling. Defining the cross-talk between these levels will be vital to further understanding their contribution to the processes that differentially modulate each class of synapse. Crucial to the advancement of this work is identification of genetic targets regulated at the transcriptional level by factors such as MECP2, MEF2 and Npas4. Parallel examination of elevated and/or depressed targets should allow identification of key E/I switches. Likewise, the continuing definition of mRNA-binding proteins such as FMRP, Staufen and Pumilio, and their interactors, will be vital in determining how differential protein translation preferentially subserves different synaptic classes. Finally, much progress has been made in elucidating the functional E/I shift toward hyperexcitation in FXS, with robust examination of both the Gq and GABAAR theories. While each of these views of FXS provides insight into the E/I imbalance that contributes to the disease state, a number of key questions remain. Are hyperexcitation and/or hypoinhibition apparent in all circuits, or are these features circuit specific? How do the alterations in excitation and inhibition intersect; are they fully independent, or is there evidence of interacting compensation? Do transmissive changes act in concert, or is there mutual exclusivity? Can developmentally specific timing be assigned to these circuit alterations? Are the E/I shifts maintained? Perhaps most importantly from a clinical perspective, could combinatorial therapies targeting both glutamatergic and GABAergic systems in FXS prove most beneficial? Answers to these questions will help in understanding the mechanistic bases of disorders that affect brain development and should provide direct avenues to the most effective disease interventions.
Conflict of Interest Statement
The authors declare that this body of work was completed in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the Broadie Lab, especially Neil Dani and Emma Rushton, for insightful discussions during manuscript preparation. This work was supported by a FRAXA Research Foundation postdoctoral fellowship to Cheryl L. Gatto and R01 grant MH084989 to Kendal Broadie.
References
Akerman, C. J., and Cline, H. T. (2007). Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 30, 382–389.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Ariani, F., Hayek, G., Rondinella, D., Artuso, R., Mencarelli, M. A., Spanhol-Rosseto, A., Pollazzon, M., Buoni, S., Spiga, O., Ricciardi, S., Meloni, I., Longo, I., Mari, F., Broccoli, V., Zappella, M., and Renieri, A. (2008). FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 83, 89–93.
Ashley, C. T. Jr., Wilkinson, K. D., Reines, D., and Warren, S. T. (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566.
Bailey, D. B. Jr., Hatton, D. D., Skinner, M., and Mesibov, G. (2001a). Autistic behavior, FMR1 protein, and developmental trajectories in young males with fragile X syndrome. J. Autism Dev. Disord. 31, 165–174.
Bailey, D. B. Jr., Hatton, D. D., Tassone, F., Skinner, M., and Taylor, A. K. (2001b). Variability in FMRP and early development in males with fragile X syndrome. Am. J. Ment. Retard. 106, 16–27.
Bakker, C., Verheij, C., Willemsen, R., and van der Helm, R. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33.
Barbaro, J., and Dissanayake, C. (2009). Autism spectrum disorders in infancy and toddlerhood: a review of the evidence on early signs, early identification tools, and early diagnosis. J. Dev. Behav. Pediatr. 30, 447–459.
Barbosa, A. C., Kim, M. S., Ertunc, M., Adachi, M., Nelson, E. D., McAnally, J., Richardson, J. A., Kavalali, E. T., Monteggia, L. M., Bassel-Duby, R., and Olson, E. N. (2008). MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. U.S.A. 105, 9391–9396.
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214.
Bear, M. F. (2005). Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav. 4, 393–398.
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377.
Belichenko, P. V., Oldfors, A., Hagberg, B., and Dahlstrom, A. (1994). Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 5, 1509–1513.
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., and Francke, U. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258.
Ben-Ari, Y. (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat. rev. 3, 728–739.
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412.
Bolduc, F. V., Bell, K., Cox, H., Broadie, K. S., and Tully, T. (2008). Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat. Neurosci. 11, 1143–1145.
Brakeman, P. R., Lanahan, A. A., O’Brien, R., Roche, K., Barnes, C. A., Huganir, R. L., and Worley, P. F. (1997). Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386, 284–288.
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., Jin, X., Feng, Y., Wilkinson, K. D., Keene, J. D., Darnell, R. B., and Warren, S. T. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487.
Bunyan, D. J., and Robinson, D. O. (2008). Multiple de novo mutations in the MECP2 gene. Genet. Test. 12, 373–375.
Buskin, J. N., and Hauschka, S. D. (1989). Identification of a myocyte nuclear factor that binds to the muscle-specific enhancer of the mouse muscle creatine kinase gene. Mol. Cell. Biol. 9, 2627–2640.
Casanova, M. F. (2006). Neuropathological and genetic findings in autism: the significance of a putative minicolumnopathy. Neuroscientist 12, 435–441.
Casanova, M. F., Buxhoeveden, D., and Gomez, J. (2003). Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. Neuroscientist 9, 496–507.
Casanova, M. F., Buxhoeveden, D. P., Switala, A. E., and Roy, E. (2002). Minicolumnar pathology in autism. Neurology 58, 428–432.
Castren, M., Tervonen, T., Karkkainen, V., Heinonen, S., Castren, E., Larsson, K., Bakker, C. E., Oostra, B. A., and Akerman, K. (2005). Altered differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 17834–17839.
Centonze, D., Rossi, S., Mercaldo, V., Napoli, I., Ciotti, M. T., De Chiara, V., Musella, A., Prosperetti, C., Calabresi, P., Bernardi, G., and Bagni, C. (2008). Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 63, 963–973.
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437.
Chang, S., Bray, S. M., Li, Z., Zarnescu, D. C., He, C., Jin, P., and Warren, S. T. (2008). Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 4, 256–263.
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65.
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., Armstrong, D. L., Percy, A. K., and Pozzo-Miller, L. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233.
Chen, G., Li, W., Zhang, Q. S., Regulski, M., Sinha, N., Barditch, J., Tully, T., Krainer, A. R., Zhang, M. Q., and Dubnau, J. (2008). Identification of synaptic targets of Drosophila pumilio. PLoS Comput. Biol. 4, e1000026. doi:10.1371/journal.pcbi.1000026.
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Chih, B., Engelman, H., and Scheiffele, P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328.
Choonara, Y. E., Pillay, V., du Toit, L. C., Modi, G., Naidoo, D., Ndesendo, V. M., and Sibambo, S. R. (2009). Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int. J. Mol. Sci. 10, 2510–2557.
Chuang, S. C., Zhao, W., Bauchwitz, R., Yan, Q., Bianchi, R., and Wong, R. K. (2005). Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J. Neurosci. 25, 8048–8055.
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., and Sudhof, T. C. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931.
Colonnier, M. (1968). Synaptic patterns on different cell types in the different laminae of the cat visual cortex. An electron microscope study. Brain Res. 9, 268–287.
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., and Greenough, W. T. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404.
Comoletti, D., Flynn, R., Jennings, L. L., Chubykin, A., Matsumura, T., Hasegawa, H., Sudhof, T. C., and Taylor, P. (2003). Characterization of the interaction of a recombinant soluble neuroligin-1 with neurexin-1beta. J. Biol. Chem. 278, 50497–50505.
