Loss of Atoh1 from neurons regulating hypoxic and hypercapnic chemoresponses causes neonatal respiratory failure in mice
- Baylor College of Medicine, United States
- Texas Children’s Hospital, United States
Abstract
Atoh1-null mice die at birth from respiratory failure, but the precise cause has remained elusive. Loss of Atoh1 from various components of the respiratory circuitry (e.g. the retrotrapezoid nucleus (RTN)) has so far produced at most 50% neonatal lethality. To identify other Atoh1-lineage neurons that contribute to postnatal survival, we examined parabrachial complex neurons derived from the rostral rhombic lip (rRL) and found that they are activated during respiratory chemochallenges. Atoh1-deletion from the rRL does not affect survival, but causes apneas and respiratory depression during hypoxia, likely due to loss of projections to the preBötzinger Complex and RTN. Atoh1 thus promotes the development of the neural circuits governing hypoxic (rRL) and hypercapnic (RTN) chemoresponses, and combined loss of Atoh1 from these regions causes fully penetrant neonatal lethality. This work underscores the importance of modulating respiratory rhythms in response to chemosensory information during early postnatal life.
https://doi.org/10.7554/eLife.38455.001eLife digest
Breathing seems very simple: humans and other animals do it all the time without even thinking about it. Yet, many different cell types coordinate rhythmic breathing movements. Some cells set the breathing rhythm, motor neurons control the muscles, and other cells sense blood oxygen and carbon dioxide levels. Information about oxygen and carbon dioxide is necessary to trigger faster and deeper breaths when there is too little oxygen, for example, at high altitude. Or when there is too much carbon dioxide, for example, during exercise.
At birth, most newborns can breathe as fast as needed because key genes oversee the development of all the cells involved in breathing. Learning more about these genes and what they do could lead to better understanding of why some newborns are at risk for sudden infant death or crib death. The Atoh1 gene, for example, helps carbon dioxide-sensing cells called retrotrapezoid neurons develop. Mice born without the Atoh1 gene are unable to breathe normally and die at birth. But when the gene is only deleted from these carbon dioxide-sensing cells in mice, just half of them die. This suggests that Atoh1 in other cells may also be important for breathing.
Now, Van der Heijden and Zoghbi show that the Atoh1 gene also helps develop another set of cells that are essential for breathing called the parabrachial complex. These cells receive information from oxygen sensors and relay the information to cells that set breathing rhythms. Mice missing parabrachial complex cells do not breathe faster when oxygen levels in the air are low. Mice lacking Atoh1 from both the parabrachial complex cells and the retrotrapezoid cells have breathing problems and die at birth.
Van der Heijden and Zoghbi show that the Atoh1 gene is essential for two cell types that make mice breathe faster when oxygen or carbon dioxide levels change. Together these two cell types are necessary for survival. The experiments also may provide insights into what goes wrong in babies who experience sudden infant death. Mutations in genes that are important to both cell types increase the risk of these infant deaths. Newborn babies with mutations in such key developmental genes will be at risk when in low oxygen or high carbon dioxide environments because their breathing systems are still maturing.
https://doi.org/10.7554/eLife.38455.002Introduction
Hard-wired, transcriptionally defined neural circuit development is often complemented by synaptic plasticity that is driven by feedback from experience. Yet circuits giving rise to vital functions, such as respiration, have no time for such trial and error: the animal must be able to maintain its own O2/CO2 homeostasis from the moment it is born. This ability likely arises from a detailed genetic blueprint in the hindbrain respiratory circuit. Indeed, mapping the expression domains of key transcription factors in the developing hindbrain reveals a checkerboard pattern, with rostro-caudal and dorso-ventral stripes crisscrossing the entire region (Gray, 2008, 2013; Pagliardini et al., 2008; Pasqualetti et al., 2007). The complexity of the circuit and the relative inaccessibility of some its individual components have made it difficult to tease out the specific contributions of various neuronal populations to neonatal survival.
For example, mice lacking the transcription factor Atonal homolog 1 (Atoh1) die at birth of respiratory failure even though they generate some respiratory movements (Ben-Arie et al., 1997; Rose et al., 2009b; Tupal et al., 2014). The precise cause of this fully penetrant lethality has eluded a number of studies, which have nonetheless deepened our understanding of Atoh1’s contributions to the respiratory circuit. Atoh1 is expressed in the developing hindbrain along the entire rostro-caudal rhombic lip, where it promotes the development of several nuclei involved in respiratory control (Gray, 2013; Rose et al., 2009a, 2009b; Wang et al., 2005). Atoh1 is also expressed in postmitotic neurons of two paramotor nuclei: the intertrigeminal region (ITR) and the retrotrapezoid nucleus (RTN) (Figure 1A and B) (Huang et al., 2012; Rose et al., 2009b). The chemosensitive RTN sends excitatory projections to the preBötzinger complex (preBötC), which generates inspiratory rhythms (Guyenet and Bayliss, 2015; Guyenet et al., 2010; Kumar et al., 2015; Onimaru and Homma, 2003). Atoh1 loss from the RTN impairs respiratory responses to hypercapnia, but, rather remarkably, causes only partial neonatal lethality (Huang et al., 2012).
Figure 1 with 2 supplements see all
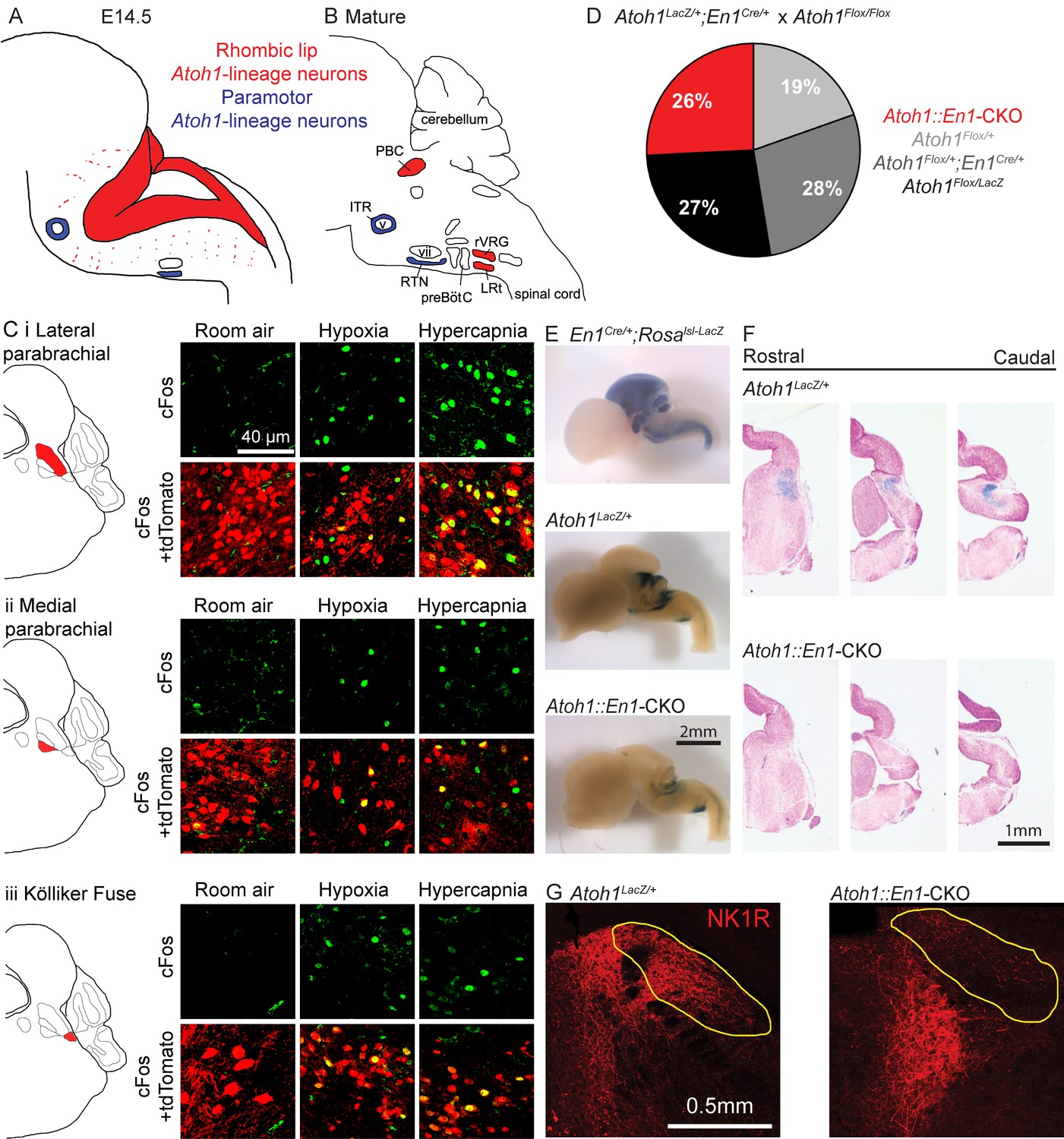
Atoh1-lineage parabrachial complex neurons are activated during chemochallenges but are not essential for neonatal survival.
