Traumatic Brain Injury Upregulates Phosphodiesterase Expression in the Hippocampus
 Nicole M. Wilson
Nicole M. Wilson David J. Titus
David J. Titus Anthony A. Oliva Jr.
Anthony A. Oliva Jr. Concepcion Furones
Concepcion Furones Coleen M. Atkins
Coleen M. Atkins- The Miami Project to Cure Paralysis, Department of Neurological Surgery, University of Miami Miller School of Medicine, Miami, FL, USA
Traumatic brain injury (TBI) results in significant impairments in hippocampal synaptic plasticity. A molecule critically involved in hippocampal synaptic plasticity, 3′,5′-cyclic adenosine monophosphate, is downregulated in the hippocampus after TBI, but the mechanism that underlies this decrease is unknown. To address this question, we determined whether phosphodiesterase (PDE) expression in the hippocampus is altered by TBI. Young adult male Sprague Dawley rats received sham surgery or moderate parasagittal fluid-percussion brain injury. Animals were analyzed by western blotting for changes in PDE expression levels in the hippocampus. We found that PDE1A levels were significantly increased at 30 min, 1 h and 6 h after TBI. PDE4B2 and 4D2 were also significantly increased at 1, 6, and 24 h after TBI. Additionally, phosphorylation of PDE4A was significantly increased at 6 and 24 h after TBI. No significant changes were observed in levels of PDE1B, 1C, 3A, 8A, or 8B between 30 min to 7 days after TBI. To determine the spatial profile of these increases, we used immunohistochemistry and flow cytometry at 24 h after TBI. PDE1A and phospho-PDE4A localized to neuronal cell bodies. PDE4B2 was expressed in neuronal dendrites, microglia and infiltrating CD11b+ immune cells. PDE4D was predominantly found in microglia and infiltrating CD11b+ immune cells. To determine if inhibition of PDE4 would improve hippocampal synaptic plasticity deficits after TBI, we treated hippocampal slices with rolipram, a pan-PDE4 inhibitor. Rolipram partially rescued the depression in basal synaptic transmission and converted a decaying form of long-term potentiation (LTP) into long-lasting LTP. Overall, these results identify several possible PDE targets for reducing hippocampal synaptic plasticity deficits and improving cognitive function acutely after TBI.
Introduction
Every year in the United States an estimated 1.7 million people sustain a traumatic brain injury (TBI) and nearly 70–80% of those who survive report chronic learning and memory deficits (Lew et al., 2006; Faul et al., 2010). The hippocampus is critically involved in learning and memory and is highly susceptible to damage after TBI. Even when not directly damaged, hippocampal atrophy and neuronal loss are often observed in TBI patients (Bigler et al., 1997; Tomaiuolo et al., 2004). This progressive hippocampal atrophy is also observed in experimental models of TBI, and is accompanied by a loss of dentate hilar cells and neurons in the CA3 region as well as synaptic loss (Maxwell et al., 2003; Hall et al., 2005; Scheff et al., 2005; Witgen et al., 2005). In addition to these gross morphological changes, in experimental models of TBI there are deficits in hippocampal basal synaptic transmission and long-term potentiation (LTP; Miyazaki et al., 1992; Titus et al., 2013b). Finding molecular targets to attenuate the damage caused by TBI and improve hippocampal synaptic plasticity is of critical importance to assist the estimated 3–5 million people currently living with cognitive disabilities from TBI in the United States (Langlois et al., 2006; Zaloshnja et al., 2008).
Targeting TBI-induced LTP deficits is a promising strategy for improving learning and memory (Atkins, 2011). During hippocampal LTP and long-term memory formation, 3′,5′-cyclic adenosine monophosphate (cAMP) signaling is critical to activate the transcription factor cAMP-response element binding protein (CREB) and mediate gene transcription required for long-term memory formation (Frey et al., 1993; Bourtchuladze et al., 1994). We have found that in an experimental model of TBI, cAMP levels are depressed and CREB activation is impaired during a learning task (Titus et al., 2013a,b). The exact molecular mechanisms leading to the decrease in cAMP levels and impairments in CREB activation after TBI are unknown. cAMP is synthesized from ATP by adenylyl cyclases (ACs) and cAMP signaling is tightly regulated in discrete spatial-temporal microdomains through hydrolysis by phosphodiesterases (PDEs; Houslay, 2010). Whether cAMP signaling in the hippocampus is decreased after TBI by either changes in ACs or PDEs is unknown and this knowledge could guide the development of therapeutic strategies for treating the cognitive consequences of TBI.
One strategy for improving cAMP signaling is to inhibit PDEs. However, an important consideration is that the PDE superfamily is a large family of enzymes with 11 different PDE families (Bender and Beavo, 2006; Omori and Kotera, 2007). Within each of the PDE families there are multiple subfamilies encoded by individual genes, with several splice variants within each subfamily. Eight of the PDE families hydrolyze cAMP: PDEs 1, 2, 3, 10, and 11 hydrolyze both cAMP and cGMP, and PDEs 4, 7, and 8 hydrolyze only cAMP (Maurice et al., 2014). Of these PDEs, PDE1, 3, 4, 8, and 10 have been found in the hippocampus (Johansson et al., 2012). The PDE1 family consists of three subfamilies, PDE1A, 1B, and 1C, which are encoded by separate genes. PDE1 is commonly referred to as a Ca2+/calmodulin-stimulated PDE, and the activity of PDE1 can be enhanced through increases in calcium and calmodulin signaling (Omori and Kotera, 2007; Heckman et al., 2015). The PDE3 family consists of two subfamilies, PDE3A and 3B, which are encoded by two separate genes. While PDE3 hydrolyzes cAMP and cGMP, the ability for PDE3 to hydrolyze cAMP is inhibited in the presence of cGMP (Bender and Beavo, 2006). The PDE8 family is a cAMP-specific PDE that consists of two subfamilies, PDE8A and 8B (Martinez and Gil, 2014). The PDE10 family is another dual-specific PDE which is encoded by one gene, PDE10A (Kelly and Brandon, 2009). Given the wide array of inhibitors that target each of these families, it is important to understand how TBI affects the expression and activity of each these enzymes to target the relevant PDE (Menniti et al., 2006; Spina, 2008; Titus et al., 2015b).
Of the cAMP-degrading PDEs, the most notable one in the context of learning and memory is the PDE4 family (Li et al., 2011; Heckman et al., 2015). The PDE4 family is encoded by four genes, PDE4A, 4B, 4C, and 4D. With the exception of PDE4C, all of these subfamilies are expressed in the brain, immune system and cardiovascular system (Henkel-Tigges and Davis, 1990; Reneerkens et al., 2009). A unique feature of the PDE4 family is that they are further classified into four groups: long, short, super-short, and dead-short isoforms (Titus et al., 2015a). This classification is based on the presence, or absence, of upstream conserved regions (UCR) near the N-terminus, which allow for differential post-translational regulation of these isoforms (Hansen and Zhang, 2015). The PDE4A family has six isoforms: PDE4A1, 4A4/5, 4A7, 4A8, 4A10, and 4A11. The PDE4B family has five isoforms: PDE4B1-5. The PDE4D family has 10 isoforms: 4D1–7, 4D9, 4D10, and 4D11. Identifying the spatial and temporal expression pattern of these PDEs in the hippocampus after brain injury could facilitate the development of a more targeted approach for reducing TBI-induced cognitive deficits.
Targeting cAMP-degrading PDEs acutely after trauma has yielded promising results in experimental models of spinal cord injury and cerebral ischemia (Kato et al., 1995; Schaal et al., 2012). However, pan-PDE inhibitors have broad effects given the widespread expression of PDE isoforms. In the context of experimental TBI, the anti-inflammatory and neuroprotective effects of pan-PDE4 inhibitors are overshadowed by the vasodilation and hemorrhagic effects when administered acutely after trauma (Atkins et al., 2012, 2013). This emphasizes the need for more specific targets when treating the acute phase of TBI. In a previous study, we reported that TBI alters PDE expression in the ipsilateral parietal cortex acutely after trauma, but whether similar changes occur in the hippocampus is unknown (Oliva et al., 2012). Determining which PDEs are upregulated after trauma could provide more specific drug targets for reducing neuronal death, inflammation and atrophy in the hippocampus after TBI. In this study we investigated changes in PDE1, 3, 4, 8, and 10 after TBI using western blot analysis, immunohistochemistry and flow cytometry analysis of the hippocampus.
Materials and Methods
Fluid-Percussion Injury Surgery
One hundred adult male Sprague Dawley rats were used in this study (2–3 mos, 300–350 gm, Charles River Laboratories). All experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and with approval from the University of Miami Animal Care and Use Committee. Animals were anesthetized (3% isoflurane, 70% N2O, 30% O2, 5 min) and received a 4.8 mm diameter craniotomy at –3.8 mm posterior to bregma and 2.5 mm lateral to the midline over the right parietal cortex. A plastic female Luer Lock adapter (18 gage) was affixed at the craniotomy site with cyanoacrylate and dental cement. Animals were allowed to recover for 12–16 h while fasting with water ad libitum. Animals were re-anesthetized (induction for 5 min with 3% isoflurane, 70% N2O, 30% O2, maintenance during surgery with 1% isoflurane, 70% N2O, 30% O2), then intubated, mechanically ventilated (Stoelting) and given rocuronium (10 mg/kg, intra-arterial) and penicillin/benzathine (20,000 IU/kg, intramuscular). Head and body temperature were maintained between 36.6 and 37.2°C using rectal and temporalis muscle thermistors connected to feedback-regulated heating lamps. Physiological parameters (blood pO2 and pCO2, blood pH, and mean arterial blood pressure) were monitored via a tail artery catheter and maintained at normal levels throughout the surgery. Animals were prospectively randomized into sham or TBI surgery groups. Brain trauma was produced with a fluid-pulse (16 ms duration, 2.0 ± 0.2 atmospheric pressure) at the craniotomy site. Sham-operated animals received all surgical procedures identical to the TBI animals with the exception of the fluid-pulse. At the end of the surgery, animals received buprenorphine (0.01 mg/kg, subcutaneously). Criteria for exclusion from the study were: mortality, >15% loss of body weight, non-resolving infection at the surgical site, inability to feed or drink, motor paralysis, listlessness, self-mutilation, excessive grooming leading to loss of dermal layers, spontaneous vocalization when touched or poor grooming habits. No animals were removed from the study. To determine the number of animals needed for the study, a power analysis was prospectively performed to detect a 50% difference in PDE protein expression between groups with western blot analysis at 80% power and significance level of 0.05 (Oliva et al., 2012). An n value of 6 animals/group was obtained. Investigators were blind to the animal surgery treatment for the electrophysiology analyses.