Cook, E. H. Jr., Courchesne, R. Y., Cox, N. J., Lord, C., Gonen, D., Guter, S. J., Lincoln, A., Nix, K., Haas, R., Leventhal, B. L., and Courchesne, E. (1998). Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am. J. Hum. Genet. 62, 1077–1083.
Cornish, K., Turk, J., and Hagerman, R. (2008). The fragile X continuum: new advances and perspectives. J. Intellect. Disabil. Res. 52, 469–482.
Cornish, K. M., Munir, F., and Cross, G. (2001). Differential impact of the FMR-1 full mutation on memory and attention functioning : a neuropsychological perspective. J. Cogn. Neurosci. 13, 144–150.
Craig, A. M., Graf, E. R., and Linhoff, M. W. (2006). How to build a central synapse: clues from cell culture. Trends Neurosci. 29, 8–20.
Craig, A. M., and Kang, Y. (2007). Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 17, 43–52.
Cummings, D. M., Andre, V. M., Uzgil, B. O., Gee, S. M., Fisher, Y. E., Cepeda, C., and Levine, M. S. (2009). Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J. Neurosci. 29, 10371–10386.
Curia, G., Papouin, T., Seguela, P., and Avoli, M. (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1515–1520.
D’Antuono, M., Merlo, D., and Avoli, M. (2003). Involvement of cholinergic and gabaergic systems in the fragile X knockout mice. Neuroscience 119, 9–13.
D’Hulst, C., Atack, J. R., and Kooy, R. F. (2009a). The complexity of the GABAA receptor shapes unique pharmacological profiles. Drug Discov. Today 14, 866–875.
D’Hulst, C., Heulens, I., Brouwer, J. R., Willemsen, R., De Geest, N., Reeve, S. P., De Deyn, P. P., Hassan, B. A., and Kooy, R. F. (2009b). Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 1253, 176–183.
D’Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., and Kooy, R. F. (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245.
D’Hulst, C., and Kooy, R. F. (2007). The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 30, 425–431.
D’Hulst, C., and Kooy, R. F. (2009). Fragile X syndrome: from molecular genetics to therapy. J. Med. Genet. 46, 577–584.
Dahlhaus, R., and El-Husseini, A. (2009). Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 208, 96–105.
Dahlhaus, R., Hines, R. M., Eadie, B. D., Kannangara, T. S., Hines, D. J., Brown, C. E., Christie, B. R., and El-Husseini, A. (2009). Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 20, 305–322.
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565.
Dani, V. S., and Nelson, S. B. (2009). Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J. Neurosci. 29, 11263–11270.
Davis, G. W. (2006). Homeostatic control of neural activity: from phenomenology to molecular design. Annu. Rev. Neurosci. 29, 307–323.
Delahanty, R. J., Kang, J. Q., Brune, C. W., Kistner, E. O., Courchesne, E., Cox, N. J., Cook, E. H. Jr., Macdonald, R. L., and Sutcliffe, J. S. (2009). Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol. Psychiatry. doi:10.1038/mp.2009.118. [Epub ahead of print].
del Gaudio, D., Fang, P., Scaglia, F., Ward, P. A., Craigen, W.J., Glaze, D. G., Neul, J. L., Patel, A., Lee, J. A., Irons, M., Berry, S. A., Pursley, A. A., Grebe, T. A., Freedenberg, D., Martin, R. A., Hsich, G. E., Khera, J. R., Friedman, N. R., Zoghbi, H. Y., Eng, C. M., Lupski, J. R., Beaudet, A. L., Cheung, S. W., and Roa, B. B. (2006). Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 8, 784–792.
Desai, N. S., Casimiro, T. M., Gruber, S. M., and Vanderklish, P. W. (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745.
de Vrij, F. M., Levenga, J., van der Linde, H. C., Koekkoek, S. K., De Zeeuw, C. I., Nelson, D. L., Oostra, B. A., and Willemsen, R. (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 31, 127–132.
Di Bonaventura, C., Mari, F., Pierallini, A., Mecarelli, O., Randi, F., Manfredi, M., Prencipe, M., and Giallonardo, A. T. (2006). Status epilepticus in a patient with fragile X syndrome: electro-clinical features and peri-ictal neuroimaging. Epileptic Disord. 8, 195–199.
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939.
Dolen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S., and Bear, M. F. (2007). Correction of Fragile X syndrome in mice. Neuron 56, 955–962.
Dubnau, J., Chiang, A. S., Grady, L., Barditch, J., Gossweiler, S., McNeil, J., Smith, P., Buldoc, F., Scott, R., Certa, U., Broger, C., and Tully, T. (2003). The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr. Biol. 13, 286–296.
Dudanova, I., Tabuchi, K., Rohlmann, A., Sudhof, T. C., and Missler, M. (2007). Deletion of alpha-neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J. Comp. Neurol. 502, 261–274.
Dudek, F. E., and Sutula, T. P. (2007). Epileptogenesis in the dentate gyrus: a critical perspective. Prog. Brain Res. 163, 755–773.
Dugre-Brisson, S., Elvira, G., Boulay, K., Chatel-Chaix, L., Mouland, A. J., and DesGroseillers, L. (2005). Interaction of Staufen1 with the 5’ end of mRNA facilitates translation of these RNAs. Nucleic Acids Res. 33, 4797–4812.
Duncan, R. S., Hwang, S. Y., and Koulen, P. (2005). Effects of Vesl/Homer proteins on intracellular signaling. Exp. Biol. Med. 30, 527–535.
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., Nygren, G., Rastam, M., Gillberg, I. C., Anckarsater, H., Sponheim, E., Goubran-Botros, H., Delorme, R., Chabane, N., Mouren-Simeoni, M. C., de Mas, P., Bieth, E., Roge, B., Heron, D., Burglen, L., Gillberg, C., Leboyer, M., and Bourgeron, T. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27.
El Idrissi, A., Ding, X. H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146.
Essrich, C., Lorez, M., Benson, J. A., Fritschy, J. M., and Luscher, B. (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1, 563–571.
Estes, P. S., O’Shea, M., Clasen, S., and Zarnescu, D. C. (2008). Fragile X protein controls the efficacy of mRNA transport in Drosophila neurons. Mol. Cell. Neurosci. 39, 170–179.
Fatemi, S. H., Halt, A. R., Stary, J. M., Kanodia, R., Schulz, S. C., and Realmuto, G. R. (2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810.
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and protein levels for GABA(A)alpha4, alpha5, beta1 and GABA (B)R1 receptors are altered in brains from subjects with autism. J. Autism Dev. Disord. 40, 743–750.
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., and Thuras, P. D. (2009). GABA(A) receptor downregulation in brains of subjects with autism. J. Autism Dev. Disord. 39, 223–230.
Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., Cook, E. H., Jr., Skinner, C., Schwartz, C. E., and Sommer, S. S. (2006). High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 409, 10–13.
Flavell, S. W., Cowan, C. W., Kim, T. K., Greer, P. L., Lin, Y., Paradis, S., Griffith, E. C., Hu, L. S., Chen, C., and Greenberg, M. E. (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012.
Fritschy, J. M. (2008). Epilepsy, E/I balance and GABA(A) receptor plasticity. Front. Mol. Neurosci. 1:5. doi:10.3389/neuro.02.005.2008.
Fritschy, J. M., Harvey, R. J., and Schwarz, G. (2008). Gephyrin: where do we stand, where do we go? Trends Neurosci. 31, 257–264.