(A) Schematic of Atoh1-expression cells in the developing brainstem (E14.5). Red area represents proliferating cells in the rhombic lip. Blue cells are postmitotic neurons in paramotor nuclei. (B) Schematic of neural populations in the brainstem respiratory circuitry. Red nuclei represent rhombic lip, Atoh1-lineage neurons important for respiratory control. Blue nuclei are Atoh1-lineage neurons in two paramotor nuclei. (C) Atoh1-lineage neurons (tdTomato+) in three sub compartments of the parabrachial complex (i) lateral parabrachial, (ii) medial parabrachial and (iii) Kölliker Fuse) express the neural activity marker cFos selectively after a one-hour-exposure to hypoxia (10% O2, balanced N2) or hypercapnia (5% CO2, 21% O2, balanced N2). (D) Atoh1::En1-CKO mice are born and survive in Mendelian ratios. (E) X-gal staining in En1Cre/+;Rosalsl-LacZ reporter allele, Atoh1LacZ/+ and Atoh1::En1-CKO E14.5 embryos to visualize En1Cre expression and Atoh1-lineage cells in the developing brain. (F) Serial sections of X-gal-stained brains at the level of the pons in Atoh1LacZ/+ and Atoh1::En1-CKO mice. No Atoh1-lineage pontine PBC neurons develop in Atoh1::En1-CKO mice at E14.5. (G) Stain for NK1R receptor that is highly expressed in Atoh1-lineage PBC neurons. Loss of NK1R expression in Atoh1::En1-CKO mice at P21. Abbreviations: PBC, parabrachial complex; ITR, intertrigeminal region; RTN, retrotrapezoid nucleus; rVRG, rostral ventral respiratory group; LRt, lateral reticular; preBötC, preBötzinger complex; v, trigeminal motor nucleus; vii, facial motor nucleus.
We therefore set out to find other Atoh1-lineage neurons contributing to neonatal survival and further delineate Atoh1’s function in respiratory development using intersectional genetics. We found that loss of Atoh1-lineage neurons developing from the rostral rhombic lip (rRL) impairs both respiratory rhythm and chemoresponsiveness. Some of these neurons are specifically activated during respiratory chemoresponses and project to the Atoh1-lineage paramotor nuclei (ITR and RTN) as well as the preBötC. Yet only the combined deletion of Atoh1 from the rRL and RTN recapitulated the fully penetrant lethality of Atoh1-null mice. This confirms that developmentally defined neural lineages have distinct roles in respiratory control and that, in neonatal mice, integration of chemosensory information is essential for survival.
Results
Rostral rhombic lip neurons are activated during chemochallenges
Atoh1 is expressed along the entire rostro-caudal rhombic lip of the developing hindbrain (Figure 1A, red), where it functions as a proneural transcription factor. Loss of Atoh1 results in loss of proliferating cells in the rhombic lip. Among the rhombic lip derived Atoh1-lineage are three populations of neurons that have been implicated in respiratory control: the parabrachial complex (PBC), the rostral ventral respiratory group (rVRG) and the lateral reticular nucleus (LRt) (Figure 1B, red) (Rose et al., 2009b; Tupal et al., 2014). Two paramotor nuclei express Atoh1 during the postmitotic phase, and its expression is essential for their proper migration and connectivity from the RTN to the preBötC (Figure 1A and B, blue) (Huang et al., 2012; Rose et al., 2009b).
To date, the Atoh1-lineage PBC neurons are the only Atoh1-lineage neurons whose role in respiratory control and neonatal survival was not assessed. We first tested whether Atoh1-lineage PBC neurons might have a role in respiratory chemoresponses. To test this we labeled the Atoh1-lineage with tdTomato using a Cre-dependent reporter allele (Atoh1Cre/+;Rosalsl-tdTomato/+ mice) and exposed these mice to either room air, hypoxia, or hypercapnia prior to staining for the neural activity marker cFos. We found tdTomato+, cFos+ double-positive cells in the medial and lateral parabrachial region as well as in the Kölliker Fuse after exposure to either hypoxia or hypercapnia, but not room air (Figure 1C). This confirms that Atoh1-lineage PBC neurons are activated by changes in O2 and CO2 and might play a role in respiratory function.
There are no previous reports that PBC neurons are intrinsically chemosensitive, so these neurons are likely activated by upstream neurons that are chemosensitive. Previous studies showed that Atoh1-null mice lose the substance P receptor NK1R in the PBC region (Rose et al., 2009b). We found that indeed all NK1R-expressing PBC neurons were Atoh1-lineage neurons (Figure 1—figure supplement 1A). We also looked whether Atoh1-lineage PBC neurons expressed calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase‐activating polypeptide (PACAP), because these peptides have been implicated in playing a role for hypercapnic and hypoxic responses respectively (Arata et al., 2013; Cummings et al., 2004; Kaur et al., 2017; Yokota et al., 2015). We found that all counted Atoh1-lineage neurons in the lateral PBC expressed CGRP and that some expressed PACAP (Figure 1—figure supplement 1A). Together, these results suggest that Atoh1-lineage neurons might be important for respiratory responses through signaling with one or both of these neuro peptides.
Next, we assessed whether these Atoh1-lineage neurons are required for neonatal survival. As PBC neurons develop from the rRL, we deleted Atoh1 only from this domain using an Engrailed-1-driven Cre-line (En1Cre). We crossed Atoh1Flox/Flox females (Shroyer et al., 2007) to males heterozygous for En1Cre/+ and Atoh1LacZ/+, which is a functional Atoh1-null allele that can be used to trace Atoh1-expressing neurons throughout development (Ben-Arie et al., 2000). Atoh1Flox/LacZ;En1Cre/+ mice (hereafter Atoh1::En1-CKO) were born in Mendelian ratios (25/97 surviving pups, Figure 1D). We stained E14.5 embryos with X-gal and confirmed that Atoh1::En1-CKO lost all Atoh1-expressing neurons from the rRL, while leaving other Atoh1-lineage neurons intact (Figure 1E and F). Anatomical analysis of postnatal (P21) animals confirmed that this conditional deletion of Atoh1 resulted in loss of NK1R-expressing PBC neurons (Figure 1G). Thus, despite losing these Atoh1-lineage PBC neurons, these animals survive, showing that Atoh1 expression in the rRL is not necessary for neonatal survival.
Atoh1::En1-CKO mice developed severe ataxia, dystonia and tremor in the second to third week after birth and died shortly after weaning (P22-25), probably because the motor phenotypes impair their ability to get proper amounts of food and water. These phenotypes were likely the result of loss of Atoh1-lineage cerebellar neurons including glutamatergic deep cerebellar nuclei and cerebellar granule cells (Figure 1—figure supplement 2A) (Ben-Arie et al., 1997; Wurst et al., 1994). Unlike Atoh1-lineage PBC neurons, however, these Atoh1-lineage cerebellar neurons are not activated during respiratory chemochallenges and are thus less likely to be important for respiratory chemoresponses (Figure 1—figure supplement 2B and C).