Western Blot Analysis
At 30 min, 1 h, 3 h, 6 h, 24 h, or 7 days after TBI or sham surgery, animals were deeply anesthetized (3% isoflurane, 70% N2O, 30% O2, 5 min) and decapitated (n = 6/time point for TBI animals, n = 3/time point for sham animals). The ipsilateral hippocampus was rapidly dissected on ice, snap frozen with liquid nitrogen and stored at –80°C. Tissue was homogenized with a Dounce homogenizer (15 s, 4°C) in: 15 mM Tris pH 7.6, 250 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 0.1 mM Na3VO4, 50 mM NaF, 2 mM Na4P2O7, 1.25 μg/ml pepstatin A, 10 μg/ml leupeptin, 25 μg/ml aprotinin, and 1x phosphatase inhibitor cocktail set II (EMD Millipore). Each hippocampus was homogenized in 750 μl of buffer. Total protein was determined using Coomassie Plus assay (Bio-Rad Laboratories). Homogenates were boiled with sample buffer (9 min, 95°C). Equal amounts of protein per lane (60 μg/sample) were electrophoresed on 12.5% SDS-PAGE gels. Proteins were transferred to Immobilon-P membranes (EMD Millipore) and membranes were incubated with the following primary antibodies: β-actin (AC-15, 1:10,000, Sigma–Aldrich), PDE1A (sc-50480, 1:4,000, Santa Cruz Biotechnology), PDE1B (ab14600, 1:500, Abcam; Giachini et al., 2011), PDE1C (sc-67323, 1:500, Santa Cruz Biotechnology; Haering et al., 2015), PDE3A (sc-20792, 1:250, Santa Cruz Biotechnology; Soler et al., 2015), phospho-PDE4A (GTX14610, 1:2,000, GeneTex), PDE4A5 (ab42094, 1:2,000, Abcam; Carito et al., 2012), PDE4A8 (GTX14606, 1:1,000, GeneTex), PDE4B (sc-25812, 1:500, Santa Cruz Biotechnology; Suhasini et al., 2015), PDE4D (sc-25814, 1:500, Santa Cruz Biotechnology; Kunal et al., 2012), phospho-PDE4D (ab59212, 1:1,000, Abcam), PDE8A (sc-30059, 1:500, Santa Cruz Biotechnology; Dong et al., 2010), PDE8B (sc-17234, 1:500, Santa Cruz Biotechnology; Shimizu-Albergine et al., 2012), and PDE10A (sc-67298, 1:250, Santa Cruz Biotechnology; Giralt et al., 2013). These antibodies were chosen based on previously published studies and resulted in bands that corresponded to the appropriate, apparent molecular weights. Identification of specific PDE isoforms was based on known molecular weights. Secondary antibodies conjugated to horseradish peroxidase were used for detection (1:1,000, Cell Signaling Technology). Epitopes were visualized with enhanced chemiluminescence or enhanced chemiluminescence plus (GE Healthcare) and x-ray film (Phenix Research Products). Quantification of films was performed using ImageJ 1.48v (NIH). Levels of each protein were normalized to β-actin within each sample and then to the average of sham levels. No significant differences in PDE levels were observed in sham animals at the different survival time points (data not shown), so the three sham animals at each recovery time point were pooled into one sample for analysis to reduce animal numbers. Representative western blots show sham animals taken from 3 and 24 h post-recovery.
Immunohistochemistry
Animals were deeply anesthetized (3% isoflurane, 70% N2O, 30% O2, 5 min) at 24 h after sham surgery (n = 3) or TBI (n = 3) and perfused with saline (75 mL) and then with 4% paraformaldehyde in PBS (350 mL, 4°C). Brains were sectioned with a vibratome (50 μm thick) and free floating sections were blocked with PBS containing 5% normal goat serum, 0.2% fish skin gelatin and 0.3% TX-100. Sections were incubated with the following primary antibodies: PDE1A (sc-50480, 1 μg/ml, Santa Cruz Biotechnology), phospho-PDE4A (GTX14610, 1 μg/ml, GeneTex), PDE4B2 (ABS181, 2 μg/ml, Millipore; Ghosh et al., 2012), PDE4D (ABS22, 2 μg/ml, Millipore; Kuroiwa et al., 2012), MAP2 (M9942, 2 μg/ml, Sigma–Aldrich) and NeuN (MAB377, 4 μg/ml, Millipore). Primary PDE antibodies were selected based on our western blot data demonstrating that these antibodies recognized proteins of the appropriate known molecular weights. Secondary antibodies used were conjugated to Alexa 488, Alexa 546 or Alexa 647 (Invitrogen). Cell nuclei were visualized using Hoechst 33342 (Invitrogen).
Images were obtained with a FluoView FV1000 laser scanning confocal microscope (Olympus America) equipped with a 10X 0.4 NA air objective, 20X 0.85 NA oil-immersion objective and 60X 1.42 NA oil-immersion objective, and an LD laser (405 nm), multi-line argon laser and HeNe(G) laser. Sections from different animals were processed in parallel and at least three sections from each animal were imaged.
Flow Cytometry
At 24 h after sham surgery (n = 6) or TBI (n = 6), animals were deeply anesthetized (3% isoflurane, 70% N2O, 30% O2, 5 min) and transcardially perfused with PBS (120 mL, 4°C). The ipsilateral hippocampus was dissected at 4°C. Tissue was mechanically dissociated into single cell suspension and cells were labeled with CD45 Alexa 647 (202212, 1.25 μg/ml, BioLegend) and CD11b v450 (53-4321-80, 1 μg/ml, eBioscience). Dead cells were excluded using LIVE/DEAD Fixable Near-IR dead cell stain (L10119, 1 μl/ml, Life Technologies). Cells were fixed and permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization kit (554714, BD Biosciences). Cells were intracellularly labeled with phospho-PDE4A (GTX14610, 2 μg/ml, GeneTex), PDE4B2 (ABS181, 2 μg/ml, EMD Millipore) or PDE4D (sc-25814, 2 μg/ml, Santa Cruz Biotechnology). PDE staining was detected with PE-conjugated secondary antibodies (12-4739-81, 10 μg/ml, eBioscience). Flow cytometry data was acquired on a BD LSR II flow cytometer with four emission lasers at 407, 488, 532, and 640 nm. Data collection was performed using BD FACSDiva 8.0.1 (BD Biosciences) and analyzed with Kaluza 1.2 software (Beckman Coulter).
Electrophysiology
At 24 h after sham surgery (n = 13) or TBI (n = 15), animals were deeply anesthetized (3% isoflurane, 70% N2O, 30% O2, 5 min) and then decapitated. The ipsilateral hippocampus was dissected and sliced with a vibratome at 4°C (Leica Microsystems). Slices from the middle third of the hippocampus were collected (400 μm thick) in artificial cerebral spinal fluid (aCSF): 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 10 mM D-glucose, 2 mM CaCl2, 1 mM MgCl2 saturated with 95% O2/5% CO2. Slices recovered at room temperature for at least 60 min. Slices were transferred to a submerged recording chamber and perfused at 2.5–3 ml/min with aCSF at 31°C (Warner Instruments). Field excitatory postsynaptic potentials (fEPSPs) were recorded from the stratum radiatum of area CA1 with glass electrodes filled with 2 M NaCl (1–3 MΩ). The Schaffer collateral pathway was stimulated with a platinum-iridium concentric bipolar electrode (tip diameter 25 μm, FHC). Electrophysiological responses were recorded using a Multiclamp 700B amplifier (Axon Instruments) and pClamp 10.4 software (Axon Instruments). Recordings were low-pass filtered at 2 kHz and digitized at 20 kHz (Digidata 1440A, Molecular Devices). Input–output (I–O) curves were generated with stepwise current increases from 20 to 180 μA. Paired-pulse facilitation (PPF) was measured with 50–250 ms stimulation intervals, delivered at a current intensity of 40–50% of the maximum fEPSP. For baseline responses prior to LTP induction, fEPSPs were recorded at a current intensity of 40–50% of the maximum fEPSP, delivered at 0.033 Hz for at least 20 min. Long-term potentiation (LTP) was induced with high frequency stimulation (HFS) of 100 Hz for 1 s at the current intensity used for baseline stimulation. Rolipram (3 μM) or vehicle (0.3% DMSO) were bath applied in aCSF beginning 10 min prior to LTP induction and for 30 min after tetanization. The amount of depolarization during tetanization was analyzed by integrating the entire HFS response (total) or integrating the last 50 ms of depolarization (steady-state; Klann et al., 1998). Synaptic fatigue was calculated by normalizing each fEPSP during tetanization to the first fEPSP of the HFS (Rutten et al., 2008b).
Data Analysis
Statistical comparisons were made using GraphPad Prism 6.05 or SigmaPlot 12.0 software. Western blot data and tetanization responses were analyzed using a one-way ANOVA and Tukey’s HSD correction for multiple comparisons. Flow cytometry data was analyzed using an unpaired Student’s t-test. I–O responses, PPF, and LTP data were analyzed using a repeated measures two-way ANOVA and Tukey’s HSD correction for multiple comparisons. Significance was designated at p < 0.05. Results presented are mean ± SEM.
Results
We have previously reported that cAMP levels are decreased in the hippocampus from 15 min to 4 h after TBI (Atkins et al., 2007). To determine whether this decrease is associated with changes in levels of PDE, the enzyme that degrades cAMP, we evaluated the hippocampus by western blot analysis for PDE expression. We chose to evaluate PDE1, 3, 4, 8, and 10 since these are either cAMP-specific or degrade both cAMP and cGMP and are found in the brain or inflammatory cells that could infiltrate the brain after injury (Boswell-Smith et al., 2006). At 30 min, 1 h, 3 h, 6 h, 24 h, or 7 days after moderate parasagittal fluid-percussion brain injury or sham surgery, animals were analyzed by western blotting for changes in PDE expression in the ipsilateral, injured hippocampus (Figure 1). We found that PDE1A was significantly increased as early as 30 min after TBI, and remained elevated at 1 and 6 h post-injury. PDE1B and 1C levels were unchanged after TBI between 30 min to 7 days post-injury. No significant changes in expression were also observed for PDE3A, 8A, or 8B. PDE10A levels were unchanged with the exception of a small decrease at 1 h post-injury.
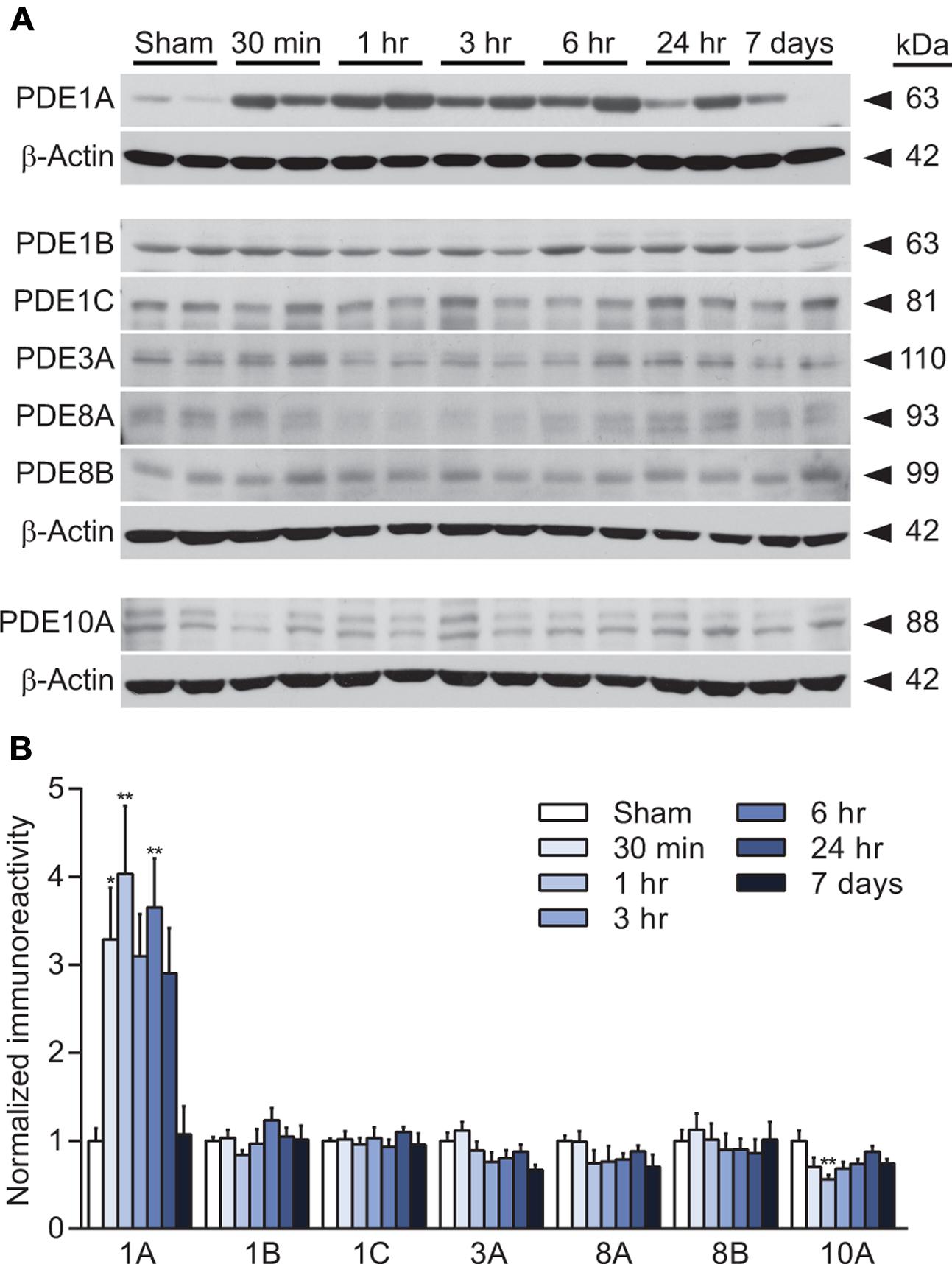
FIGURE 1. PDE expression changes after TBI in the hippocampus. (A,C,E) Representative western blots of the ipsilateral hippocampus analyzed for PDE1A, 1B, 1C, 3A, 8A, 8B, and 10A levels. Each of the corresponding β-actin western blots are shown below the PDE western blots. (B,D,F) Densitometry results. Significant increases in PDE1A were observed at 30 min, 1 h and 6 h post-injury. PDE10A decreased at 1 h post-injury. n = 6/group, ∗p < 0.05, ∗∗p < 0.01 vs. Sham; one-way ANOVA with Tukey’s HSD correction for multiple comparisons.