Gabel, L. A., Won, S., Kawai, H., McKinney, M., Tartakoff, A. M., and Fallon, J. R. (2004). Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J. Neurosci. 24, 10579–10583.
Galvez, R., Gopal, A. R., and Greenough, W. T. (2003). Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 971, 83–89.
Gantois, I., Vandesompele, J., Speleman, F., Reyniers, E., D’Hooge, R., Severijnen, L. A., Willemsen, R., Tassone, F., and Kooy, R. F. (2006). Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis. 21, 346–357.
Garber, K. B., Visootsak, J., and Warren, S. T. (2008). Fragile X syndrome. Eur. J. Hum. Genet. 16, 666–672.
Gatto, C. L., and Broadie, K. (2008). Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135, 2637–2648.
Gatto, C. L., and Broadie, K. (2009a). Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front. Neural Circuits 3:8. doi:10.3389/neuro.04.008.2009.
Gatto, C. L., and Broadie, K. (2009b). The fragile X mental retardation protein in circadian rhythmicity and memory consolidation. Mol. Neurobiol. 39, 107–129.
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered up states reflect network hyperexcitability in the mouse model of Fragile X Syndrome. J. Neurophysiol. 100, 2615–2626.
Gillespie, D. C., Kim, G., and Kandler, K. (2005). Inhibitory synapses in the developing auditory system are glutamatergic. Nat. Neurosci. 8, 332–338.
Glaze, D. G. (2002). Neurophysiology of Rett syndrome. Ment. Retard Dev. Disabil. Res. Rev. 8, 66–71.
Glykys, J., and Mody, I. (2006). Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J. Neurophysiol. 95, 2796–2807.
Goetze, B., Tuebing, F., Xie, Y., Dorostkar, M. M., Thomas, S., Pehl, U., Boehm, S., Macchi, P., and Kiebler, M. A. (2006). The brain-specific double-stranded RNA-binding protein Staufen2 is required for dendritic spine morphogenesis. J. Cell Biol. 172, 221–231.
Gossett, L. A., Kelvin, D. J., Sternberg, E. A., and Olson, E. N. (1989). A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol. Cell. Biol. 9, 5022–5033.
Gould, E. L., Loesch, D. Z., Martin, M. J., Hagerman, R. J., Armstrong, S. M., and Huggins, R. M. (2000). Melatonin profiles and sleep characteristics in boys with fragile X syndrome: a preliminary study. Am. J. Med. Genet. 95, 307–315.
Graf, E. R., Zhang, X., Jin, S. X., Linhoff, M. W., and Craig, A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026.
Gray, E. G. (1959). Axo-somatic and axo-dendritic synapses of the cerebral cortex: an electron microscope study. J. Anat. 93, 420–433.
Greenough, W. T., Klintsova, A. Y., Irwin, S. A., Galvez, R., Bates, K. E., and Weiler, I. J. (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc. Natl. Acad. Sci. U.S.A. 98, 7101–7106.
Gruss, M., and Braun, K. (2001). Alterations of amino acids and monoamine metabolism in male Fmr1 knockout mice: a putative animal model of the human fragile X mental retardation syndrome. Neural Plast. 8, 285–298.
Gruss, M., and Braun, K. (2004). Age- and region-specific imbalances of basal amino acids and monoamine metabolism in limbic regions of female Fmr1 knock-out mice. Neurochem. Int. 45, 81–88.
Gulyas, A. I., Megias, M., Emri, Z., and Freund, T. F. (1999). Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J. Neurosci. 19, 10082–10097.
Gutierrez, R. C., Hung, J., Zhang, Y., Kertesz, A. C., Espina, F. J., and Colicos, M. A. (2009). Altered synchrony and connectivity in neuronal networks expressing an autism-related mutation of neuroligin 3. Neuroscience 162, 208–221.
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Hagerman, R. (2008b). Commonalities in the neurobiology between autism and fragile X. J. Intellect. Disabil. Res. 52, 817.
Hagerman, P. J., and Stafstrom, C. E. (2009). Origins of epilepsy in fragile x syndrome. Epilepsy Curr. 9, 108–112.
Hall, S. S., Burns, D. D., Lightbody, A. A., and Reiss, A. L. (2008). Longitudinal changes in intellectual development in children with Fragile X syndrome. J. Abnorm. Child. Psychol. 36, 927–939.
Harris, S. W., Hessl, D., Goodlin-Jones, B., Ferranti, J., Bacalman, S., Barbato, I., Tassone, F., Hagerman, P. J., Herman, H., and Hagerman, R. J. (2008). Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 113, 427–438.
Hatton, D. D., Hooper, S. R., Bailey, D. B., Skinner, M. L., Sullivan, K. M., and Wheeler, A. (2002). Problem behavior in boys with fragile X syndrome. Am. J. Med. Genet. 108, 105–116.
Hines, R. M., Wu, L., Hines, D. J., Steenland, H., Mansour, S., Dahlhaus, R., Singaraja, R. R., Cao, X., Sammler, E., Hormuzdi, S. G., Zhuo, M., and El-Husseini, A. (2008). Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J. Neurosci. 28, 6055–6067.
Hinton, V. J., Brown, W. T., Wisniewski, K., and Rudelli, R. D. (1991). Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 41, 289–294.
Hirtz, D., Thurman, D. J., Gwinn-Hardy, K., Mohamed, M., Chaudhuri, A. R., and Zalutsky, R. (2007). How common are the “common” neurologic disorders? Neurology 68, 326–337.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750.
Hung, A. Y., Futai, K., Sala, C., Valtschanoff, J. G., Ryu, J., Woodworth, M. A., Kidd, F. L., Sung, C. C., Miyakawa, T., Bear, M. F., Weinberg, R. J., and Sheng, M. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708.
Huppke, P., Kohler, K., Brockmann, K., Stettner, G. M., and Gartner, J. (2007). Treatment of epilepsy in Rett syndrome. Eur. J. Paediatr. Neurol. 11, 10–16.
Irwin, S. A., Christmon, C. A., Grossman, A. W., Galvez, R., Kim, S. H., Degrush, B. J., Weiler, I. J., and Greenough, W. T. (2005). Fragile X mental retardation protein levels increase following complex environment exposure in rat brain regions undergoing active synaptogenesis. Neurobiol. Learn. Mem. 83, 180–187.
Irwin, S. A., Swain, R. A., Christmon, C. A., Chakravarti, A., Weiler, I. J., and Greenough, W. T. (2000). Evidence for altered Fragile-X mental retardation protein expression in response to behavioral stimulation. Neurobiol. Learn. Mem. 73, 87–93.
Jacob, F. D., Ramaswamy, V., Andersen, J., and Bolduc, F. V. (2009a). Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur. J. Hum. Genet. 17, 1577–1581.
Jacob, T. C., Wan, Q., Vithlani, M., Saliba, R. S., Succol, F., Pangalos, M. N., and Moss, S. J. (2009b). GABA(A) receptor membrane trafficking regulates spine maturity. Proc. Natl. Acad. Sci. U.S.A. 106, 12500–12505.
Jacobs, M. P., Leblanc, G. G., Brooks-Kayal, A., Jensen, F. E., Lowenstein, D. H., Noebels, J. L., Spencer, D. D., and Swann, J. W. (2009). Curing epilepsy: progress and future directions. Epilepsy Behav. 14, 438–445.