Rostral rhombic lip neurons contribute to respiratory rhythms
Given that Atoh1::En1-CKO mice survive the early neonatal period, we were able to examine their respiration using unrestrained whole-body plethysmography (UWBP) at three weeks of age (Figure 2A). In room air, Atoh1::En1-CKO mice had a greater number of sigh-induced and spontaneous apneas, sighs, and irregular respiratory rhythms than their control littermates (Figure 2B). No other respiratory parameters were affected (Figure 2—figure supplement 1).
Figure 2 with 1 supplement see all
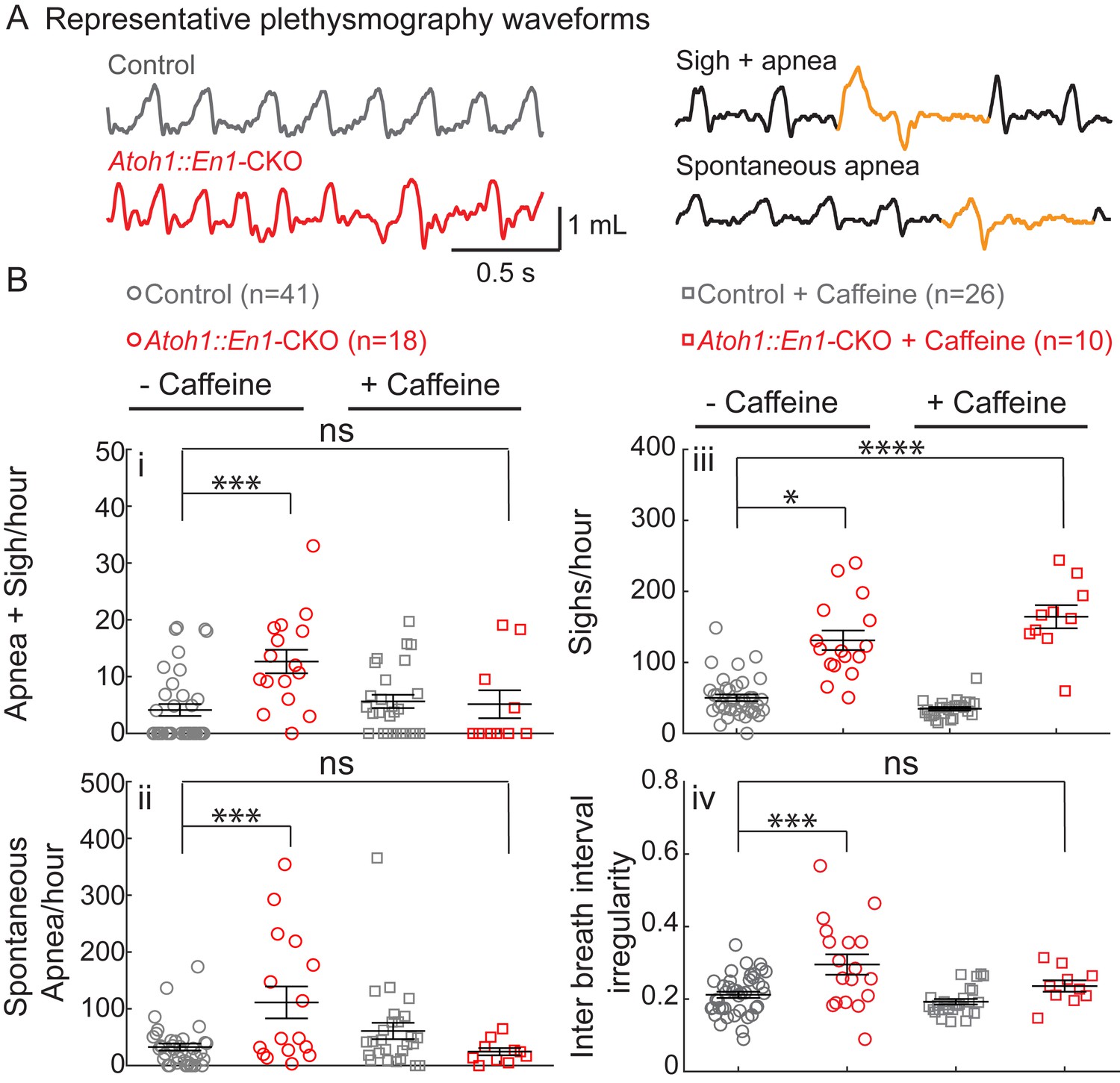
Atoh1::En1-CKO mice have many apneas causing irregular breathing rhythms that can be rescued by caffeine treatment in room air.
(A) Representative plethysmography traces from a control and Atoh1::En1-CKO mouse. Example traces of apnea and sigh. (Bi) Atoh1::En1-CKO mice have more apneas following sighs and (Bii) spontaneous apneas per hour than control littermates. Caffeine treatment rescues apneas. (Biii) Atoh1::En1-CKO mice have more sighs per hour than control littermates, which was not be rescued by caffeine. (Biv) Atoh1::En1-CKO mice breathe more irregularly than control littermates, which can be rescued with caffeine treatment. Inter breath interval irregularity was defined as: absolute (breath length(n + 1) – breath length(n)/breath length(n). Significance was determined using a Two-way ANOVA (genotype*treatment), Tukey-Kramer post-hoc. *p<0.05. **p<0.01. ***p<0.001. ****p<0.0001. Error bars represent: mean ± SEM.
-
Figure 2—source data 1
Raw plethysmography data room air recordings.
- https://doi.org/10.7554/eLife.38455.008
Apneas, sighs, and rhythmic irregularity are hallmarks of immature respiration that can occur in some human infants (Abu-Shaweesh and Martin, 2008; Martin et al., 2004; Abu-Shaweesh, 2004). When infants present with apnea of prematurity (AOP) in a clinical setting, they are treated with caffeine to stabilize their breathing rhythms (Aranda et al., 1977; Natarajan et al., 2007). We therefore tested whether caffeine treatment could rescue respiratory rhythms in Atoh1::En1-CKO mice by administering caffeine through the drinking water of lactating dams from P2 onward. This was sufficient to detect caffeine levels in the blood plasma of the pups (treated: 6.15 ± 2.1 mg/L caffeine; untreated: 0.15 ± 0.04 mg/L caffeine; p=0.02, two-tailed t-test). These levels are similar to those observed in infants treated with caffeine (Natarajan et al., 2007).
Caffeine treatment normalized apnea frequency and irregular breathing rhythms, but not sighs (Figure 2B), showing that caffeine is sufficient to stabilize irregular breathing rhythms in our mice similar to human infants. Much to our surprise, this method of caffeine treatment also significantly decreased minute ventilation in control mice, as a result of both decreased tidal volume and breathing frequency (Figure 2—figure supplement 1). Although Atoh1::En1-CKO mice also showed a decrease in tidal volume, they seemingly compensated by increasing their rate of respiration, resulting in normal minute ventilation compared to control conditions.
Rostral rhombic lip neurons are essential for respiratory chemoresponsiveness
As Atoh1-lineage PBC neurons are specifically activated during hypoxia and hypercapnia, we hypothesized that these neurons also contribute to respiratory chemoresponses. We therefore assessed whether Atoh1::En1-CKO mice showed abnormal responses to hypoxia and whether the caffeine treatment rescued their irregular respiratory rhythms by restoring proper chemoresponses. Control mice had a short initial increase in tidal volume and breathing frequency that returned to baseline within several minutes of exposure to hypoxia (Figure 3A and B). This resulted in a bimodal response in minute ventilation (Figure 3C), similar to what others have reported in juvenile mice (Haddad and Mellins, 1984; Waters and Gozal, 2003). Atoh1::En1-CKO mice also showed a brief initial increase in tidal volume (Figure 3A), but this was accompanied by a rapid, sustained decrease in respiratory rate that repressed minute ventilation during hypoxia (Figure 3D). Interestingly, while caffeine treatment in control mice did prolong the respiratory response to hypoxia by limiting respiratory depression during exposure to hypoxic gas (Figure 3C), it did not improve respiratory chemoresponses in Atoh1::En1-CKO mice (Figure 3D). Respiratory depression in response to hypoxia resembles the suppression of fetal breathing movements during hypoxia in prenatal mammals (Gluckman and Johnston, 1987; Haddad and Mellins, 1984; Abu-Shaweesh, 2004; Waters and Gozal, 2003), underscoring how loss of rRL neurons recapitulates many aspects of immature breathing control.