PDE4 is the major cAMP-degrading enzyme in the brain and the predominant PDE in inflammatory cells (Boswell-Smith et al., 2006). PDE4A, 4B, and 4D are present in the brain, whereas PDE4C is expressed only at very low levels in the brain (Perez-Torres et al., 2000; Lakics et al., 2010; Johansson et al., 2012). PDE4A and 4D are regulated by phosphorylation, and we found that phosphorylation of PDE4A, but not 4D, was significantly increased at 6 and 24 h after TBI (Figures 2A,E). Total levels of both PDE4A5 and 4A8 were unaltered after TBI (Figures 2A,B). Levels of PDE4B1/3 were modestly, but significantly decreased at 6 and 24 h after TBI (Figures 2C,D). Similarly, PDE4B4 was decreased at 24 h after injury. In contrast, both PDE4B2 and 4D2 were significantly increased at 1, 6, and 24 h after TBI (Figures 2D,F). PDE4D4 and 4D3 were also elevated at 24 h and 7 days post-injury, respectively (Figures 2E,F).
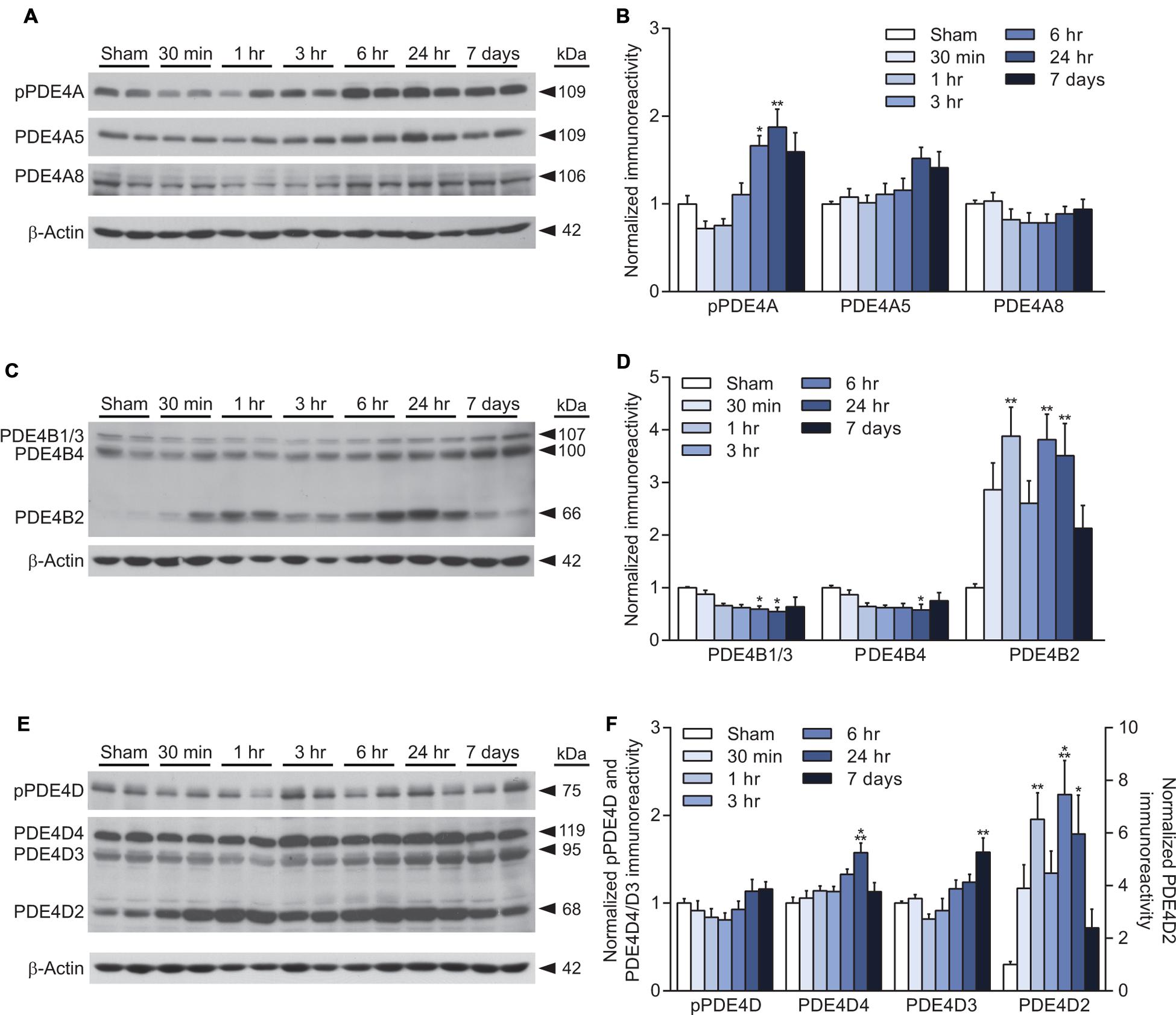
FIGURE 2. Increased PDE4B2 and 4D2 expression in the hippocampus after TBI. (A,C,E) Representative western blots of the ipsilateral hippocampus analyzed for changes in PDE4A, 4B, and 4D. (B,D,F) Densitometric analysis. Significant increases in phospho-PDE4A, and total levels of PDE4B2, 4D4, 4D3, and 4D2 were observed. Levels of PDE4B1/3 and 4B4 decreased after TBI. n = 6/group, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. Sham; one-way ANOVA with Tukey’s HSD correction for multiple comparisons.
To determine the localization of these changes, we performed immunohistochemistry at 24 h after TBI for PDE1A, phospho-PDE4A, PDE4B2, and PDE4D4, the isoforms that showed the largest changes in expression after TBI. PDE1A was found in nuclei and neurites of neurons (Figure 3). Phospho-PDE4A localized predominately to neuronal nuclei (Figure 4). In contrast, PDE4B2 was expressed in dendrites as determined by co-localization with MAP2, but not in the cell bodies or nuclei (Figure 5). PDE4D immunoreactivity was present in scattered cells throughout the hippocampus, suggestive of immune cell infiltration (Figure 6).
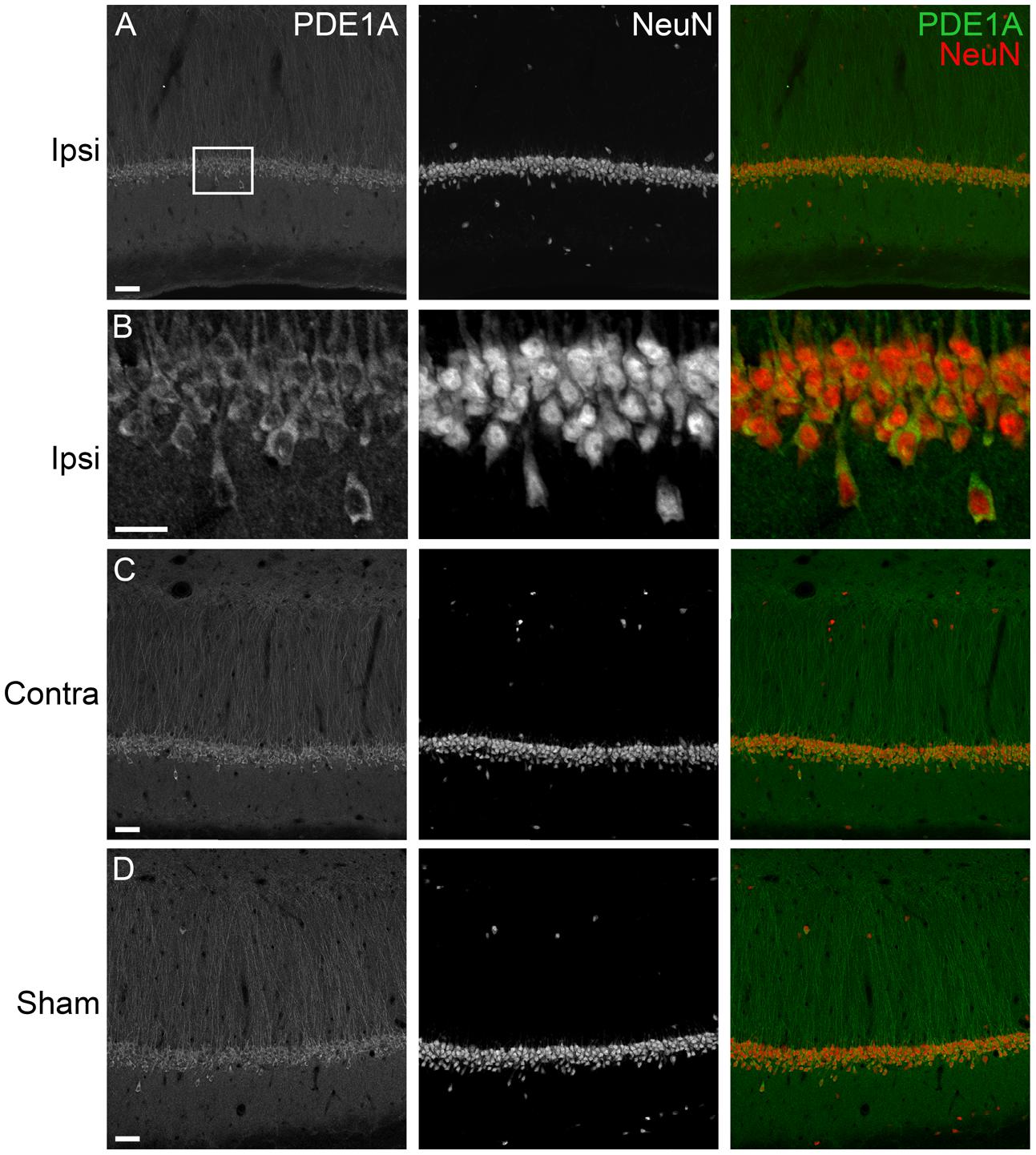
FIGURE 3. Immunohistochemistry of PDE1A in the hippocampus. (A) The ipsilateral hippocampus was immunostained for PDE1A at 24 h after TBI. PDE1A (green) colocalized to NeuN-positive cells (red) in area CA1. Scale bar 50 μm. (B) Higher magnification of the ipsilateral CA1 pyramidal cell layer (box). Scale bar 20 μm. (C) Expression of PDE1A in the contralateral hippocampus in area CA1. Scale bar 50 μm. (D) PDE1A immunohistochemistry in area CA1 from a sham animal. Scale bar 50 μm.