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., Soderstrom, H., Giros, B., Leboyer, M., Gillberg, C., and Bourgeron, T. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29.
Job, C., and Eberwine, J. (2001). Identification of sites for exponential translation in living dendrites. Proc. Natl. Acad. Sci. U.S.A. 98, 13037–13042.
Kang, J. Q., and Macdonald, R. L. (2009). Making sense of nonsense GABA(A) receptor mutations associated with genetic epilepsies. Trends. Mol. Med. 15, 430–438.
Kang, Y., Zhang, X., Dobie, F., Wu, H., and Craig, A. M. (2008). Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J. Biol. Chem. 283, 2323–2334.
Kato, A., Ozawa, F., Saitoh, Y., Fukazawa, Y., Sugiyama, H., and Inokuchi, K. (1998). Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J. Biol. Chem. 273, 23969–23975.
Kau, A. S., Tierney, E., Bukelis, I., Stump, M. H., Kates, W. R., Trescher, W. H., and Kaufmann, W. E. (2004). Social behavior profile in young males with fragile X syndrome: characteristics and specificity. Am. J. Med. Genet. 126A, 9–17.
Kehrer, C., Maziashvili, N., Dugladze, T., and Gloveli, T. (2008). Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 1:6. doi:10.3389/neuro.02.006.2008.
Keith, D., and El-Husseini, A. (2008). Excitation control: balancing PSD-95 function at the synapse. Front. Mol. Neurosci. 1:4. doi:10.3389/neuro.02.004.2008.
Kilman, V., van Rossum, M. C., and Turrigiano, G. G. (2002). Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J. Neurosci. 22, 1328–1337.
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., Lally, E., Weiss, L. A., Najm, J., Kutsche, K., Descartes, M., Holt, L., Braddock, S., Troxell, R., Kaplan, L., Volkmar, F., Klin, A., Tsatsanis, K., Harris, D. J., Noens, I., Pauls, D. L., Daly, M. J., MacDonald, M. E., Morton, C. C., Quade, B. J., and Gusella, J. F. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207.
Kim, K. C., and Kim, H. K. (2006). Role of Staufen in dendritic mRNA transport and its modulation. Neurosci. Lett. 397, 48–52.
Kim, Y. K., Furic, L., Desgroseillers, L., and Maquat, L. E. (2005). Mammalian Staufen1 recruits Upf1 to specific mRNA 3’UTRs so as to elicit mRNA decay. Cell 120, 195–208.
Kirov, G., Gumus, D., Chen, W., Norton, N., Georgieva, L., Sari, M., O’Donovan, M. C., Erdogan, F., Owen, M. J., Ropers, H. H., and Ullmann, R. (2008). Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet. 17, 458–465.
Kirsch, J., Langosch, D., Prior, P., Littauer, U. Z., Schmitt, B., and Betz, H. (1991). The 93-kDa glycine receptor-associated protein binds to tubulin. J. Biol. Chem. 266, 22242–22245.
Kluger, G., Bohm, I., Laub, M. C., and Waldenmaier, C. (1996). Epilepsy and fragile X gene mutations. Pediatr. Neurol. 15, 358–360.
Kneussel, M., Brandstatter, J. H., Laube, B., Stahl, S., Muller, U., and Betz, H. (1999). Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J. Neurosci. 19, 9289–9297.
Kullmann, D. M. (2010). Neurological channelopathies. Annu. Rev. Neurosci. 33, [Epub ahead of print]
Kumar, R. A., and Christian, S. L. (2009). Genetics of autism spectrum disorders. Curr. Neurol. Neurosci. Rep. 9, 188–197.
Kwon, H., Menon, V., Eliez, S., Warsofsky, I. S., White, C. D., Dyer-Friedman, J., Taylor, A. K., Glover, G. H., and Reiss, A. L. (2001). Functional neuroanatomy of visuospatial working memory in fragile X syndrome: relation to behavioral and molecular measures. Am. J. Psychiatry 158, 1040–1051.
Laggerbauer, B., Ostareck, D., Keidel, E. M., Ostareck-Lederer, A., and Fischer, U. (2001). Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338.
Landa, R. J., Holman, K. C., and Garrett-Mayer, E. (2007). Social and communication development in toddlers with early and later diagnosis of autism spectrum disorders. Arch. Gen. Psychiatry 64, 853–864.
Lanfranchi, S., Cornoldi, C., Drigo, S., and Vianello, R. (2009). Working memory in individuals with Fragile X syndrome. Child Neuropsychol. 15, 105–119.
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., Raynaud, M., Ronce, N., Lemonnier, E., Calvas, P., Laudier, B., Chelly, J., Fryns, J. P., Ropers, H. H., Hamel, B. C., Andres, C., Barthelemy, C., Moraine, C., and Briault, S. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557.
Lebeau, G., Maher-Laporte, M., Topolnik, L., Laurent, C. E., Sossin, W., Desgroseillers, L., and Lacaille, J. C. (2008). Staufen1 regulation of protein synthesis-dependent long-term potentiation and synaptic function in hippocampal pyramidal cells. Mol. Cell. Biol. 28, 2896–2907.
Levi, S., Logan, S. M., Tovar, K. R., and Craig, A. M. (2004). Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J. Neurosci. 24, 207–217.
Levinson, J. N., Chery, N., Huang, K., Wong, T. P., Gerrow, K., Kang, R., Prange, O., Wang, Y. T., and El-Husseini, A. (2005). Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J. Biol. Chem. 280, 17312–17319.
Levinson, J. N., and El-Husseini, A. (2005a). Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron 48, 171–174.
Levinson, J. N., and El-Husseini, A. (2005b). New players tip the scales in the balance between excitatory and inhibitory synapses. Mol. Pain 1, 12.
Li, J., Ashley, J., Budnik, V., and Bhat, M. A. (2007). Crucial role of Drosophila neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron 55, 741–755.
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151.
Li, R. W., Yu, W., Christie, S., Miralles, C. P., Bai, J., Loturco, J. J., and De Blas, A. L. (2005). Disruption of postsynaptic GABA receptor clusters leads to decreased GABAergic innervation of pyramidal neurons. J. Neurochem. 95, 756–770.
Li, Z., Zhang, Y., Ku, L., Wilkinson, K. D., Warren, S. T., and Feng, Y. (2001). The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29, 2276–2283.
Liao, L., Park, S. K., Xu, T., Vanderklish, P., and Yates, J. R. III (2008). Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15281–15286.
Lin, Y., Bloodgood, B. L., Hauser, J. L., Lapan, A. D., Koon, A. C., Kim, T. K., Hu, L. S., Malik, A. N., and Greenberg, M. E. (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204.
Liu, G. (2004). Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat. Neurosci. 7, 373–379.
Liu, Y., Zhang, L. I., and Tao, H. W. (2007). Heterosynaptic scaling of developing GABAergic synapses: dependence on glutamatergic input and developmental stage. J. Neurosci. 27, 5301–5312.
Lohmann, C., and Bonhoeffer, T. (2008). A role for local calcium signaling in rapid synaptic partner selection by dendritic filopodia. Neuron 59, 253–260.