Figure 3 with 1 supplement see all

Atoh1::En1-CKO mice have abnormal respiratory chemoresponses that cannot be rescued by caffeine.
Normalized changes in tidal volume (TV) (A), respiratory frequency (Vf) (B), and minute ventilation (VE) (C and D) during hypoxic challenge (10% O2, balanced N2). Atoh1::En1-CKO mice show respiratory repression during hypoxia (B) that cannot be rescued by caffeine (D). Normalized changes in TV (E), Vf (F), and VE (G and H) during hypercapnic chemochallenge (5% CO2, 21% O2, balanced N2). Atoh1::En1-CKO mice have attenuated response to hypercapnia (E and F) that cannot be rescued by caffeine (H). Significance was determined using a t-test (2-tailed) at each individual time point, *p<0.0011 (0.05/44 for Bonferroni correction). Error bars represent mean ± SEM.
-
Figure 3—source data 1
Raw plethysmography data respiratory chemoresponses.
- https://doi.org/10.7554/eLife.38455.012
We next assessed how Atoh1::En1-CKO mice responded to hypercapnia. We found that control littermates showed a rapid increase in tidal volume and breathing frequency during hypercapnia, whereas respiratory chemoresponses of Atoh1::En1-CKO mice were severely attenuated (Figure 3E and F). Caffeine treatment delayed the return to baseline minute ventilation in control mice, but did not improve the hypercapnic chemoresponses of Atoh1::En1-CKO littermates (Figure 3G and H).
Thus, the mechanism by which caffeine rescues apneas, sighs, and irregular respiratory rhythms in Atoh1::En1-CKO mice, cannot be through normalizing chemoresponses and these results confirm that Atoh1-lineage rRL neurons contribute to respiratory chemoresponses to low oxygen and high carbon dioxide.
The cerebellar cortex is not essential for respiratory chemoresponses
As neither Atoh1-lineage deep cerebellar nuclei nor cerebellar granule cells were activated during respiratory chemochallenges, we hypothesized that the abnormal chemoresponses, seen upon deletion of Atoh1 from the rRL, were not due to cerebellar dysfunction. To date, there is no Cre-line that can specifically delete Atoh1 from the pontine PBC without affecting the cerebellum, or vice versa. Therefore, we tested our hypothesis by silencing neurotransmission of Purkinje cells (PCs), which are the sole output of the cerebellar cortex. PCs receive input from granule cells and directly project onto deep cerebellar nuclei. These Pcp2Cre/+;Slc32a1Flox/Flox mice have no defects in cell types other than PCs, and loss of PC signaling results in abnormal motor control including ataxia and poor performance on the rotarod (White et al., 2014). Despite this abnormal cerebellar function, Pcp2Cre/+;Slc32a1Flox/Flox mice do not have more apneas or sighs than control littermates during room air breathing, although they have slightly more irregular respiratory rhythms than their control littermates (Figure 3—figure supplement 1Aiv). No other respiratory parameters (Figure 3—figure supplement 1A) such as respiratory chemoresponses to either hypoxia or hypercapnia were altered upon silencing of PCs (Figure 3—figure supplement 1B). Thus, abnormal cerebellar function or ataxia does not explain the abnormal respiratory chemoresponses observed in Atoh1::En1-CKO mice.
Rostral rhombic lip neurons are important for respiratory control in P7 mice
Atoh1::En1-CKO mice display irregular breathing rhythms, sighs, and apneas, as well as respiratory depression in response to hypoxia and attenuated respiratory response to hypercapnia. As noted above, these features characterize immature respiration in some human infants, although they usually resolve on their own with time (Abu-Shaweesh and Martin, 2008; Abu-Shaweesh, 2004). Our results suggest that rRL neurons might play a prominent role in the postnatal maturation of respiratory control.
To test this hypothesis, we evaluated respiratory control in one-week-old (P7) Atoh1::En1-CKO mice and control littermates (Figure 4A). At this age, Atoh1::En1-CKO mice display about twice as many apneas as their control littermates, but do not sigh more (Figure 4B). We also observed that P7 Atoh1::En1-CKO mice have longer inhalation times and shallower breaths, resulting in a slower breathing rhythm and smaller minute ventilation (Figure 4B).
Figure 4
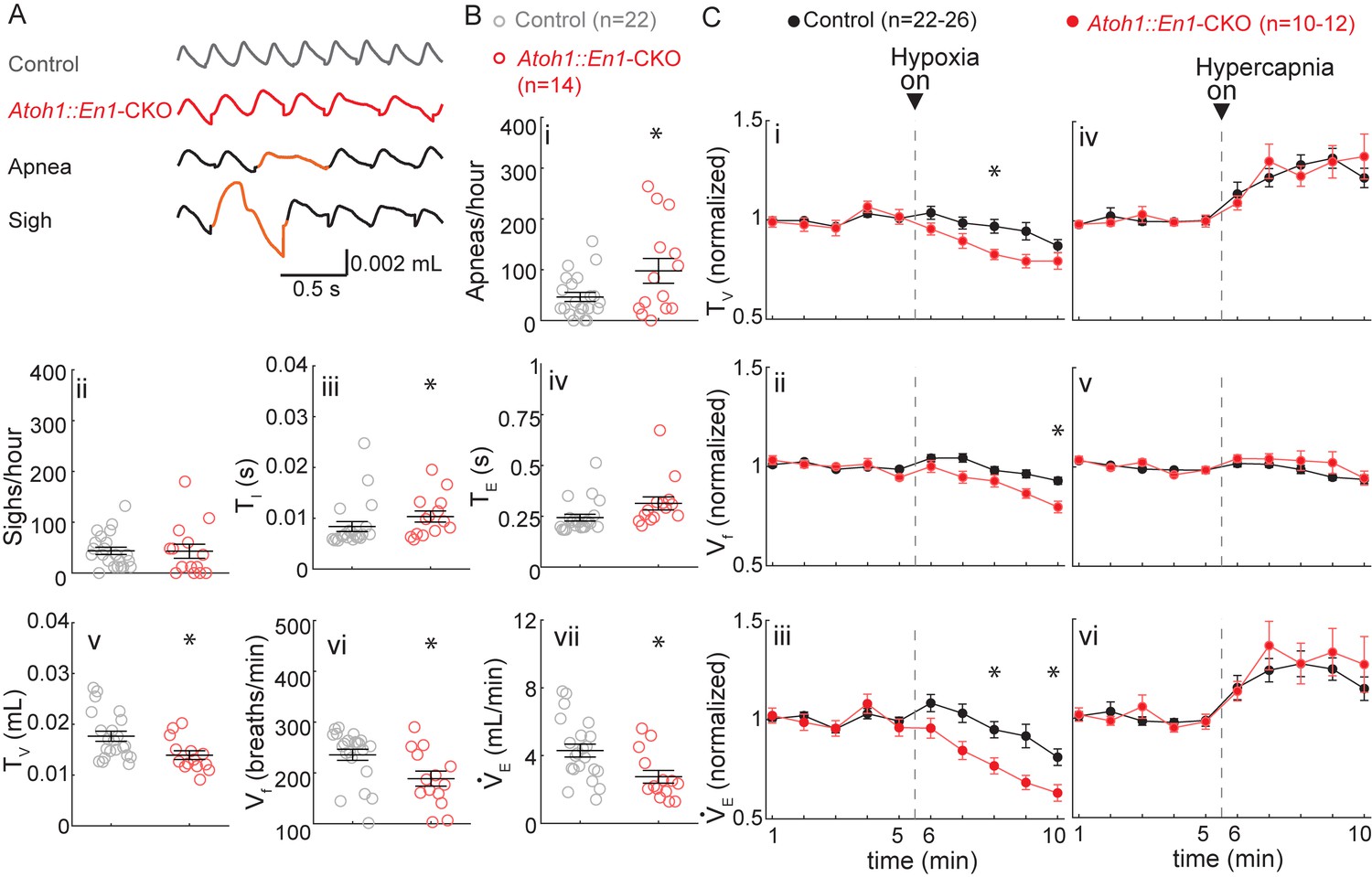
P7 Atoh1::En1-CKO mice have abnormal respiratory control.