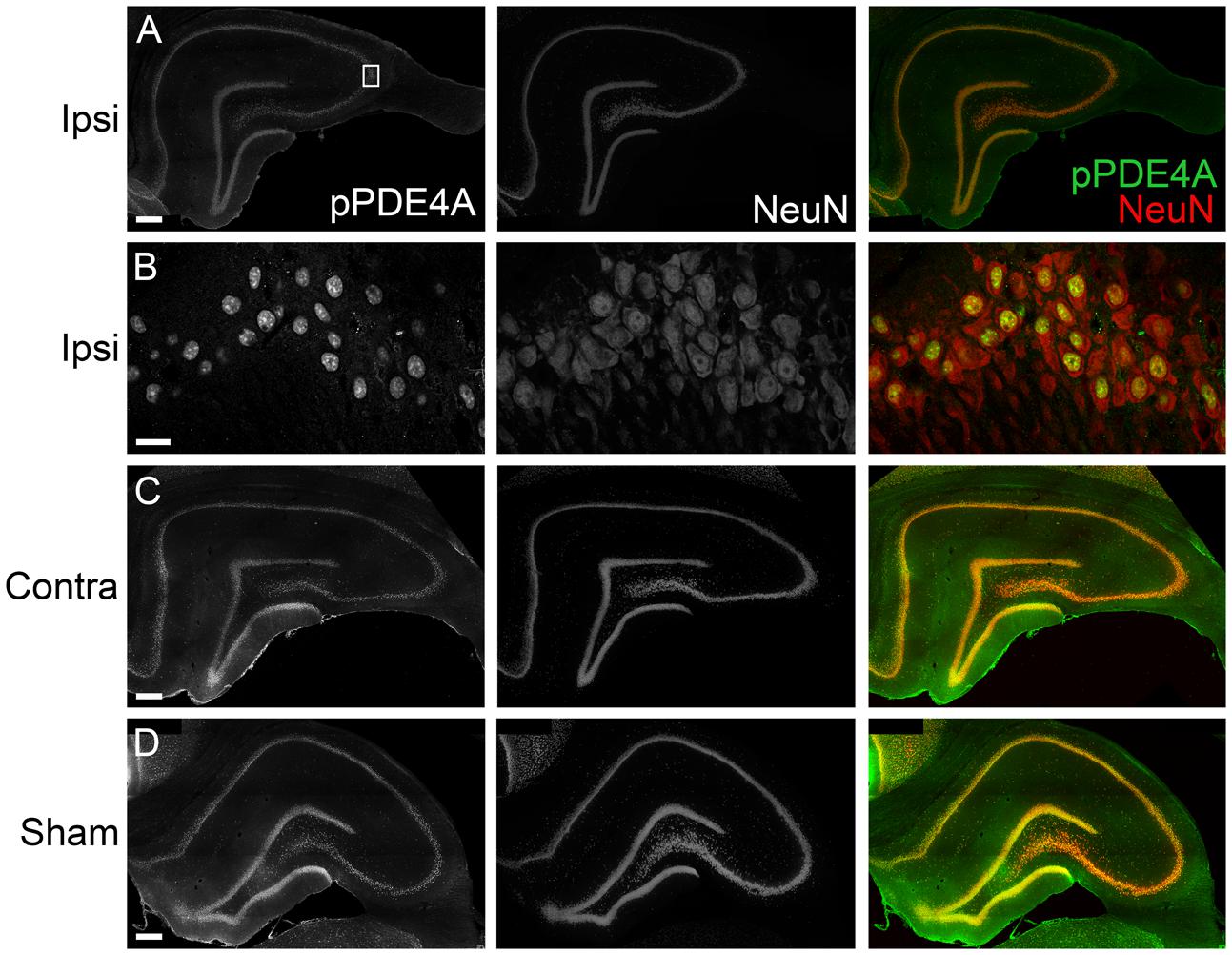
FIGURE 4. Immunohistochemistry of phospho-PDE4A in the hippocampus. (A) At 24 h after TBI, phosphorylated PDE4A (green) was found throughout all subregions of the ipsilateral hippocampus and colocalized with NeuN-positive cells (red). Scale bar 100 μm. (B) Higher magnification of the ipsilateral CA3 pyramidal cell layer (box). Scale bar 20 μm. (C) Expression of phospho-PDE4A in the contralateral hippocampus. Scale bar 100 μm. (D) Phospho-PDE4A immunohistochemistry in the sham, non-injured hippocampus. Scale bar 100 μm.
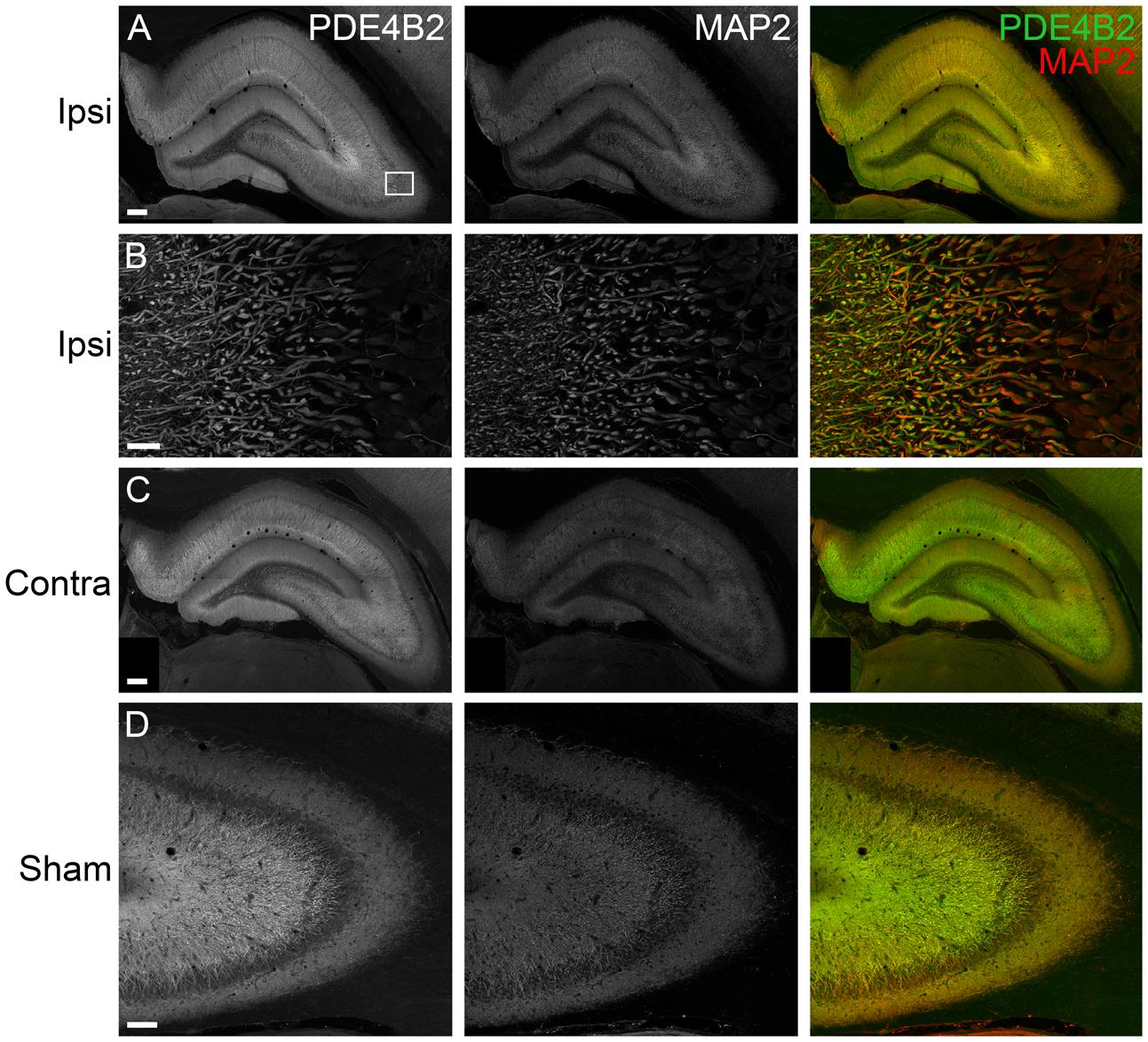
FIGURE 5. Immunohistochemistry of PDE4B2 in the hippocampus at 24 h after TBI. (A) PDE4B2 (green) colocalized to MAP2-positive dendrites (red) throughout all subregions of the ipsilateral hippocampus. Scale bar 200 μm. (B) Higher magnification of the ipsilateral CA3 region (box). Scale bar 20 μm. (C) Expression of PDE4B2 in dendrites in the contralateral hippocampus. Scale bar 200 μm. (D) PDE4B2 immunohistochemistry in area CA3 from a sham animal. Scale bar 100 μm.
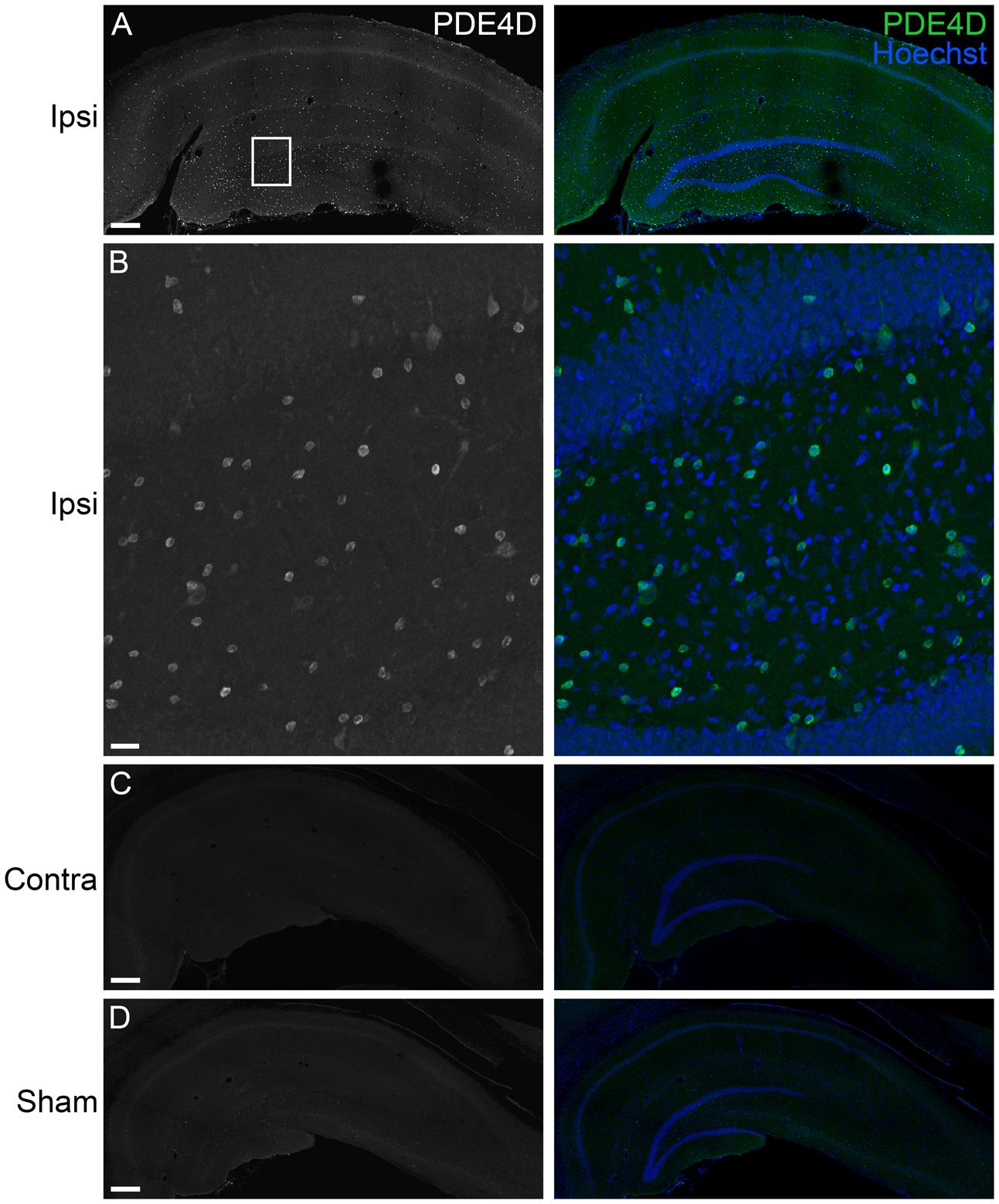
FIGURE 6. Immunohistochemistry of PDE4D in the hippocampus at 24 h after TBI. (A) PDE4D (green) was found in scattered cells throughout all regions of the ipsilateral hippocampus. Hoescht counterstaining was used to identify the hippocampal pyramidal cell layers (blue). Scale bar 250 μm. (B) Higher magnification of the ipsilateral dentate gyrus (box). Scale bar 20 μm. (C) Expression of PDE4D was at very low levels in the contralateral hippocampus. Scale bar 250 μm. (D) PDE4D was present at very low levels in the sham, non-injured hippocampus. Scale bar 250 μm.