Lu, R., Wang, H., Liang, Z., Ku, L., O’Donnell, W, T., Li, W., Warren, S. T., and Feng, Y. (2004). The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc. Natl. Acad. Sci. U.S.A. 101, 15201–15206.
Ma, D. Q., Whitehead, P. L., Menold, M. M., Martin, E. R., Ashley-Koch, A. E., Mei, H., Ritchie, M. D., Delong, G. R., Abramson, R. K., Wright, H. H., Cuccaro, M. L., Hussman, J. P., Gilbert, J. R., and Pericak-Vance, M. A. (2005). Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am. J. Hum. Genet. 77, 377–388.
Maffei, A., and Fontanini, A. (2009). Network homeostasis: a matter of coordination. Curr. Opin. Neurobiol. 19, 168–173.
Mari, F., Azimonti, S., Bertani, I., Bolognese, F., Colombo, E., Caselli, R., Scala, E., Longo, I., Grosso, S., Pescucci, C., Ariani, F., Hayek, G., Balestri, P., Bergo, A., Badaracco, G., Zappella, M., Broccoli, V., Renieri, A., Kilstrup-Nielsen, C., and Landsberger, N. (2005). CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 14, 1935–1946.
Massey, P. V., and Bashir, Z. I. (2007). Long-term depression: multiple forms and implications for brain function. Trends Neurosci. 30, 176–184.
Mee, C. J., Pym, E. C., Moffat, K. G., and Baines, R. A. (2004). Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J. Neurosci. 24, 8695–8703.
Megias, M., Emri, Z., Freund, T. F., and Gulyas, A. I. (2001). Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102, 527–540.
Mencarelli, M. A., Spanhol-Rosseto, A., Artuso, R., Rondinella, D., De Filippis, R., Bahi-Buisson, N., Nectoux, J., Rubinsztajn, R., Bienvenu, T., Moncla, A., Chabrol, B., Villard, L., Krumina, Z., Armstrong, J., Roche, A., Pineda, M., Gak, E., Mari, F., Ariani, F., and Renieri, A. (2010). Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J. Med. Genet. 47, 49–53.
Menon, K. P., Andrews, S., Murthy, M., Gavis, E. R., and Zinn, K. (2009). The translational repressors Nanos and Pumilio have divergent effects on presynaptic terminal growth and postsynaptic glutamate receptor subunit composition. J. Neurosci. 29, 5558–5572.
Menon, K. P., Sanyal, S., Habara, Y., Sanchez, R., Wharton, R. P., Ramaswami, M., and Zinn, K. (2004a). The translational repressor Pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eIF-4E. Neuron 44, 663–676.
Menon, V., Leroux, J., White, C. D., and Reiss, A. L. (2004b). Frontostriatal deficits in fragile X syndrome: relation to FMR1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 101, 3615–3620.
Mercaldo, V., Descalzi, G., and Zhuo, M. (2009). Fragile X mental retardation protein in learning-related synaptic plasticity. Mol. Cells 28, 501–507.
Miano, S., Bruni, O., Elia, M., Scifo, L., Smerieri, A., Trovato, A., Verrillo, E., Terzano, M. G., and Ferri, R. (2008). Sleep phenotypes of intellectual disability: a polysomnographic evaluation in subjects with Down syndrome and Fragile-X syndrome. Clin. Neurophysiol. 119, 1242–1247.
Michel, C. I., Kraft, R., and Restifo, L. L. (2004). Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J. Neurosci. 24, 5798–5809.
Mientjes, E. J., Nieuwenhuizen, I., Kirkpatrick, L., Zu, T., Hoogeveen-Westerveld, M., Severijnen, L., Rife, M., Willemsen, R., Nelson, D. L., and Oostra, B. A. (2006). The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 21, 549–555.
Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R. E., Gottmann, K., and Sudhof, T. C. (2003). Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948.
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J., Zwaigenbaum, L., Fernandez, B., Roberts, W., Szatmari, P., and Scherer, S. W. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297.
Monteggia, L. M., and Kavalali, E. T. (2009). Rett syndrome and the impact of MeCP2 associated transcriptional mechanisms on neurotransmission. Biol. Psychiatry 65, 204–210.
Morales, J., Hiesinger, P. R., Schroeder, A. J., Kume, K., Verstreken, P., Jackson, F. R., Nelson, D. L., and Hassan, B. A. (2002). Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972.
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., Armstrong, D., Arancio, O., Sweatt, J. D., and Zoghbi, H. Y. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327.
Moser, S. J., Weber, P., and Lutschg, J. (2007). Rett syndrome: clinical and electrophysiologic aspects. Pediatr. Neurol. 36, 95–100.
Muddashetty, R. S., Kelic, S., Gross, C., Xu, M., and Bassell, G. J. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348.
Mulvihill, B, W. M., Kirby, R. S., Pettygrove, S., Cunniff, C., Meaney, F. J., Miller, L., Robinson, C., Quintana, G., Kaiser, M. Y., Lee, L. C., Landa, R., Newschaffer, C., Constantino, J., Fitzgerald, R., Daniels, J., Giarelli, E., Pinto-Martin, J., Levy, S. E., Charles, J., Nicholas, J., Durkin, M., Rice, C., Baio, J., Braun, K. V., Yeargin-Allsopp, M., Hepburn, M., Garner, N., Mancilla, K. C., Ratchford, A., Castillo, Y., Kolotos, M., Fitzgerald, R., Bell, P., Meade, R., King, L., Arneson, C., Washington, A., Graham, S., Lance, R., Grummon, N., Plummer, L., Jones, L., Wojcik, J., and Doernberg, N. (2009). Prevalence of autism spectrum disorders – Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill. Summ. 58, 1–20.
Munir, F., Cornish, K. M., and Wilding, J. (2000). Nature of the working memory deficit in fragile-X syndrome. Brain Cogn. 44, 387–401.
Musumeci, S. A., Hagerman, R. J., Ferri, R., Bosco, P., Dalla Bernardina, B., Tassinari, C. A., De Sarro, G. B., and Elia, M. (1999). Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40, 1092–1099.
Nagarajan, R. P., Hogart, A. R., Gwye, Y., Martin, M. R., and LaSalle, J. M. (2006). Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 1, e1–e11.
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., Weinberg, R. J., Worley, P. F., and Sheng, M. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582.
Nakayama, K., Kiyosue, K., and Taguchi, T. (2005). Diminished neuronal activity increases neuron-neuron connectivity underlying silent synapse formation and the rapid conversion of silent to functional synapses. J. Neurosci. 25, 4040–4051.
Nimchinsky, E. A., Oberlander, A. M., and Svoboda, K. (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146.
Noh, J., Seal, R. P., Garver, J. A., Edwards, R. H., and Kandler, K. (2010). Glutamate co-release at GABA/glycinergic synapses is crucial for the refinement of an inhibitory map. Nat. Neurosci. 13, 232–238.
Nosyreva, E. D., and Huber, K. M. (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of Fragile X Syndrome. J. Neurophysiol. 95, 3291–3295.
O’Donnell, W. T., and Warren, S. T. (2002). A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 25, 315–338.
O’Hare, A. (2009). Autism spectrum disorder: diagnosis and management. Arch. Dis. Child. 94, 161–168.
Pan, L., Zhang, Y. Q., Woodruff, E., and Broadie, K. (2004). The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 14, 1863–1870.