(A) Representative plethysmography traces from a control and Atoh1::En1-CKO mouse. Example traces of apnea and sigh. (B) One-week-old Atoh1::En1-CKO mice have significantly more apneas (i), longer inspiratory time (TI) (iii), smaller tidal volume (TV) (v), slower respiratory rhythms (Vf) (vi), and lower minute ventilation (VE) (vii). Number of sighs (ii) and expiratory time (TE) (iv) were not different. Significance for room air breathing parameters were determined using a t-test (2-tailed). *p<0.05. (C) One-week-old Atoh1::En1-CKO mice have enhanced respiratory depression in response to hypoxia through an enhanced decrease in TV (i) and Vf (ii), resulting in a decreased VE(iii). There were no significant differences in the respiratory chemoresponses to hypercapnia (iv to vi). Significance was determined using a t-test (2-tailed) at each individual time point, *p<0.01. Error bars represent mean ± SEM.
-
Figure 4—source data 1
Raw data P7 plethysmography recordings.
- https://doi.org/10.7554/eLife.38455.014
In response to hypoxia, control mice showed the bimodal respiratory response we observed in P21 Atoh1::En1-CKO mice with an initial increase in tidal volume and respiratory frequency, followed by respiratory depression (Figure 4Ci to Ciii). Nevertheless, the decrease in tidal volume and respiratory frequency was initiated earlier during the hypoxic exposure and was more pronounced in Atoh1::En1-CKO mice. In contrast, there were no differences in respiratory response to hypercapnia between Atoh1::En1-CKO mice and control littermates at P7 (Figure 4Civ to Cvi).
Additionally, these results underscore our findings that cerebellar dysfunction is not the main driver of respiratory abnormalities in Atoh1::En1-CKO mice, the cerebellum is not yet developed at this age: cerebellar granule cells do not form their first functional synapses with Purkinje cells until P8 (White and Sillitoe, 2013). Thus, at P7 wild-type cerebelli are functionally more similar to those of Atoh1::En1-CKO mice, and if the cerebellum caused respiratory dysfunction in Atoh1::En1-CKO mice we would expect the differences between P7 control mice and Atoh1::En1-CKO mice to be smaller, not larger.
Our results show that P7 Atoh1::En1-CKO mice have abnormal respiratory control in room air and in response to hypoxia. This suggests that although some respiratory phenotypes of P21 Atoh1::En1-CKO mice (high number of sighs and abnormal hypercapnic chemoresponses) might indeed result from abnormal postnatal maturation, others (slow breathing rhythms and small tidal volume) might be self-resolving with maturation. Yet at all ages tested rRL neurons are essential to prevent apneas and respiratory depression during hypoxia.
Rostral rhombic lip neurons project to Atoh1-lineage paramotor nuclei and the preBötC
Oxygen sensors in the carotid body communicate with the nucleus tractus solitarius (NTS) to integrate the sensory information into the central respiratory circuit, which is necessary for hypoxic chemoresponses (Accorsi-Mendonça et al., 2015; Dutschmann et al., 2008; Ferreira et al., 2015; Mayer et al., 2015; Song et al., 2011). There is ample evidence that NTS neurons directly project to the PBC (Bianchi et al., 1995; Dutschmann et al., 2008; Roman et al., 2016; Song et al., 2011) and our results suggest that the Atoh1-lineage PBC neurons are necessary to prevent respiratory depression in response to hypoxia. Yet it is unknown whether they modulate respiratory rhythms through activation of downstream rhythmogenic or chemosensitive nuclei, or act directly as premotor neurons.
To trace the projections from the Atoh1-lineage rRL neurons, we made use of an intersectional reporter allele (Ai65) that expresses tdTomato only after removal of both an FRT-flanked and a loxP-flanked stop-cassette (Madisen et al., 2015). We generated an Atoh1FlpO knock-in mouse line that expresses FlpO recombinase in place of Atoh1 under the Atoh1 promoter (Figure 5—figure supplement 1). This mouse line can be used to remove the first stop-cassette in the reporter allele, exclusively in Atoh1-lineage neurons (Figure 5A); the second stop-cassette is removed using En1Cre, so that only neurons in the Atoh1;En1 intersectional domain will be labeled with tdTomato (Figure 5A). We confirmed that the cell bodies of Atoh1-lineage parabrachial neurons were labeled red (Figure 5B) and tdTomato+ puncta overlapped with the synaptic marker synapsin, thus representing synapses on downstream neurons (Figure 5C). We then assessed whether any tdTomato+ puncta were found in key respiratory nuclei or motor nuclei involved in respiratory motor rhythms (Summarized in Figure 5D). We found tdTomato+ puncta in the Atoh1-lineage paramotor nuclei: the intertrigeminal region (ITR) and the chemosensitive retrotrapezoid nucleus (RTN) (Figure 5E and F). We also found projections towards the rhythmogenic preBötC (Figure 5G), but detected no feedback projections to the nucleus tractus solitarius (NTS) (Figure 5H). We found no tdTomato+ puncta in any of the motor nuclei in the respiratory circuitry (Figure 5I–L). This shows that the rRL neurons selectively innervate downstream neurons in the respiratory circuit that are important for chemoresponsive adaptations in the respiratory rhythm, but do not function directly as premotor neurons. Loss of activation from the PBC might subdue the respiratory responses, impairing adaptation to hypercapnia and hypoxia.
Figure 5 with 1 supplement see all
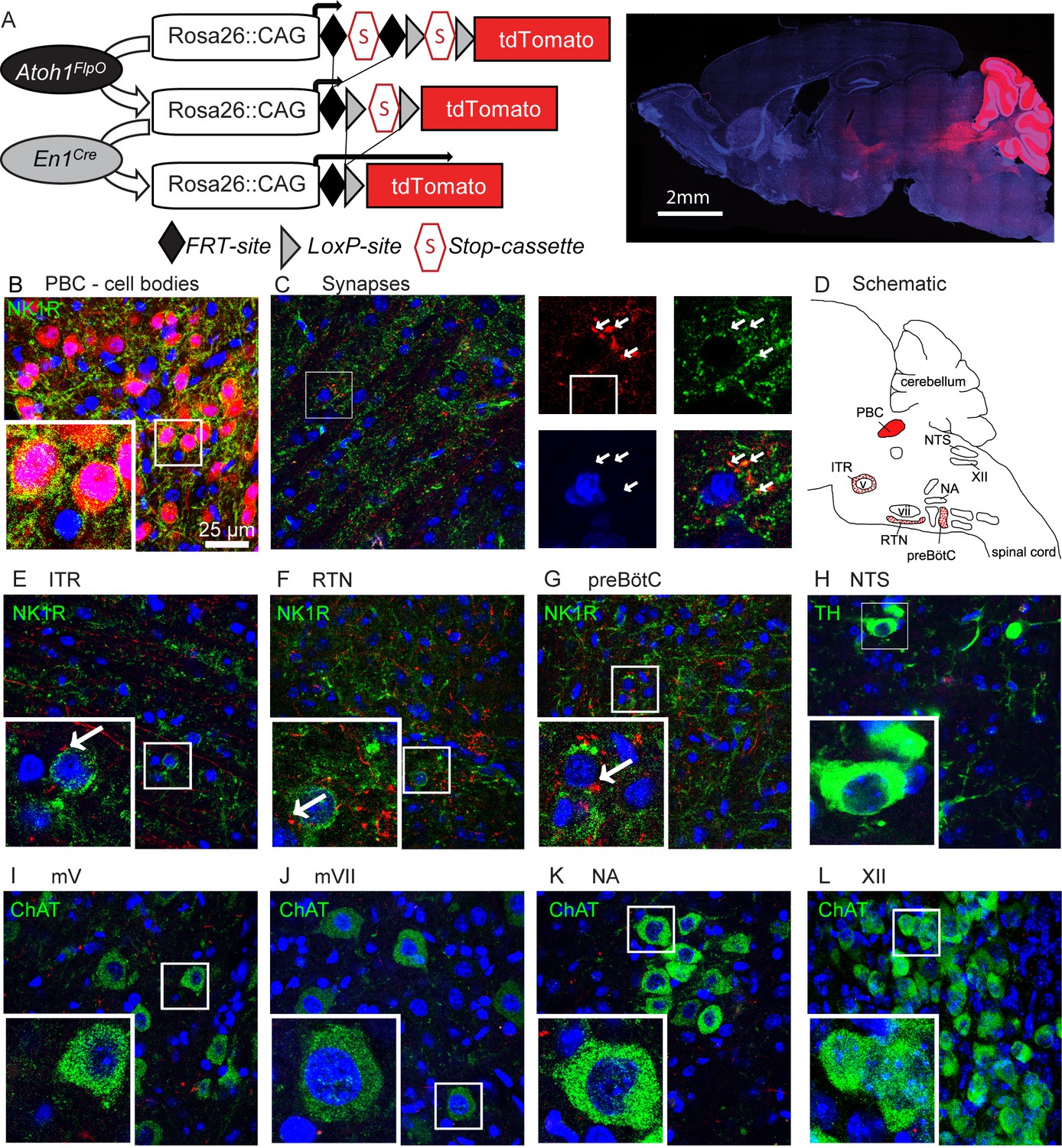
Rostral rhombic lip neurons project to paramotor nuclei and the preBötC.