To more definitively determine the cellular expression of PDE4D, we performed flow cytometry (Figure 7). We also selected PDE4B2 and phospho-PDE4A for analysis since these were also significantly elevated in the injured hippocampus at 24 h post-TBI and have been identified in immune cells (Shepherd et al., 2004; Ghosh et al., 2012). Phospho-PDE4A, PDE4B2, and 4D were localized to microglia and infiltrating CD11b+/CD45+ immune cells in the hippocampus at 24 h post-injury.
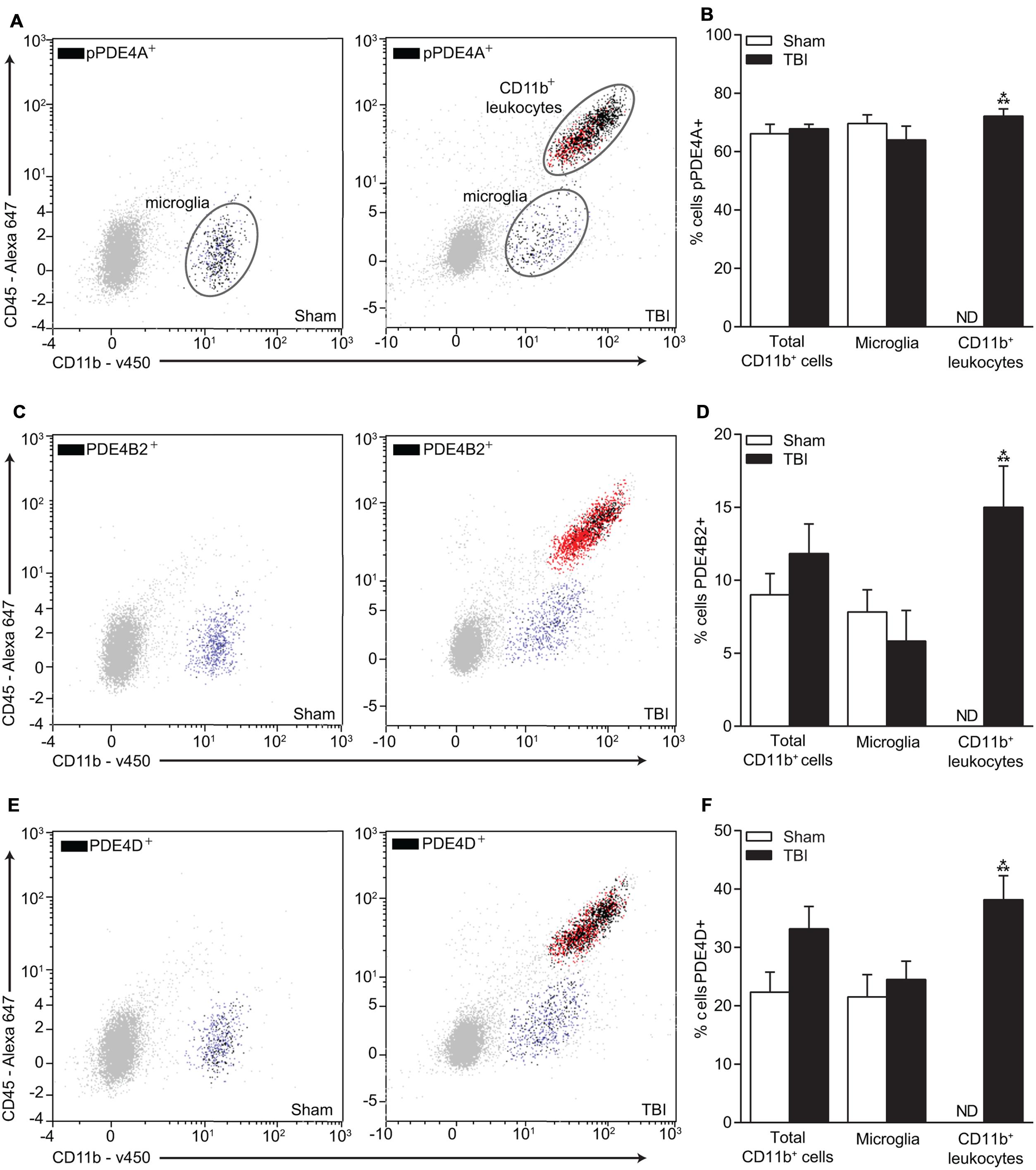
FIGURE 7. Flow cytometry of microglia and infiltrating CD11b+ immune cells and co-localization with phospho-PDE4A, PDE4B2, and 4D. (A) Phospho-PDE4A, (C) PDE4B2, and (E) PDE4D expression were localized to microglia and infiltrating immune cells in the injured hippocampus at 24 h post-injury. There was a significant increase in (B) phospho-PDE4A, (D) PDE4B2 and (F) PDE4D-positive infiltrating CD11b+/CD45+ immune cells. n = 6/group, ∗∗∗p < 0.001 vs. Sham; unpaired Student’s t-test.
Next, to determine if upregulation of PDE4 expression is involved in hippocampal synaptic plasticity deficits after TBI, we assessed whether administration of a pan-PDE4 inhibitor would rescue the deficits in hippocampal LTP induced by TBI. Hippocampal LTP in area CA1 induced with a single tetanus was significantly impaired in slices from TBI animals (Figures 8A,B). Rolipram (3 μM) applied to the hippocampal slices rescued the deficits in LTP expression [time × animal treatment interaction F(177,2115) = 1.60, p < 0.001; Figures 8A,B]. Both total and steady-state depolarization levels as well as synaptic fatigue during tetanization were comparable between all groups, suggesting that the impairment in LTP after TBI was not due to differences in depolarization during the tetanus (Figures 8C,D). To evaluate whether changes in basal synaptic transmission were involved in the rescue of hippcampal LTP, we analyzed input–output (I–O) responses. I–O curves were significantly depressed in slices from TBI animals as compared to sham animals [stimulation intensity × animal treatment interaction F(24,288) = 2.76, p < 0.001]. This depression in basal synaptic transmission was partially rescued with rolipram [main effect of animal treatment F(3,288) = 8.12, p < 0.001; Figures 9A,B]. PPF was depressed after TBI and also partially rescued with rolipram treatment [main effect of animal treatment F(3,144) = 3.35, p < 0.05; Figure 9C]. These results indicate that hippocampal basal synaptic transmission and LTP expression in area CA1 were impaired acutely after TBI and rescued with the pan-PDE4 inhibitor, rolipram.
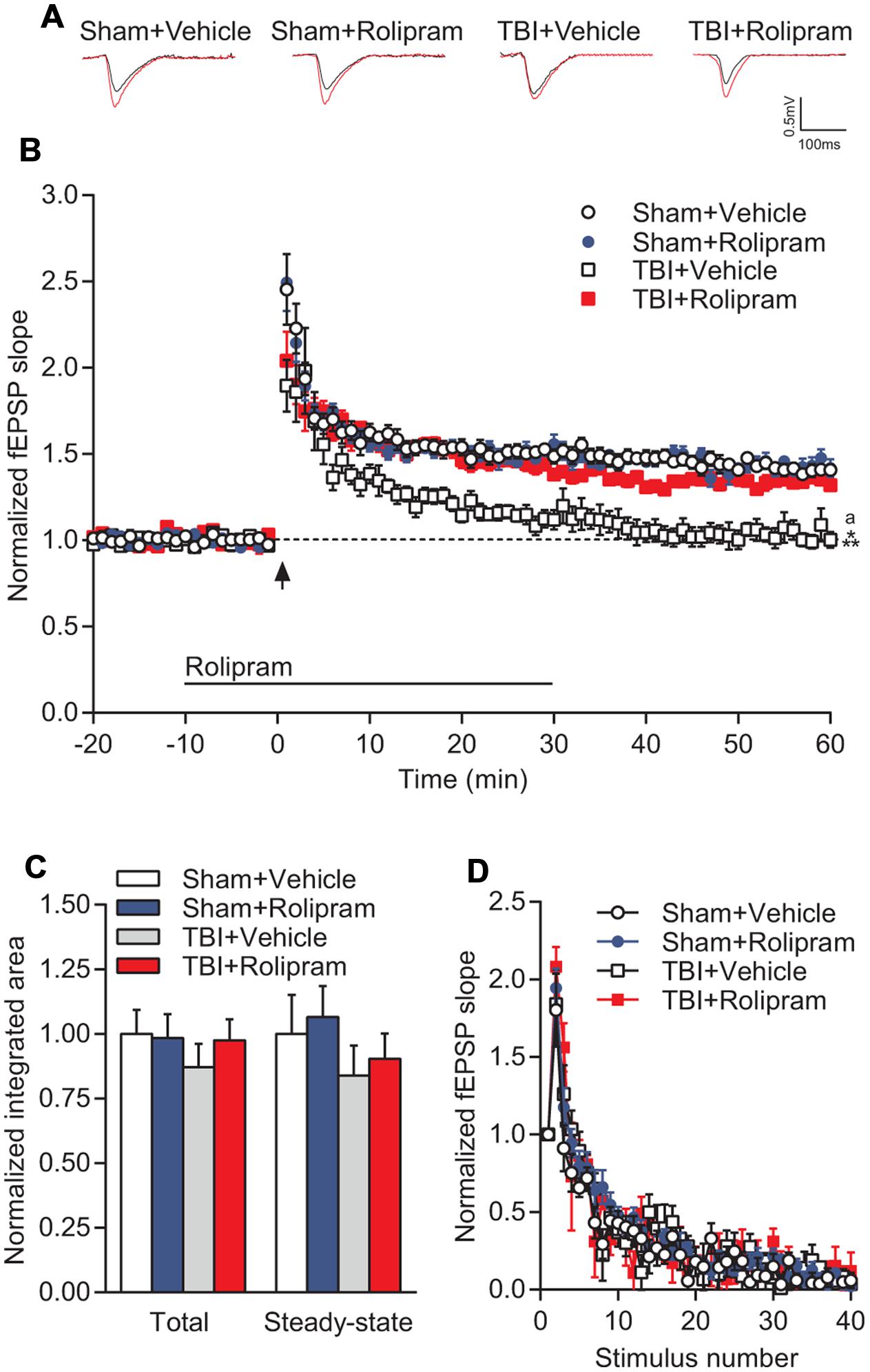
FIGURE 8. PDE4 inhibition significantly rescued TBI-induced LTP deficits in area CA1 of the hippocampus. (A) Representative traces of the fEPSP before (black) and 60 min after (red) tetanization. (B) fEPSP slopes were normalized to baseline prior to LTP induction. Rolipram (3 μM) or vehicle (0.3% DMSO) were bath applied 10 min prior to and for 30 min after tetanization (bar). Hippocampal LTP was induced with 1 × 100 Hz tetanization, 1 s long (arrow). (C) Total and steady-state depolarization levels during tetanization. (D) Synaptic fatigue during tetanization. Sham + Vehicle n = 10 slices/9 animals, Sham + Rolipram n = 14 slices/12 animals, TBI+Vehicle n = 8 slices/7 animals, TBI+Rolipram n = 8 slices/8 animals; ∗∗∗p < 0.001 vs. Sham + Vehicle or Sham + Rolipram, ap < 0.05 vs. TBI + Rolipram; repeated-measures two-way ANOVA with Tukey’s HSD correction for multiple comparisons.