Park, S., Park, J. M., Kim, S., Kim, J. A., Shepherd, J. D., Smith-Hicks, C. L., Chowdhury, S., Kaufmann, W., Kuhl, D., Ryazanov, A. G., Huganir, R. L., Linden, D. J., and Worley, P. F. (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83.
Pathak, H. R., Weissinger, F., Terunuma, M., Carlson, G. C., Hsu, F. C., Moss, S. J., and Coulter, D. A. (2007). Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J. Neurosci. 27, 14012–14022.
Penagarikano, O., Mulle, J. G., and Warren, S. T. (2007). The pathophysiology of fragile x syndrome. Annu. Rev. Genomics Hum. Genet. 8, 109–129.
Percy, A. K., and Lane, J. B. (2005). Rett syndrome: model of neurodevelopmental disorders. J. Child Neurol. 20, 718–721.
Pfeiffer, B. E., and Huber, K. M. (2007). Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J. Neurosci. 27, 3120–3130.
Pfeiffer, B. E., and Huber, K. M. (2009). The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567.
Porter, B. E., Judkins, A. R., Clancy, R. R., Duhaime, A., Dlugos, D. J., and Golden, J. A. (2003). Dysplasia: a common finding in intractable pediatric temporal lobe epilepsy. Neurology 61, 365–368.
Prange, O., Wong, T. P., Gerrow, K., Wang, Y. T., and El-Husseini, A. (2004). A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. U.S.A. 101, 13915–13920.
Qin, M., Kang, J., Burlin, T. V., Jiang, C., and Smith, C. B. (2005). Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J. Neurosci. 25, 5087–5095.
Qiu, L. F., Hao, Y. H., Li, Q. Z., and Xiong, Z. Q. (2008). Fragile X syndrome and epilepsy. Neurosci. Bull. 24, 338–344.
Rakhade, S. N., and Jensen, F. E. (2009). Epileptogenesis in the immature brain: emerging mechanisms. Nat. Rev. Neurol. 5, 380–391.
Reid, C. A., Berkovic, S. F., and Petrou, S. (2009). Mechanisms of human inherited epilepsies. Prog. Neurobiol. 87, 41–57.
Repicky, S. E., and Broadie, K. (2008). Metabotropic glutamate receptor mediated use-dependent down-regulation of synaptic excitability involves the Fragile X mental retardation protein. J. Neurophysiol. 102, 672–687.
Rheims, S., Minlebaev, M., Ivanov, A., Represa, A., Khazipov, R., Holmes, G. L., Ben-Ari, Y., and Zilberter, Y. (2008). Excitatory GABA in rodent developing neocortex in vitro. J. Neurophysiol. 100, 609–619.
Rippon, G., Brock, J., Brown, C., and Boucher, J. (2007). Disordered connectivity in the autistic brain: challenges for the “new psychophysiology”. Int. J. Psychophysiol. 63, 164–172.
Rogers, S. J., Wehner, D. E., and Hagerman, R. (2001). The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 22, 409–417.
Roper, S. N. (1998). In utero irradiation of rats as a model of human cerebrocortical dysgenesis: a review. Epilepsy Res. 32, 63–74.
Roper, S. N., Eisenschenk, S., and King, M. A. (1999). Reduced density of parvalbumin- and calbindin D28-immunoreactive neurons in experimental cortical dysplasia. Epilepsy Res. 37, 63–71.
Roussignol, G., Ango, F., Romorini, S., Tu, J. C., Sala, C., Worley, P. F., Bockaert, J., and Fagni, L. (2005). Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 25, 3560–3570.
Rowen, L., Young, J., Birditt, B., Kaur, A., Madan, A., Philipps, D. L., Qin, S., Minx, P., Wilson, R. K., Hood, L., and Graveley, B. R. (2002). Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics 79, 587–597.
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267.
Rudelli, R. D., Brown, W. T., Wisniewski, K., Jenkins, E. C., Laure-Kamionowska, M., Connell, F., and Wisniewski, H. M. (1985). Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 67, 289–295.
Rujescu, D., Ingason, A., Cichon, S., Pietilainen, O. P., Barnes, M. R., Toulopoulou, T., Picchioni, M., Vassos, E., Ettinger, U., Bramon, E., Murray, R., Ruggeri, M., Tosato, S., Bonetto, C., Steinberg, S., Sigurdsson, E., Sigmundsson, T., Petursson, H., Gylfason, A., Olason, P. I., Hardarsson, G., Jonsdottir, G. A., Gustafsson, O., Fossdal, R., Giegling, I., Moller, H. J., Hartmann, A. M., Hoffmann, P., Crombie, C., Fraser, G., Walker, N., Lonnqvist, J., Suvisaari, J., Tuulio-Henriksson, A., Djurovic, S., Melle, I., Andreassen, O. A., Hansen, T., Werge, T., Kiemeney, L. A., Franke, B., Veltman, J., Buizer-Voskamp, J. E., Sabatti, C., Ophoff, R. A., Rietschel, M., Nothen, M. M., Stefansson, K., Peltonen, L., St Clair, D., Stefansson, H., and Collier, D. A. (2009). Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 18, 988–996.
Sabaratnam, M., Vroegop, P. G., and Gangadharan, S. K. (2001). Epilepsy and EEG findings in 18 males with fragile X syndrome. Seizure 10, 60–63.
Sala, C., Futai, K., Yamamoto, K., Worley, P. F., Hayashi, Y., and Sheng, M. (2003). Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J. Neurosci. 23, 6327–6337.
Sala, C., Piech, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130.
Schrader, N., Kim, E. Y., Winking, J., Paulukat, J., Schindelin, H., and Schwarz, G. (2004). Biochemical characterization of the high affinity binding between the glycine receptor and gephyrin. J. Biol. Chem. 279, 18733–18741.
Schutt, J., Falley, K., Richter, D., Kreienkamp, H. J., and Kindler, S. (2009). Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J. Biol. Chem. 284, 25479–25487.
Schweizer, C., Balsiger, S., Bluethmann, H., Mansuy, I. M., Fritschy, J. M., Mohler, H., and Luscher, B. (2003). The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 24, 442–450.
Selby, L., Zhang, C., and Sun, Q. Q. (2007). Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 412, 227–232.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., Armstrong, D., Paylor, R., and Zoghbi, H. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254.
Shao, Y., Cuccaro, M. L., Hauser, E. R., Raiford, K. L., Menold, M. M., Wolpert, C. M., Ravan, S. A., Elston, L., Decena, K., Donnelly, S. L., Abramson, R. K., Wright, H. H., DeLong, G. R., Gilbert, J. R., and Pericak-Vance, M. A. (2003). Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am. J. Hum. Genet. 72, 539–548.
Sheng, M., and Kim, E. (2000). The Shank family of scaffold proteins. J. Cell. Sci. 113(Pt 11), 1851–1856.
Shi, J., Shibayama, A., Liu, Q., Nguyen, V. Q., Feng, J., Santos, M., Temudo, T., Maciel, P., and Sommer, S. S. (2005). Detection of heterozygous deletions and duplications in the MECP2 gene in Rett syndrome by Robust Dosage PCR (RD-PCR). Hum. Mutat. 25, 505.