(A) Intersectional strategy to label only those neurons with a history of Atoh1 (FlpO) and En1 (Cre) expression. Representative whole-brain image of P21 Atoh1FlpO;En1Cre;Ai65/+ mouse on the right. (B) Cell bodies in the PBC are tdTomato+. (C) tdTomato+ puncta overlap with the synapse-marker synapsin. (D) Schematic of the respiratory circuitry. Solid red PBC is the only respiratory nucleus expressing tdTomato. Dotted nuclei are the nuclei in which tdTomato+ projections were observed. TdTomato+ puncta near cell bodies were observed in the (E) ITR, (F) RTN, and (G) preBötC. No tdTomato+puncta were observed in the (H) NTS, or in respiratory motor nuclei (I) mV, (J) mVII, (K), NA, (L), XII. Abbreviations: PBC, parabrachial complex; ITR, intertrigeminal region; RTN, retrotrapezoid nucleus; preBötC, preBötzinger Complex; NTS, nucleus tractus solitarius; mV, trigeminal motor nucleus; mVII, facial motor nucleus; NA, nucleus ambiguus; XII, hypoglossal motor nucleus.
Atoh1-lineage intertrigeminal region neurons are not required for neonatal survival or normal respiratory chemoresponses
We found that rRL neurons project to the Atoh1-lineage intertrigeminal region (ITR) (Figure 5E), suggesting that part of the Atoh1::En1-CKO phenotype might be through loss of modulation of the ITR region. Loss of Atoh1 from the ITR was previously assessed only in Atoh1::Phox2b-CKO mice that also have impaired RTN development (Huang et al., 2012; Ruffault et al., 2015).
To assess the function of Atoh1-lineage ITR neurons, we generated an ITR-specific conditional knockout mouse using the HoxA2::CreTG mouse line that expresses Cre only in r2 neurons (Awatramani et al., 2003). We crossed homozygous Atoh1Flox/Flox females to males that were doubly heterozygous for HoxA2::CreTG and Atoh1LacZ to obtain Atoh1Flox/LacZ;HoxA2::CreTG/+ mice, hereafter called Atoh1::HoxA2-CKO mice. In situ hybridization for Atoh1 RNA confirmed that conditional deletion only from the ITR (Figure 6—figure supplement 1A). ITR neurons were still present but mislocalized on the lateral side of the trigeminal motor nucleus instead of migrating medially at E14.5 (Figure 6—figure supplement 1B and C).
Because Atoh1::HoxA2-CKO mice survived the neonatal period (Figure 6—figure supplement 2), we could test their breathing behavior using unrestrained whole-body plethysmography (UWBP) at three weeks of age. We found that Atoh1::HoxA2-CKO mice had more sigh-induced and spontaneous apneas, without a change in the number of sighs or inter breath interval irregularity (Figure 6Ai–Aiv). Interestingly, Atoh1::HoxA2-CKO mice did have a smaller tidal volume per breath, resulting in a smaller minute ventilation than control littermates (Figure 6Avi and Aviii), phenotypes that were observed neither in our Atoh1::En1-CKO mice nor in Atoh1::Phox2b-CKO mice. Nevertheless, Atoh1::HoxA2-CKO mice did not have abnormal respiratory chemoresponses (Figure 6B). Atoh1 is thus essential for normal ITR development, but loss of Atoh1 from the ITR does not impair neonatal survival or respiratory chemoresponses. The abnormal chemoresponses caused by loss of Atoh1 from the En1- or Phox2b-domain are thus caused by loss of rRL or abnormal development of RTN neurons, respectively.
Figure 6 with 2 supplements see all
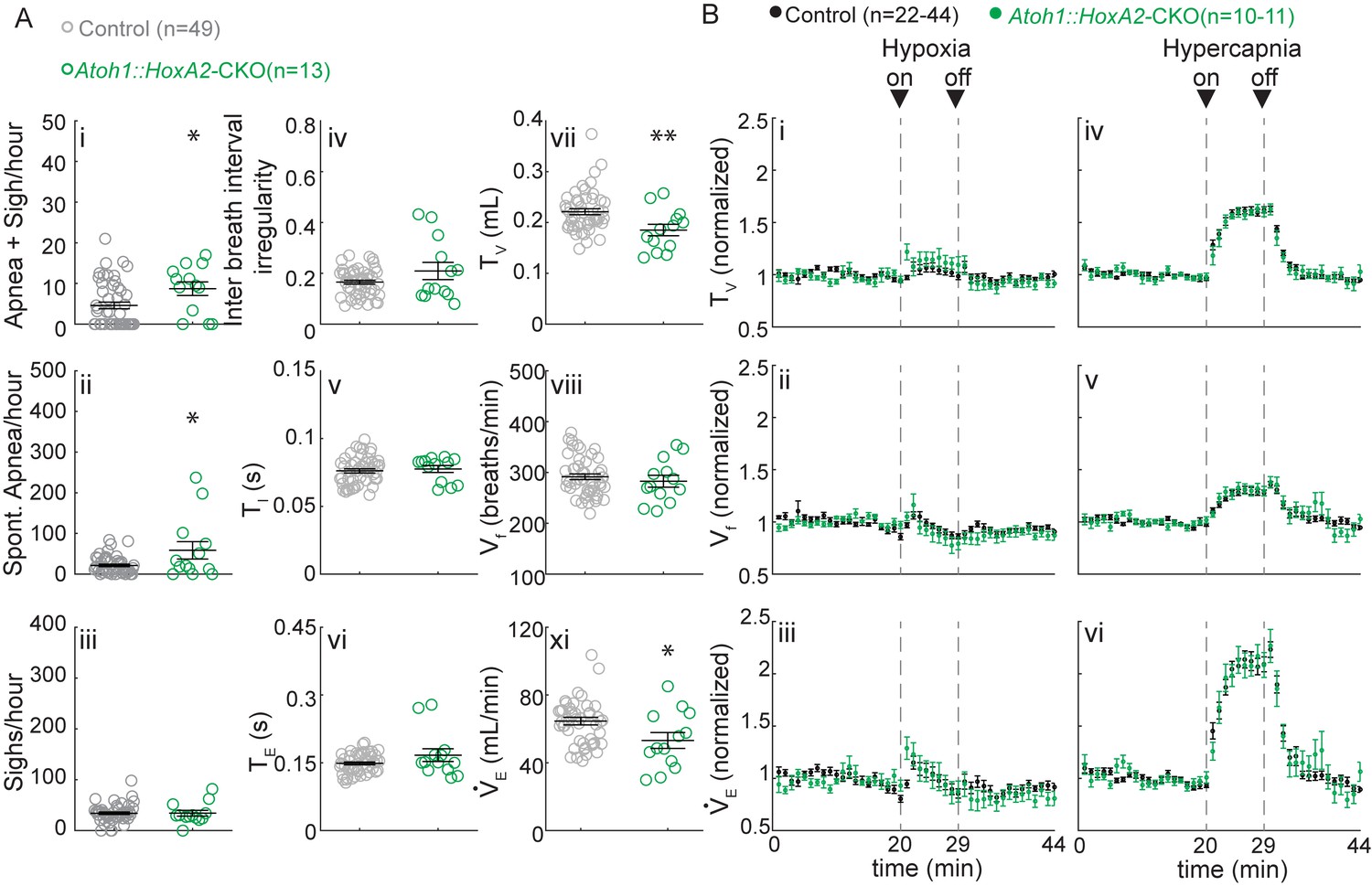
Atoh1::HoxA2-CKO mice have normal chemoresponses.