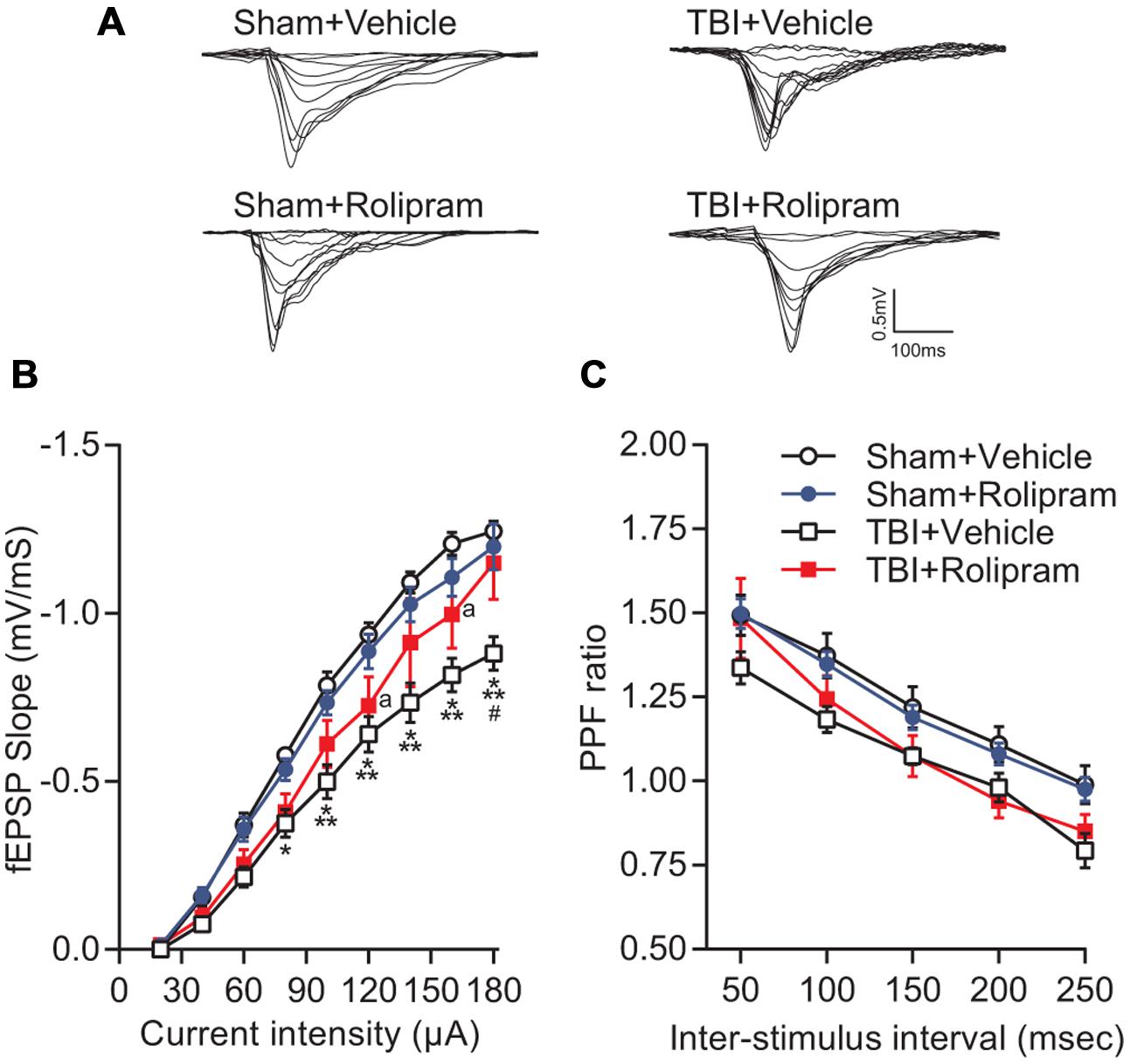
FIGURE 9. Effects of rolipram on hippocampal synaptic transmission at 24 h post-TBI. (A) Representative traces of the fEPSP evoked during input–output (I–O) responses in stratum radiatum of area CA1 with Schaffer collateral stimulation. (B) The I–O curve was significantly shifted downward in hippocampal slices from TBI animals as compared to sham animals. Rolipram (3 μM) significantly enhanced the fEPSP slope in slices from TBI animals. ∗p < 0.05, ∗∗∗p < 0.001 vs. Sham + Vehicle, #p < 0.01 vs. TBI + Rolipram, ap < 0.05 vs. Sham + Vehicle. (C) A decrease in PPF was observed in TBI + Vehicle-treated slices (p < 0.05 TBI + Vehicle vs. Sham + Vehicle) that was not significant in TBI + Rolipram-treated slices. Data represent the ratio of the second fEPSP slope to the first fEPSP slope. Sham + Vehicle n = 10 slices/9 animals, Sham + Rolipram n = 14 slices/12 animals, TBI+Vehicle n = 8 slices/7 animals, TBI + Rolipram n = 8 slices/8 animals; repeated-measures two-way ANOVA with Tukey’s HSD correction for multiple comparisons.
Discussion
Downregulation of the cAMP signaling pathway following TBI significantly alters hippocampal synaptic plasticity, however, the underlying mechanisms are unknown. The PDE enzymes play a critical role in the regulation of cAMP levels, and alterations in PDE expression and phosphorylation may be involved in the depression of cAMP levels following TBI. Therefore, in the present study we evaluated whether expression of PDEs such as PDE1, 3, 4, 8, and 10, which are present in the central nervous system and hydrolyze cAMP, were altered acutely after TBI (Bender and Beavo, 2006; Omori and Kotera, 2007; Lakics et al., 2010). While not all PDE isoforms showed altered expression levels, several were significantly changed. PDE1A, 4B2, and 4D2, and phosphorylation of PDE4A were significantly upregulated, whereas PDE4B1/3, 4B4, and 10A levels decreased acutely after TBI. The PDEs 1B, 1C, 3A, 4A5, 4A8, 8A, and 8B, and phospho-PDE4D were unchanged acutely after TBI. Although an a priori power analysis was conducted to ensure the sample sizes were sufficient to detect significant changes in PDE levels after TBI, for these particular PDEs the negative results should be interpreted cautiously.
Phosphodiesterases are key enzymes involved in regulating cAMP levels in the central nervous system. Understanding the spatio-temporal alterations in PDE isoform expression levels after injury is important for targeting the relevant isoform as well as treating within the appropriate therapeutic time window. In a previous study, we found that there were significant temporal changes in PDE expression within the ipsilateral cortex acutely after TBI (Oliva et al., 2012). Like the present results found in the injured hippocampus, TBI induced upregulation of PDE1A, 4B2, and 4D2 in the ipsilateral cortex. However, not all PDEs were regulated similarly between the cortex and hippocampus. For example, PDE10A was upregulated in the cortex, but downregulated in the hippocampus acutely after TBI. PDE4A5 and 4A8 were downregulated in the cortex, but unaltered in the hippocampus. Furthermore, both PDE4D4 and 4D3 were upregulated at 24 h after TBI in the hippocampus, but unchanged in the cortex. These discrepancies suggest that developing a therapeutic to target a specific PDE and particular functional outcome requires consideration of both the temporal and spatial regulation of that particular PDE isoform.
The upregulation of PDE1A in the hippocampus after TBI suggests that beyond decreasing cAMP levels, cGMP levels may also be affected by TBI. PDE1 is a family of Ca2+/calmodulin-dependent PDEs encoded by three genes (A–C) and involved in the regulation of both cGMP and cAMP (Bender and Beavo, 2006). In the human brain, PDE1A is distributed in the parietal cortex and hippocampus, but at lower levels than PDE1B or 1C (Lakics et al., 2010). In the present study, expression of PDE1A was significantly increased, while PDE1B and 1C were unchanged after TBI. Although the PDE1 family uses both cyclic nucleotides as a substrate, PDE1A displays a higher affinity toward cGMP than cAMP (Bender and Beavo, 2006). Conversely, PDE10A which is also a dual-substrate PDE, was downregulated in the hippocampus. These opposing changes may be involved in the lack of changes in basal cGMP levels reported in the hippocampus after TBI (Temple et al., 2001). However, NMDA-stimulated levels of cGMP are actually increased in the ipsilateral hippocampus after TBI and further studies are required to determine if cGMP-selective PDEs expressed in the brain such as PDE9 are altered by TBI (Temple et al., 2001). Together these results support the potential therapeutic use of PDE1 inhibitors such as vinpocetine, amantadine and caffeine, which have shown promise to improve cognitive impairments in preclinical models of TBI and in humans (Dixon et al., 1999; Li et al., 2008; Amen et al., 2011).
In particular, the PDE4 family was highly regulated in the hippocampus after TBI. PDE4B2, 4D2, 4D3, and 4D4 expression levels and phosphorylation of PDE4A were increased following TBI, while PDE4B1/3 and 4B4 levels were decreased. Due to the similar molecular weights of PDEB1 and B3, we were unable to differentiate which particular isoform decreased after TBI. The changes in PDE4 isoforms were transient with most returning to sham, non-injured levels by 7 days post-injury. The upregulation of PDE4 isoforms correlates loosely with the decrease in cAMP levels and PKA activation after TBI (Atkins et al., 2007). However, a causal link between an upregulation of PDE4 expression and decreased cAMP levels has not been established. Alternatively, changes in AC activity or expression could be involved in the decrease in cAMP levels after TBI. Accordingly, reduction in type 1 AC enzyme levels have been reported within the hippocampus of Alzheimer’s disease patients, aged rodents and after ischemia, and are correlated with impairments in cAMP signal transduction (Araki et al., 1995; Yamamoto et al., 2000; Nagakura et al., 2002; Ramos et al., 2003; Mons et al., 2004). Further studies are needed to determine if AC levels are also altered by TBI or whether the increase in PDE expression after TBI also results in increased PDE activity.
The PDE4 subfamily, like other PDEs, is subject to post-translational modification to allow for rapid feedback in controlling the spatial and temporal dynamics of cAMP signaling (Houslay, 2010). Unlike the other PDE4 isoforms, the long isoforms have a UCR1 domain that contains a PKA phosphorylation site that enhances PDE4 activity (Mackenzie et al., 2002). The PKA phosphorylation site allows for cAMP signaling to negatively feedback on itself through enhanced activation of long PDE4A isoforms. PDE4A5 and 4A8 are both long isoforms with similar molecular weights, which precluded definitively identifying which isoform was increased in phosphorylation after TBI. Phospho-PDE4A localized to both hippocampal neurons and infiltrating CD11b+ leukocytes. While this increase in phospho-PDE4A is indicative of an increase in PKA-mediated PDE4A activity, the timing of this change does not correlate with changes seen in cAMP and phospho-PKA, which are depressed in the hippocampus during this time frame (Atkins et al., 2007). However, it is possible that there could be changes in cAMP and PKA signaling in microdomains mediating this effect that were missed with the western blot analysis or immunohistochemistry. Subcellular, compartmentalized signaling changes have been reported when assessing differences between PDE4B and 4D signaling (Blackman et al., 2011). An alternative possibility was that another protein kinase mediated this phosphorylation, such as MAPK-activated protein kinase 2, which has been reported to phosphorylate a serine near the PKA phosphorylation site of PDE4A5 (Mackenzie et al., 2011).
In contrast to the results with phospho-PDE4A, the increase in PDE4B2 and 4D2 expression correlated with the decrease in cAMP levels after TBI (Atkins et al., 2007). Within 1 h after trauma, PDE4B2 and 4D2 were significantly elevated in the injured hippocampus and remained elevated for up to 24 h post-injury. Although the immunohistochemistry results indicated that PDE4B2 and phospho-PDE4A localized to neurons, flow cytometry revealed expression of PDE4B2 and phospho-PDE4A in microglia and infiltrating CD11b+/CD45+ leukocytes as well. The absence of obvious expression in microglia or infiltrating immune cells with immunohistochemistry suggests that phospho-PDE4A- and PDE4B2-positive immune cells may be present near the contusion site between the parietal cortex and hippocampus which was likely sampled in the western blot analysis. Immunohistochemistry of this damaged area can be hindered by autofluorescence of red blood cells present at the contusion site. These findings are supported by other studies which have found that both isoforms are differentially distributed throughout the brain and also found in the immune system. PDE4B and 4D are present in monocytes, macrophages and neutrophils and regulate pro-inflammatory mediators such as tumor necrosis factor and neutrophil infiltration (Jin and Conti, 2002; Ariga et al., 2004; Shepherd et al., 2004). Interestingly, phospho-PDE4A and PDE4D were expressed in a larger proportion of infiltrating CD11b+/CD45+ cells as compared to PDE4B2. While the increase in phospho-PDE4A, PDE4B2, and PDE4D in immune cells may contribute to inflammatory signaling after TBI, the upregulation of PDE4B2 in hippocampal dendrites suggests that this molecule may have multiple roles in TBI and may also be involved in learning and memory deficits after TBI.