Singh, K., Gaur, P., and Prasad, S. (2007). Fragile x mental retardation (Fmr-1) gene expression is down regulated in brain of mice during aging. Mol. Biol. Rep. 34, 173–181.
Singh, R., Sutherland, G. R., and Manson, J. (1999). Partial seizures with focal epileptogenic electroencephalographic patterns in three related female patients with fragile-X syndrome. J. Child Neurol. 14, 108–112.
Smyk, M., Obersztyn, E., Nowakowska, B., Nawara, M., Cheung, S. W., Mazurczak, T., Stankiewicz, P., and Bocian, E. (2008). Different-sized duplications of Xq28, including MECP2, in three males with mental retardation, absent or delayed speech, and recurrent infections. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 799–806.
Sola, M., Bavro, V. N., Timmins, J., Franz, T., Ricard-Blum, S., Schoehn, G., Ruigrok, R. W., Paarmann, I., Saiyed, T., O’Sullivan, G. A., Schmitt, B., Betz, H., and Weissenhorn, W. (2004). Structural basis of dynamic glycine receptor clustering by gephyrin. EMBO J. 23, 2510–2519.
Song, J. Y., Ichtchenko, K., Sudhof, T. C., and Brose, N. (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. U.S.A. 96, 1100–1105.
Spigelman, I., Li, Z., Banerjee, P. K., Mihalek, R. M., Homanics, G. E., and Olsen, R. W. (2002). Behavior and physiology of mice lacking the GABAA-receptor delta subunit. Epilepsia 43(Suppl. 5), 3–8.
Sprovieri, T., Conforti, F. L., Fiumara, A., Mazzei, R., Ungaro, C., Citrigno, L., Muglia, M., Arena, A., and Quattrone, A. (2009). A novel mutation in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene associated with a severe Rett phenotype. Am. J. Med. Genet. 149A, 722–725.
Steward, O., and Schuman, E. M. (2001). Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 24, 299–325.
Sullivan, K., Hatton, D., Hammer, J., Sideris, J., Hooper, S., Ornstein, P., and Bailey, D. Jr. (2006). ADHD symptoms in children with FXS. Am. J. Med. Genet. 140, 2275–2288.
Sun, M., Liu, L., Zeng, X., Xu, M., Liu, L., Fang, M., and Xie, W. (2009). Genetic interaction between Neurexin and CAKI/CMG is important for synaptic function in Drosophila neuromuscular junction. Neurosci. Res. 64, 362–371.
Sung, Y. J., Dolzhanskaya, N., Nolin, S. L., Brown, T., Currie, J. R., and Denman, R. B. (2003). The fragile X mental retardation protein FMRP binds elongation factor 1A mRNA and negatively regulates its translation in vivo. J. Biol. Chem. 278, 15669–15678.
Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., and Warren, S. T. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400.
Sutton, M. A., and Schuman, E. M. (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127, 49–58.
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., and Sudhof, T. C. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76.
Tabuchi, K., and Sudhof, T. C. (2002). Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics 79, 849–859.
Tang, S. J., Meulemans, D., Vazquez, L., Colaco, N., and Schuman, E. (2001). A role for a rat homolog of staufen in the transport of RNA to neuronal dendrites. Neuron 32, 463–475.
Tervonen, T. A., Louhivuori, V., Sun, X., Hokkanen, M. E., Kratochwil, C. F., Zebryk, P., Castren, E., and Castren, M. L. (2009). Aberrant differentiation of glutamatergic cells in neocortex of mouse model for fragile X syndrome. Neurobiol. Dis. 33, 250–259.
Tessier, C. R., and Broadie, K. (2008). Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135, 1547–1557.
Tessier, C. R., and Broadie, K. (2009). Activity-dependent modulation of neural circuit synaptic connectivity. Front. Mol. Neurosci. 2:8. doi:10.3389/neuro.02.008.2009.
Thind, K. K., Yamawaki, R., Phanwar, I., Zhang, G., Wen, X., and Buckmaster, P. S. (2010). Initial loss but later excess of GABAergic synapses with dentate granule cells in a rat model of temporal lobe epilepsy. J. Comp. Neurol. 518, 647–667.
Todd, P. K., and Mack, K. J. (2000). Sensory stimulation increases cortical expression of the fragile X mental retardation protein in vivo. Brain Res. 80, 17–25.
Todd, P. K., Mack, K. J., and Malter, J. S. (2003). The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. U.S.A. 100, 14374–14378.
Tretter, V., Jacob, T. C., Mukherjee, J., Fritschy, J. M., Pangalos, M. N., and Moss, S. J. (2008). The clustering of GABA(A) receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J. Neurosci. 28, 1356–1365.
Tretter, V., and Moss, S. J. (2008). GABA(A) receptor dynamics and constructing GABAergic synapses. Front. Mol. Neurosci. 1:7. doi:10.3389/neuro.02.007.2008.
Tsiouris, J. A., and Brown, W. T. (2004). Neuropsychiatric symptoms of fragile X syndrome: pathophysiology and pharmacotherapy. CNS Drugs 18, 687–703.
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., Doan, A., Aakalu, V. K., Lanahan, A. A., Sheng, M., and Worley, P. F. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592.
Tuchman, R., Moshe, S. L., and Rapin, I. (2009). Convulsing toward the pathophysiology of autism. Brain Dev. 31, 95–103.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896.
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. rev. 5, 97–107.
Tyagarajan, S. K., and Fritschy, J. M. (2010). GABA(A) receptors, gephyrin and homeostatic synaptic plasticity. J. Physiol. (Lond.) 588, 101–106.
Van Esch, H., Bauters, M., Ignatius, J., Jansen, M., Raynaud, M., Hollanders, K., Lugtenberg, D., Bienvenu, T., Jensen, L. R., Gecz, J., Moraine, C., Marynen, P., Fryns, J. P., and Froyen, G. (2005). Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 77, 442–453.
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., Zhang, W., Sudhof, T. C., and Brose, N. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754.
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 83, 449–456.
Ventura, R., Pascucci, T., Catania, M. V., Musumeci, S. A., and Puglisi-Allegra, S. (2004). Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav. Pharmacol. 15, 433–442.
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., Reiner, O., Richards, S., Victoria, M. F., Zhang, F. P., Eussen, B. E., van Ommen, G-J.B., Blonden, L. A. J., Riggins, G. J., Chastain, J. L., Kunst, C. B., Galjaard, H., Caskey, C. T., Nelson, D. L., Oostra, B. A., and Warren, S. T. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914.
Vessey, J. P., Schoderboeck, L., Gingl, E., Luzi, E., Riefler, J., Di Leva, F., Karra, D., Thomas, S., Kiebler, M. A., and Macchi, P. (2010). Mammalian Pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc. Natl. Acad. Sci. U.S.A. 107, 3222–3227.
Villace, P., Marion, R. M., and Ortin, J. (2004). The composition of Staufen-containing RNA granules from human cells indicates their role in the regulated transport and translation of messenger RNAs. Nucleic Acids Res. 32, 2411–2420.
Volk, L. J., Pfeiffer, B. E., Gibson, J. R., and Huber, K. M. (2007). Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J. Neurosci. 27, 11624–11634.