(A) Atoh1::HoxA2-CKO mice have more apneas following sighs (i) and spontaneous apneas (ii) than control littermates and a smaller tidal volume per breath (TV) (vi) resulting in smaller minute ventilation (VE) (Viii). Other breathing parameters were not affected. Significance for room air breathing parameters were determined using a t-test (2-tailed). *p<0.05, **p<0.01. (B) Atoh1::HoxA2-CKO mice have normal respiratory chemoresponses in hypoxia (i to iii) and hypercapnia (iv to vi). Significance was determined using a t-test (2-tailed) at each individual time point, *p<0.0011 (0.05/44 for Bonferroni correction). Error bars represent mean ± SEM.
-
Figure 6—source data 1
Raw numbers observed surviving offspring.
- https://doi.org/10.7554/eLife.38455.020
Loss of Atoh1 from neurons involved in respiratory chemoresponses results in neonatal lethality
Through studying a series of conditional knockout mice, our lab has now mapped the role of all Atoh1-lineage neurons in neonatal survival (Figure 7). There is no single Atoh1-derived population that fully accounts for the perinatal death of Atoh1-null mice. In light of the fact that Atoh1 loss from either the En1 or Phox2b domains leads to abnormal chemoresponses, and that loss of Atoh1 from the Phox2b domain results in neonatal lethality in only about half of the mice, we hypothesized that combined loss of Atoh1-derived neurons involved in respiratory chemoresponsiveness might be responsible for the 100% neonatal lethality of Atoh1-nulls.
Figure 7 with 1 supplement see all

Atoh1-dependent neurons involved in chemoresponses and rates of neonatal lethality.
https://doi.org/10.7554/eLife.38455.021To test this hypothesis, we crossed Atoh1Flox/+;En1Cre/+ males with Atoh1LacZ/+;Phox2b::CreTG/+ females to obtain mice that lack Atoh1 from both the En1- and Phox2b-intersectional domains (Atoh1Flox/LacZ;En1Cre/+;Phox2b::CreTG/+ mice, hereafter Atoh1-dCKO mice). Out of nearly four hundred offspring observed, no Atoh1-dCKO mice survived past P1 (Figure 7—figure supplement 1). We did not find any alive Atoh1-dCKO mice, but several were found dead in the cage on the day of birth. These animals had turned blue and there was no visible milk spot, suggesting that they died of respiratory failure before drinking any milk. These results show that loss of Atoh1 from chemoresponsive nuclei accounts for the 100% neonatal death seen in Atoh1-null mice (Ben-Arie et al., 1997).
Discussion
Many of the transcription factors expressed in the developing hindbrain are necessary for respiratory circuit development and survival (Gray, 2008). In some cases, loss of a particular factor causes respiratory failure that is traceable to a single respiratory nucleus: for example, neonatal death in Dbx1-null mice is caused by loss of rhythmogenic preBötC neurons (Bouvier et al., 2010; Wu et al., 2017). The broad expression domains of factors such as Atoh1, Tlx3 and Lbx1, however, have made it difficult to pinpoint their role in a specific nucleus or functional impairment (Gray, 2008; Huang et al., 2012; Pagliardini et al., 2008; Shirasawa et al., 2000). Here we used intersectional genetics to uncover a role for Atoh1 in the development of chemoresponsive neuronal populations that also express Engrailed1 and Phox2b (the PBC and RTN, respectively). Concomitant loss of Atoh1 from these two domains causes fully penetrant neonatal lethality. This study thus answers the decades-old question about the cause of respiratory failure in Atoh1-null mice: they die of an inability to modulate respiratory rhythms in response to hypoxic and hypercapnic conditions.
Several pieces of evidence suggest that the abnormal chemoresponses we observed in the Atoh1::En1-CKO mice are primarily due to loss of Atoh1-lineage parabrachial neurons. First, even though multiple nuclei develop from the intersectional domain of Atoh1 and En1, the parabrachial neurons were the only Atoh1-lineage neurons that were activated specifically during hypercapnic and hypoxic chemochallenges (Figure 1). Second, silencing the output from the cerebellar cortex does not cause abnormal respiratory chemoresponses (Figure 3—figure supplement 1). Third, differences in abnormal chemoresponses between Atoh1::En1-CKO mice and control littermates are observed long before the cerebellum fully matures or Atoh1-lineage cerebellar granule cells even make their first functional synapses (Figure 4). Lastly, our results confirm and extend previous work showing that lesioning the parabrachial nucleus impairs respiratory chemoresponses (Mizusawa et al., 1995; Song and Poon, 2009a, 2009b) and that the parabrachial nucleus projects to the ventrolateral medulla (Fulwiler and Saper, 1984; Holstege, 1988; Yokota et al., 2015). We found rostral Atoh1-lineage neurons projecting to the retrotrapezoid nucleus and preBötC, but we did not observe any projections to motor nuclei or specifically the reported connections with the hypoglossal nucleus. This discrepancy can be because we used genetic tools (Atoh1-driven) that only target a subset of PBC neurons. Furthermore, these genetic tools are less prone to potential off target effects caused by needle tracks and local injections. Therefore, we propose that Atoh1-lineage PBC neurons are an essential component in the chemosensory pathways that relays information from peripheral, and perhaps central, chemosensors to enhance the firing of central chemosensitive and rhythmogenic neurons within the respiratory network.
Nevertheless, we cannot exclude the possibility that Atoh1-lineage, deep cerebellar nuclei contribute to the observed phenotypes. Atoh1-lineage parabrachial and deep cerebellar neurons are born around the same embryonic day (E9.5-E12.5) (Rose et al., 2009a) and since we do not know what factors determine their differentiation, there are no developmental or genetic tools to target the deep cerebellar nuclei without affecting the parabrachial nucleus. Viral approaches do not work, either, as we discovered: deep cerebellar neurons send collateral projections throughout the cerebellum and the brainstem, so viral injections infect Atoh1-lineage deep cerebellar neurons regardless of the injection site. Nevertheless, the primary source of input to the deep cerebellar nuclei are the Purkinje cells, and silencing these cells did not recapitulate or alter respiratory chemoresponses. This latter finding, together with our data showing absence of chemoresponses in deep cerebellar neurons, make them unlikely to be essential for respiratory chemoresponses.
Likewise, Atoh1 loss from the intertrigeminal region had little effect on respiratory chemoresponses. In agreement with earlier studies from our lab, we found that intertrigeminal neurons depend on Atoh1 expression for normal migration, which resulted in increased number of apneas and a shallower breaths. Previous studies suggested a role for intertrigeminal neurons in the attenuation of sleep and reflex apneas (Radulovacki et al., 2003, 2004). We indeed saw an increase in sigh-induced apneas but we saw an increase in the number of spontaneous apneas in only two out of thirteen Atoh1::HoxA2-CKO mice, suggesting that the previously reported apneas were not likely caused by a central mechanism (Figure 6). Despite the changes in respiratory control in room air, we did not observe any abnormal respiratory chemoresponses in Atoh1::HoxA2-CKO mice. These results support the hypothesis that the respiratory phenotype of mice that lack Atoh1 in both ITR and RTN (the Atoh1::Phox2b-CKO mice) results from abnormal RTN development.