PDE4 is well known to be involved in hippocampal LTP and long-term memory formation (Navakkode et al., 2004; Rutten et al., 2008a,b; Li et al., 2011). General pan-PDE4 inhibitors such as rolipram reverse learning and memory impairments in animal models of Alzheimer’s disease, psychosis, stress, and ischemia (Henkel-Tigges and Davis, 1990; Imanishi et al., 1997; Bach et al., 1999; Gong et al., 2004; Chen et al., 2010; Wiescholleck and Manahan-Vaughan, 2012; Sierksma et al., 2014). In accordance with these previous studies, we found that bath application of rolipram to hippocampal slices from TBI animals reversed the decay of LTP (Navakkode et al., 2004). However, the dose of rolipram required to rescue hippocampal LTP (3 μM) at 24 h post-injury was threefold greater than the dose sufficient to rescue hippocampal LTP deficits at 2 weeks post-injury (1 μM; Titus et al., 2013a,b). Indeed, 1 μM rolipram did not rescue the deficits in hippocampal LTP at 24 h post-injury (data not shown). Given that expression of most PDE4 isoforms returned to sham, non-injured levels by 7 days post-injury, we speculate that the acute, transient upregulation of PDE4 isoforms at 24 h post-injury resulted in the necessity for a higher dose of rolipram to rescue hippocampal LTP deficits at this acute time point.
Interestingly, rolipram also partially improved the depression of basal synaptic transmission in area CA1 of the hippocampus. Basal synaptic transmission in the hippocampus is mediated primarily through AMPA-type glutamate receptors, which are well known to be regulated by PKA (Man et al., 2007). Trafficking of AMPA-type glutamate receptors into the postsynaptic membrane is increased by PKA phosphorylation of serine 845 on the GluA1 subunit (Oh et al., 2006; Middei et al., 2013). GluA1 levels have been found to be altered in some experimental models of TBI (Schumann et al., 2008; Kharlamov et al., 2011). The observed partial rescue in basal synaptic transmission may possibly be through regulation of AMPA receptor trafficking although further studies are needed to evaluate this potential mechanism.
In summary, we found that in an experimental model of TBI there was an acute upregulation of PDE1A, 4B2, 4D4, 4D3, 4D2, and phosphorylated PDE4A in the hippocampus after injury. Furthermore, we found that PDE4B2 and phospho-PDE4A were localized to neurons, microglia and infiltrating CD11b+ leukocytes in the injured hippocampus early after trauma. PDE4D was found predominantly in microglia and infiltrating CD11b+/CD45+ leukocytes. Rolipram, a pan-PDE4 inhibitor, rescued synaptic plasticity deficits in the hippocampus at 24 h after TBI. Given the role of these PDE4 subfamilies in learning, memory and inflammation, this study highlights new potential targets for reducing damage and improving hippocampal synaptic plasticity after TBI.
Author Contributions
NW contributed in experimental design, data collection and analysis, and writing of the manuscript. DT contributed in experimental design, data collection and analysis, and writing of the manuscript. AO contributed in experimental design, data collection and analysis, and writing of the manuscript. CF contributed in experimental design, data collection, and writing of the manuscript. CA contributed in experimental design, data analysis, and writing of the manuscript.
Funding
This work was supported by the National Institutes of Health/NINDS Grants R01 NS069721 and F31 NS089351, and The Miami Project to Cure Paralysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Yuan Kang, Dr. Oliver Umland and Julie Freund for technical assistance.
References
Amen, D. G., Wu, J. C., Taylor, D., and Willeumier, K. (2011). Reversing brain damage in former NFL players: implications for traumatic brain injury and substance abuse rehabilitation. J. Psychoactive Drugs 43, 1–5. doi: 10.1080/02791072.2011.566489
Araki, T., Kato, H., Fujiwara, T., and Itoyama, Y. (1995). Age-related changes in bindings of second messengers in the rat brain. Brain Res. 704, 227–232. doi: 10.1016/0006-8993(95)01117-X
Ariga, M., Neitzert, B., Nakae, S., Mottin, G., Bertrand, C., Pruniaux, M. P., et al. (2004). Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J. Immunol. 173, 7531–7538. doi: 10.4049/jimmunol.173.12.7531
Atkins, C. M. (2011). Decoding hippocampal signaling deficits after traumatic brain injury. Transl. Stroke Res. 2, 546–555. doi: 10.1007/s12975-011-0123-z
Atkins, C. M., Cepero, M. L., Kang, Y., Liebl, D. J., and Dietrich, W. D. (2013). Effects of early rolipram treatment on histopathological outcome after controlled cortical impact injury in mice. Neurosci. Lett. 532, 1–6. doi: 10.1016/j.neulet.2012.10.019
Atkins, C. M., Kang, Y., Furones, C., Truettner, J. S., Alonso, O. F., and Dietrich, W. D. (2012). Postinjury treatment with rolipram increases hemorrhage after traumatic brain injury. J. Neurosci. Res. 90, 1861–1871. doi: 10.1002/jnr.23069
Atkins, C. M., Oliva, A. A. Jr., Alonso, O. F., Pearse, D. D., Bramlett, H. M., and Dietrich, W. D. (2007). Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 208, 145–158. doi: 10.1016/j.expneurol.2007.08.011
Bach, M. E., Barad, M., Son, H., Zhuo, M., Lu, Y. F., Shih, R., et al. (1999). Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 5280–5285. doi: 10.1073/pnas.96.9.5280
Bender, A. T., and Beavo, J. A. (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58, 488–520. doi: 10.1124/pr.58.3.5
Bigler, E. D., Blatter, D. D., Anderson, C. V., Johnson, S. C., Gale, S. D., Hopkins, R. O., et al. (1997). Hippocampal volume in normal aging and traumatic brain injury. AJNR Am. J. Neuroradiol. 18, 11–23.
Blackman, B. E., Horner, K., Heidmann, J., Wang, D., Richter, W., Rich, T. C., et al. (2011). PDE4D and PDE4B function in distinct subcellular compartments in mouse embryonic fibroblasts. J. Biol. Chem. 286, 12590–12601. doi: 10.1074/jbc.M110.203604
Boswell-Smith, V., Spina, D., and Page, C. P. (2006). Phosphodiesterase inhibitors. Br. J. Pharmacol. 147(Suppl. 1), S252–S257. doi: 10.1038/sj.bjp.0706495
Bourtchuladze, R., Frenguelli, B., Blendy, J., Cioffi, D., Schutz, G., and Silva, A. J. (1994). Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68. doi: 10.1016/0092-8674(94)90400-6
Carito, V., Pingitore, A., Cione, E., Perrotta, I., Mancuso, D., Russo, A., et al. (2012). Localization of nerve growth factor (NGF) receptors in the mitochondrial compartment: characterization and putative role. Biochim. Biophys. Acta 1820, 96–103. doi: 10.1016/j.bbagen.2011.10.015
Chen, C. C., Yang, C. H., Huang, C. C., and Hsu, K. S. (2010). Acute stress impairs hippocampal mossy fiber-CA3 long-term potentiation by enhancing cAMP-specific phosphodiesterase 4 activity. Neuropsychopharmacology 35, 1605–1617. doi: 10.1038/npp.2010.33
Dixon, C. E., Kraus, M. F., Kline, A. E., Ma, X., Yan, H. Q., Griffith, R. G., et al. (1999). Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restor. Neurol. Neurosci. 14, 285–294.
Dong, H., Zitt, C., Auriga, C., Hatzelmann, A., and Epstein, P. M. (2010). Inhibition of PDE3, PDE4 and PDE7 potentiates glucocorticoid-induced apoptosis and overcomes glucocorticoid resistance in CEM T leukemic cells. Biochem. Pharmacol. 79, 321–329. doi: 10.1016/j.bcp.2009.09.001
Faul, M., Xu, L., Wald, M. M., and Coronado, V. G. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control.
Frey, U., Huang, Y. Y., and Kandel, E. R. (1993). Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260, 1661–1664. doi: 10.1126/science.8389057
Ghosh, M., Garcia-Castillo, D., Aguirre, V., Golshani, R., Atkins, C. M., Bramlett, H. M., et al. (2012). Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia 60, 1839–1859. doi: 10.1002/glia.22401
Giachini, F. R., Lima, V. V., Carneiro, F. S., Tostes, R. C., and Webb, R. C. (2011). Decreased cGMP level contributes to increased contraction in arteries from hypertensive rats: role of phosphodiesterase 1. Hypertension 57, 655–663. doi: 10.1161/HYPERTENSIONAHA.110.164327
Giralt, A., Saavedra, A., Carreton, O., Arumi, H., Tyebji, S., Alberch, J., et al. (2013). PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus 23, 684–695. doi: 10.1002/hipo.22128
Gong, B., Vitolo, O. V., Trinchese, F., Liu, S., Shelanski, M., and Arancio, O. (2004). Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Invest. 114, 1624–1634. doi: 10.1172/JCI22831
Haering, C., Kanageswaran, N., Bouvain, P., Scholz, P., Altmuller, J., Becker, C., et al. (2015). Ion transporter NKCC1, modulator of neurogenesis in murine olfactory neurons. J. Biol. Chem. 290, 9767–9779. doi: 10.1074/jbc.M115.640656
Hall, E. D., Sullivan, P. G., Gibson, T. R., Pavel, K. M., Thompson, B. M., and Scheff, S. W. (2005). Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J. Neurotrauma 22, 252–265. doi: 10.1089/neu.2005.22.252
Hansen, R. T. III, and Zhang, H. T. (2015). Phosphodiesterase-4 modulation as a potential therapeutic for cognitive loss in pathological and non-pathological aging: possibilities and pitfalls. Curr. Pharm. Des. 21, 291–302.
Heckman, P. R., Wouters, C., and Prickaerts, J. (2015). Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: a translational overview. Curr. Pharm. Des. 21, 317–331. doi: 10.2174/1381612820666140826114601
Henkel-Tigges, J., and Davis, R. L. (1990). Rat homologs of the Drosophila dunce gene code for cyclic AMP phosphodiesterases sensitive to rolipram and RO 20-1724. Mol. Pharmacol. 37, 7–10.