Vrijenhoek, T., Buizer-Voskamp, J. E., van der Stelt, I., Strengman, E., Sabatti, C., Geurts van Kessel, A., Brunner, H. G., Ophoff, R. A., and Veltman, J. A. (2008). Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 83, 504–510.
Wang, H., Wu, L. J., Kim, S. S., Lee, F. J., Gong, B., Toyoda, H., Ren, M., Shang, Y. Z., Xu, H., Liu, F., Zhao, M. G., and Zhuo, M. (2008). FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 59, 634–647.
Wassef, A., Baker, J., and Kochan, L. D. (2003). GABA and schizophrenia: a review of basic science and clinical studies. J. Clin. Psychopharmacol. 23, 601–640.
Waung, M. W., and Huber, K. M. (2009). Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr. Opin. Neurobiol. 19, 319–326.
Weaving, L. S., Christodoulou, J., Williamson, S. L., Friend, K. L., McKenzie, O. L., Archer, H., Evans, J., Clarke, A., Pelka, G. J., Tam, P. P., Watson, C., Lahooti, H., Ellaway, C. J., Bennetts, B., Leonard, H., and Gecz, J. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 75, 1079–1093.
Weiler, I. J., and Greenough, W. T. (1993). Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc. Natl. Acad. Sci. U.S.A. 90, 7168–7171.
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., Comery, T. A., Patel, B., Eberwine, J., and Greenough, W. T. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400.
Weiler, I. J., Spangler, C. C., Klintsova, A. Y., Grossman, A .W., Kim, S. H., Bertaina-Anglade, V., Khaliq, H., de Vries, F. E., Lambers, F. A., Hatia, F., Base, C. K., and Greenough, W. T. (2004). Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc. Natl. Acad. Sci. U.S.A. 101, 17504–17509.
Westmark, C. J., and Malter, J. S. (2007). FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 5, e52. doi:10.1371/journal.pbio.0050052.
Wierenga, C. J., Becker, N., and Bonhoeffer, T. (2008). GABAergic synapses are formed without the involvement of dendritic protrusions. Nat. Neurosci. 11, 1044–1052.
Wilson, B. M., and Cox, C. L. (2007). Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc. Natl. Acad. Sci. U.S.A. 104, 2454–2459.
Wood, L., Gray, N. W., Zhou, Z., Greenberg, M. E., and Shepherd, G. M. (2009). Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J. Neurosci. 29, 12440–12448.
Wulffaert, J., Van Berckelaer-Onnes, I. A., and Scholte, E. M. (2009). Autistic disorder symptoms in Rett syndrome. Autism 13, 567–581.
Xiang, H., Chen, H. X., Yu, X. X., King, M. A., and Roper, S. N. (2006). Reduced excitatory drive in interneurons in an animal model of cortical dysplasia. J. Neurophysiol. 96, 569–578.
Xiao, B., Tu, J. C., and Worley, P. F. (2000). Homer: a link between neural activity and glutamate receptor function. Curr. Opin. Neurobiol. 10, 370–374.
Yan, J., Noltner, K., Feng, J., Li, W., Schroer, R., Skinner, C., Zeng, W., Schwartz, C. E., and Sommer, S. S. (2008). Neurexin 1alpha structural variants associated with autism. Neurosci. Lett. 438, 368–370.
Yan, Q. J., Rammal, M., Tranfaglia, M., and Bauchwitz, R. P. (2005). Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066.
Yu, W., and De Blas, A. L. (2008). Gephyrin expression and clustering affects the size of glutamatergic synaptic contacts. J. Neurochem. 104, 830–845.
Yu, W., Jiang, M., Miralles, C. P., Li, R. W., Chen, G., and de Blas, A. L. (2007). Gephyrin clustering is required for the stability of GABAergic synapses. Mol. Cell. Neurosci. 36, 484–500.
Zalfa, F., Adinolfi, S., Napoli, I., Kuhn-Holsken, E., Urlaub, H., Achsel, T., Pastore, A., and Bagni, C. (2005). Fragile X mental retardation protein (FMRP) binds specifically to the brain cytoplasmic RNAs BC1/BC200 via a novel RNA-binding motif. J. Biol. Chem. 280, 33403–33410.
Zalfa, F., Eleuteri, B., Dickson, K. S., Mercaldo, V., De Rubeis, S., di Penta, A., Tabolacci, E., Chiurazzi, P., Neri, G., Grant, S. G., and Bagni, C. (2007). A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci. 10, 578–587.
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., Oostra, B., and Bagni, C. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327.
Zeng, X., Sun, M., Liu, L., Chen, F., Wei, L., and Xie, W. (2007). Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett. 581, 2509–2516.
Zhang, A., Shen, C. H., Ma, S. Y., Ke, Y., and El Idrissi, A. (2009). Altered expression of Autism-associated genes in the brain of Fragile X mouse model. Biochem. Biophys. Res. Commun. 379, 920–923.
Zhang, L., He, J., Jugloff, D. G., and Eubanks, J. H. (2008). The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 18, 294–309.
Zhang, M., Wang, Q., and Huang, Y. (2007). Fragile X mental retardation protein FMRP and the RNA export factor NXF2 associate with and destabilize Nxf1 mRNA in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10057–10062.
Zhang, Y. Q., Bailey, A. M., Matthies, H. J., Renden, R. B., Smith, M. A., Speese, S. D., Rubin, G. M., and Broadie, K. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603.
Zhang, Y. Q., Friedman, D. B., Wang, Z., Woodruff, E., 3rd, Pan, L., O’Donnell, J., and Broadie, K. (2005). Protein expression profiling of the Drosophila fragile X mutant brain reveals up-regulation of monoamine synthesis. Mol. Cell Proteomics 4, 278–290.
Zhao, M. G., Toyoda, H., Ko, S. W., Ding, H. K., Wu, L. J., and Zhuo, M. (2005). Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 25, 7385–7392.
Zhou, F. W., Chen, H. X., and Roper, S. N. (2009). Balance of inhibitory and excitatory synaptic activity is altered in fast-spiking interneurons in experimental cortical dysplasia. J. Neurophysiol. 102, 2514–2525.
Zhu, W. J., and Roper, S. N. (2000). Reduced inhibition in an animal model of cortical dysplasia. J. Neurosci. 20, 8925–8931.
Zweier, C., de Jong, E. K., Zweier, M., Orrico, A., Ousager, L. B., Collins, A.L., Bijlsma, E. K., Oortveld, M. A., Ekici, A. B., Reis, A., Schenck, A., and Rauch, A. (2009). CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 85, 655–666.
Keywords: neurodevelopment, synapse, GABA, glutamate, excitation, inhibition, E/I ratio, Fragile X syndrome
Citation: Gatto CL and Broadie K (2010) Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Syn. Neurosci. 2:4. doi: 10.3389/fnsyn.2010.00004
Received: 01 March 2010;
Paper pending published: 12 April 2010;
Accepted: 14 May 2010;
Published online: 07 June 2010
Edited by:
Susana Cohen-Cory, University of California, USAReviewed by:
Randi Hagerman, UC Davis Medical Center, USAHollis Cline, The Scripps Research Institute, USA
Copyright: © 2010 Gatto and Broadie. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Kendal Broadie, Vanderbilt University, 6270 MRB III, 465 21st Avenue South, Nashville, TN 37232, USA. e-mail: kendal.broadie@vanderbilt.edu