Houslay, M. D. (2010). Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 35, 91–100. doi: 10.1016/j.tibs.2009.09.007
Imanishi, T., Sawa, A., Ichimaru, Y., Miyashiro, M., Kato, S., Yamamoto, T., et al. (1997). Ameliorating effects of rolipram on experimentally induced impairments of learning and memory in rodents. Eur. J. Pharmacol. 321, 273–278. doi: 10.1016/S0014-2999(96)00969-7
Jin, S. L., and Conti, M. (2002). Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-α responses. Proc. Natl. Acad. Sci. U.S.A. 99, 7628–7633. doi: 10.1073/pnas.122041599
Johansson, E. M., Reyes-Irisarri, E., and Mengod, G. (2012). Comparison of cAMP-specific phosphodiesterase mRNAs distribution in mouse and rat brain. Neurosci. Lett. 525, 1–6. doi: 10.1016/j.neulet.2012.07.050
Kato, H., Araki, T., Itoyama, Y., and Kogure, K. (1995). Rolipram, a cyclic AMP-selective phosphodiesterase inhibitor, reduces neuronal damage following cerebral ischemia in the gerbil. Eur. J. Pharmacol. 272, 107–110. doi: 10.1016/0014-2999(94)00694-3
Kelly, M. P., and Brandon, N. J. (2009). Differential function of phosphodiesterase families in the brain: gaining insights through the use of genetically modified animals. Prog. Brain Res. 179, 67–73. doi: 10.1016/S0079-6123(09)17908-6
Kharlamov, E. A., Lepsveridze, E., Meparishvili, M., Solomonia, R. O., Lu, B., Miller, E. R., et al. (2011). Alterations of GABAA and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. 95, 20–34. doi: 10.1016/j.eplepsyres.2011.02.008
Klann, E., Roberson, E. D., Knapp, L. T., and Sweatt, J. D. (1998). A role for superoxide in protein kinase C activation and induction of long-term potentiation. J. Biol. Chem. 273, 4516–4522. doi: 10.1074/jbc.273.8.4516
Kunal, S. B., Killivalavan, A., and Medhamurthy, R. (2012). Involvement of Src family of kinases and cAMP phosphodiesterase in the luteinizing hormone/chorionic gonadotropin receptor-mediated signaling in the corpus luteum of monkey. Reprod. Biol. Endocrinol. 10:25. doi: 10.1186/1477-7827-10-25
Kuroiwa, M., Snyder, G. L., Shuto, T., Fukuda, A., Yanagawa, Y., Benavides, D. R., et al. (2012). Phosphodiesterase 4 inhibition enhances the dopamine D1 receptor/PKA/DARPP-32 signaling cascade in frontal cortex. Psychopharmacology (Berl.) 219, 1065–1079. doi: 10.1007/s00213-011-2436-8
Lakics, V., Karran, E. H., and Boess, F. G. (2010). Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59, 367–374. doi: 10.1016/j.neuropharm.2010.05.004
Langlois, J. A., Rutland-Brown, W., and Wald, M. M. (2006). The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378. doi: 10.1097/00001199-200609000-00001
Lew, H. L., Poole, J. H., Guillory, S. B., Salerno, R. M., Leskin, G., and Sigford, B. (2006). Persistent problems after traumatic brain injury: the need for long-term follow-up and coordinated care. J. Rehabil. Res. Dev. 43, 7–10. doi: 10.1682/JRRD.2006.05.0054
Li, W., Dai, S., An, J., Li, P., Chen, X., Xiong, R., et al. (2008). Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience 151, 1198–1207. doi: 10.1016/j.neuroscience.2007.11.020
Li, Y. F., Cheng, Y. F., Huang, Y., Conti, M., Wilson, S. P., O’Donnell, J. M., et al. (2011). Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J. Neurosci. 31, 172–183. doi: 10.1523/JNEUROSCI.5236-10.2011
MacKenzie, K. F., Wallace, D. A., Hill, E. V., Anthony, D. F., Henderson, D. J., Houslay, D. M., et al. (2011). Phosphorylation of cAMP-specific PDE4A5 (phosphodiesterase-4A5) by MK2 (MAPKAPK2) attenuates its activation through protein kinase A phosphorylation. Biochem. J. 435, 755–769. doi: 10.1042/BJ20101184
MacKenzie, S. J., Baillie, G. S., McPhee, I., MacKenzie, C., Seamons, R., McSorley, T., et al. (2002). Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br. J. Pharmacol. 136, 421–433. doi: 10.1038/sj.bjp.0704743
Man, H. Y., Sekine-Aizawa, Y., and Huganir, R. L. (2007). Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. U.S.A. 104, 3579–3584. doi: 10.1073/pnas.0611698104
Martinez, A., and Gil, C. (2014). cAMP-specific phosphodiesterase inhibitors: promising drugs for inflammatory and neurological diseases. Expert Opin. Ther. Pat. 24, 1311–1321. doi: 10.1517/13543776.2014.968127
Maurice, D. H., Ke, H., Ahmad, F., Wang, Y., Chung, J., and Manganiello, V. C. (2014). Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 13, 290–314. doi: 10.1038/nrd4228
Maxwell, W. L., Dhillon, K., Harper, L., Espin, J., Macintosh, T. K., Smith, D. H., et al. (2003). There is differential loss of pyramidal cells from the human hippocampus with survival after blunt head injury. J. Neuropathol. Exp. Neurol. 62, 272–279. doi: 10.1093/jnen/62.3.272
Menniti, F. S., Faraci, W. S., and Schmidt, C. J. (2006). Phosphodiesterases in the CNS: targets for drug development. Nat. Rev. Drug Discov. 5, 660–670. doi: 10.1038/nrd2058
Middei, S., Houeland, G., Cavallucci, V., Ammassari-Teule, M., D’amelio, M., and Marie, H. (2013). CREB is necessary for synaptic maintenance and learning-induced changes of the AMPA receptor GluA1 subunit. Hippocampus 23, 488–499. doi: 10.1002/hipo.22108
Miyazaki, S., Katayama, Y., Lyeth, B. G., Jenkins, L. W., Dewitt, D. S., Goldberg, S. J., et al. (1992). Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 585, 335–339. doi: 10.1016/0006-8993(92)91232-4
Mons, N., Segu, L., Nogues, X., and Buhot, M. C. (2004). Effects of age and spatial learning on adenylyl cyclase mRNA expression in the mouse hippocampus. Neurobiol. Aging 25, 1095–1106. doi: 10.1016/j.neurobiolaging.2003.10.014
Nagakura, A., Takagi, N., and Takeo, S. (2002). Selective reduction in type I adenylyl cyclase after microsphere embolism in rat brain. Neurosci. Lett. 317, 69–72. doi: 10.1016/S0304-3940(01)02421-1
Navakkode, S., Sajikumar, S., and Frey, J. U. (2004). The type IV-specific phosphodiesterase inhibitor rolipram and its effect on hippocampal long-term potentiation and synaptic tagging. J. Neurosci. 24, 7740–7744. doi: 10.1523/JNEUROSCI.1796-04.2004
Oh, M. C., Derkach, V. A., Guire, E. S., and Soderling, T. R. (2006). Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 281, 752–758. doi: 10.1074/jbc.M509677200
Oliva, A. A. Jr., Kang, Y., Furones, C., Alonso, O. F., Bruno, O., Dietrich, W. D., et al. (2012). Phosphodiesterase isoform-specific expression induced by traumatic brain injury. J. Neurochem. 123, 1019–1029. doi: 10.1111/jnc.12049
Omori, K., and Kotera, J. (2007). Overview of PDEs and their regulation. Circ. Res. 100, 309–327. doi: 10.1161/01.RES.0000256354.95791.f1
Perez-Torres, S., Miro, X., Palacios, J. M., Cortes, R., Puigdomenech, P., and Mengod, G. (2000). Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and [3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J. Chem. Neuroanat. 20, 349–374.
Ramos, B. P., Birnbaum, S. G., Lindenmayer, I., Newton, S. S., Duman, R. S., and Arnsten, A. F. (2003). Dysregulation of protein kinase A signaling in the aged prefrontal cortex: new strategy for treating age-related cognitive decline. Neuron 40, 835–845. doi: 10.1016/S0896-6273(03)00694-9
Reneerkens, O. A., Rutten, K., Steinbusch, H. W., Blokland, A., and Prickaerts, J. (2009). Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology (Berl.) 202, 419–443. doi: 10.1007/s00213-008-1273-x
Rutten, K., Basile, J. L., Prickaerts, J., Blokland, A., and Vivian, J. A. (2008a). Selective PDE inhibitors rolipram and sildenafil improve object retrieval performance in adult cynomolgus macaques. Psychopharmacology (Berl.) 196, 643–648. doi: 10.1007/s00213-007-0999-1
Rutten, K., Misner, D. L., Works, M., Blokland, A., Novak, T. J., Santarelli, L., et al. (2008b). Enhanced long-term potentiation and impaired learning in phosphodiesterase 4D-knockout (PDE4D) mice. Eur. J. Neurosci. 28, 625–632. doi: 10.1111/j.1460-9568.2008.06349.x
Schaal, S. M., Garg, M. S., Ghosh, M., Lovera, L., Lopez, M., Patel, M., et al. (2012). The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury. PLoS ONE 7:e43634. doi: 10.1371/journal.pone.0043634
Scheff, S. W., Price, D. A., Hicks, R. R., Baldwin, S. A., Robinson, S., and Brackney, C. (2005). Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J. Neurotrauma 22, 719–732. doi: 10.1089/neu.2005.22.719
Schumann, J., Alexandrovich, G. A., Biegon, A., and Yaka, R. (2008). Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J. Neurotrauma 25, 945–957. doi: 10.1089/neu.2008.0521
Shepherd, M. C., Baillie, G. S., Stirling, D. I., and Houslay, M. D. (2004). Remodelling of the PDE4 cAMP phosphodiesterase isoform profile upon monocyte-macrophage differentiation of human U937 cells. Br. J. Pharmacol. 142, 339–351. doi: 10.1038/sj.bjp.0705770
Shimizu-Albergine, M., Tsai, L. C., Patrucco, E., and Beavo, J. A. (2012). cAMP-specific phosphodiesterases 8A and 8B, essential regulators of Leydig cell steroidogenesis. Mol. Pharmacol. 81, 556–566. doi: 10.1124/mol.111.076125
Sierksma, A. S., Van Den Hove, D. L., Pfau, F., Philippens, M., Bruno, O., Fedele, E., et al. (2014). Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 77, 120–130. doi: 10.1016/j.neuropharm.2013.09.015
Soler, F., Fernandez-Belda, F., Perez-Schindler, J., and Hernandez-Cascales, J. (2015). Single inhibition of either PDE3 or PDE4 unmasks β2-adrenoceptor-mediated inotropic and lusitropic effects in the left but not right ventricular myocardium of rat. Eur. J. Pharmacol. 765, 429–436. doi: 10.1016/j.ejphar.2015.09.001
Spina, D. (2008). PDE4 inhibitors: current status. Br. J. Pharmacol. 155, 308–315. doi: 10.1038/bjp.2008.307
Suhasini, A. N., Wang, L., Holder, K. N., Lin, A. P., Bhatnagar, H., Kim, S. W., et al. (2015). A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia [Epub ahead of print].
Temple, M. D., Delahunty, T. M., Hamm, R. J., Phillips, L. L., Lyeth, B. G., and Povlishock, J. T. (2001). Subtle alterations in NMDA-stimulated cyclic GMP levels following lateral fluid percussion brain injury. J. Neurotrauma 18, 47–55. doi: 10.1089/089771501750055767
Titus, D. J., Furones, C., Atkins, C. M., and Dietrich, W. D. (2015a). Emergence of cognitive deficits after mild traumatic brain injury due to hyperthermia. Exp. Neurol. 263, 254–262. doi: 10.1016/j.expneurol.2014.10.020
Titus, D. J., Oliva, A. A., Wilson, N. M., and Atkins, C. M. (2015b). Phosphodiesterase inhibitors as therapeutics for traumatic brain injury. Curr. Pharm. Des. 21, 332–342. doi: 10.2174/1381612820666140826113731
Titus, D. J., Furones, C., Kang, Y., and Atkins, C. M. (2013a). Age-dependent alterations in cAMP signaling contribute to synaptic plasticity deficits following traumatic brain injury. Neuroscience 231, 182–194. doi: 10.1016/j.neuroscience.2012.12.002
Titus, D. J., Sakurai, A., Kang, Y., Furones, C., Jergova, S., Santos, R., et al. (2013b). Phosphodiesterase inhibition rescues chronic cognitive deficits induced by traumatic brain injury. J. Neurosci. 33, 5216–5226. doi: 10.1523/JNEUROSCI.5133-12.2013
Tomaiuolo, F., Carlesimo, G. A., Di Paola, M., Petrides, M., Fera, F., Bonanni, R., et al. (2004). Gross morphology and morphometric sequelae in the hippocampus, fornix, and corpus callosum of patients with severe non-missile traumatic brain injury without macroscopically detectable lesions: a T1 weighted MRI study. J. Neurol. Neurosurg. Psychiatry 75, 1314–1322. doi: 10.1136/jnnp.2003.017046
Wiescholleck, V., and Manahan-Vaughan, D. (2012). PDE4 inhibition enhances hippocampal synaptic plasticity in vivo and rescues MK801-induced impairment of long-term potentiation and object recognition memory in an animal model of psychosis. Transl. Psychiatry 2:e89. doi: 10.1038/tp.2012.17
Witgen, B. M., Lifshitz, J., Smith, M. L., Schwarzbach, E., Liang, S. L., Grady, M. S., et al. (2005). Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience 133, 1–15. doi: 10.1016/j.neuroscience.2005.01.052
Yamamoto, M., Gotz, M. E., Ozawa, H., Luckhaus, C., Saito, T., Rosler, M., et al. (2000). Hippocampal level of neural specific adenylyl cyclase type I is decreased in Alzheimer’s disease. Biochim. Biophys. Acta 1535, 60–68. doi: 10.1016/S0925-4439(00)00083-1
Keywords: cAMP, CREB, fluid-percussion, hippocampus, inflammation, phosphodiesterase, traumatic brain injury
Citation: Wilson NM, Titus DJ, Oliva AA Jr., Furones C and Atkins CM (2016) Traumatic Brain Injury Upregulates Phosphodiesterase Expression in the Hippocampus. Front. Syst. Neurosci. 10:5. doi: 10.3389/fnsys.2016.00005
Received: 30 September 2015; Accepted: 18 January 2016;
Published: 05 February 2016.
Edited by:
Akiva Cohen, University of Pennsylvania/Children’s Hospital of Philadelphia, USAReviewed by:
Ronald Davis, Baylor College of Medicine, USACandace L. Floyd, University of Alabama at Birmingham, USA
Copyright © 2016 Wilson, Titus, Oliva, Furones and Atkins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Coleen M. Atkins, catkins@med.miami.edu
†These authors have contributed equally to this work.