The current work provides evidence that there are two Atoh1-lineage nuclei that rely on Atoh1 expression for their function in respiratory chemoreflexes: the PBC and RTN. Removal of Atoh1 from both of these nuclei recapitulated the fully penetrant neonatal lethality of Atoh1-null mice, which we hypothesize is caused by combined loss of CO2-evoked glutamatergic signaling (RTN) and hypoxia-induced activation of PBC neurons. Previous studies in neonatal models have shown that arterial CO2 unloading removes respiratory drive and results in sustained apnea, in accordance with the notion that CO2 sensing is essential for neonatal breathing (Nattie, 1999; Praud et al., 1997). We suspect that, when directly or indirectly activated during respiratory chemochallenges, Atoh1-lineage neurons increase glutamatergic input to the preBötzinger complex, which is thought to form the central respiratory pattern generator. Since neither Atoh1::En1-CKO mice nor Atoh1::Phox2b-CKO mice show complete lethality nor complete loss of respiratory chemoresponses, it is likely that these two pathways of chemosensation (PBC and RTN-mediated) are partially redundant, as would make sense for any circuit that controls a process as fundamental to survival as breathing. Indeed, en bloc recordings from Atoh1-null mice reveal that slow respiratory frequency can be fully restored by application of a glutamate reuptake inhibitor and partially restored by substance P application (Rose et al., 2009b). Our results are also in agreement with previous recordings in wild-type brainstems, where loss of input from the pons and RTN to the ventrolateral medulla results in a transition from a tri-phasic to a slow gasp-like, mono-phasic rhythm (Rubin et al., 2009; Smith et al., 2009). The present study provides further evidence that pontine PBC and RTN neurons are essential for the respiratory versatility observed in vivo. Despite the normal development of preBötzinger neurons that are sufficient for in situ respiratory rhythms, excitatory projections from Atoh1-lineage neurons are necessary for normal breathing in vivo.
Caffeine treatment, along with glutamate and substance P, can stimulate endogenous, slow respiratory rhythms in rats (Ruangkittisakul et al., 2010); this is thought to be the mechanism by which caffeine reduces apneas of prematurity in newborn infants (Aranda et al., 1977; Natarajan et al., 2007). Caffeine drives this excitation mainly by acting as an antagonist to the A1 adenosine receptor that inhibits respiratory frequency (Koos et al., 2001). Decreased inhibition might compensate for the loss of Atoh1-dependent glutamatergic drive in Atoh1::En1-CKO mice, thereby rescuing the apnea. Caffeine treatment did not, however, improve the respiratory chemoresponses in Atoh1::En1-CKO mice, suggesting that increased ventilation during chemochallenge relies on more precise signaling of distinct neurons in the circuitry. This is in agreement with the observation that some, but not all, Atoh1-dependent parabrachial neurons are activated during chemochallenges (Figure 1). To our surprise, caffeine treatment actually decreased respiratory output in control animals, which might be caused by inhibition of A2 adenosine receptors that increase respiratory frequency (Koos, 2011; Koos and Chau, 1998; Koos et al., 2001). Future studies with specific adenosine agonists and antagonists are needed to elucidate the specific effects of long-term caffeine treatment on respiratory control in neonatal mice.
The breathing abnormalities of Atoh1::En1-CKO mice resemble those seen in premature infants: increased apneas and sighs, abnormal rhythms, attenuated responses to hypercapnia, and, perhaps most surprisingly, respiratory suppression in hypoxia (Martin et al., 2004; Abu-Shaweesh, 2004). These also mimic respiratory responses to hypoxia that occur in utero, when the oxygenation of fetal blood is regulated by the mother. During development, exposure to low O2 suppresses fetal movements, including breathing movements, likely to limit oxygen demand in relatively anaerobic environments. Several reports have suggested that pontine nuclei play a role in this respiratory suppression (Gluckman and Johnston, 1987; Haddad and Mellins, 1984) and in the postnatal maturation of hypoxic responses (Bissonnette and Knopp, 2001; Waters and Gozal, 2003). Nevertheless, it was not known precisely which neurons were important for adaptation to hypoxia. Given that abnormal development of pontine nuclei and mutations in En1 have been observed in several sudden infant death syndrome (SIDS) cases (Lavezzi, 2015, 2016, 2004; Weese-Mayer et al., 2004), we hypothesize that hypoxia-mediated respiratory suppression concomitant with immature responses to hypercapnia might be a major risk factor for SIDS.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers |
|---|---|---|---|
| Strain: C57BL/6 | Rosalsl-tdTomato | The Jackson Laboratory | RRID: IMSR_JAX:007914 |
| Strain: C57BL/6 | Atoh1Cre | Yang et al., 2001 | RRID: MGI:4844110 |
| Strain: C57BL/6 | En1 Cre | The Jackson Laboratory | RRID: IMSR_JAX:007916 |
| Strain: C57BL/6 | Rosalsl-LacZ | The Jackson Laboratory | RRID:IMSR_JAX:012429 |
| Strain: C57BL/6 | Atoh1LacZ | The Jackson Laboratory | RRID:IMSR_JAX:005970 |
| Strain: C57BL/6 | Atoh1Flox | The Jackson Laboratory | RRID:MGI:4420944 |
| Strain: C57BL/6 | Slc32a1Flox | The Jackson Laboratory | RRID:IMSR_JAX:012897 |
| Strain: C57BL/6 | Pcp2::CreTG | The Jackson Laboratory | RRID:IMSR_JAX:004146 |
| Strain: C57BL/6 | HoxA2::CreTG | Awatramani et al. (2003) | N/A |
| Strain: C57BL/6 | Phox2b::CreTG | The Jackson Laboratory | RRID: IMSR_JAX:016223 |
| Strain: C57BL/6 | Atoh1FlpO | This paper: Figure 5—figure supplement 1 | N/A |
| Strain: C57BL/6 | RosaFSF-LSL-tdTomato | The Jackson Laboratory | RRID:IMSR_JAX:021875 |
| Strain: C57BL/6 | Sox2::CreTG | The Jackson Laboratory | RRID:MGI:3801167 |
| Antibody | anti-cFos (rabbit polyclonal) | Santa Cruz | SC-52; RRID:AB_2106783 |
| Antibody | anti-NK1R (rabbit polyclonal) | Advanced Targeting Systems | AB-N04; RRID: AB_171801 |
| Antibody | anti-CGRP (rabbit polyclonal) | Sigma Aldrich | C8198; RRID:AB_259091 |
| Antibody | anti-PACAP (mouse monoclonal) | Abcam | ab216589 |
| Antibody | anti-synapsin (mouse monoclonal) | Synaptic Systems | 106 001; RRID:AB_2617071 |
| Antibody | anti-TH (rabbit polyclonal) | ImmunoStar | 22941; RRID:AB_572268 |
| Antibody | anti-ChAT (goat polyclonal) | EMD millipore | AB144P; RRID:AB_2079751 |
| Chemical compound, drug | Caffeine | FISHER | S25215A |
| Chemical compound, drug | X-gal | Gold Biotechnology | X4281C |
| Commercial assay or kit | Caffeine/Pentoxifylline ELISA | Neogen | 106419 |
| Software, algorithm | MATLAB | MathWorks | RRID: SCR_001622 |
| Software, algorithm | Ponemah 3 | DSI | N/A |
| Software, algorithm | FinePoint | DSI | N/A |
| Software, algorithm | ImageJ | NIH | RRID: SCR_003070 |
Mouse lines
Request a detailed protocolAll animals were housed in a Level 3, AALAS-certified facility on a 14 hr light cycle. Husbandry, housing, euthanasia, and experimental guidelines were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine. The following genetically engineered mouse lines were used: Rosalsl-tdTomato (Gt(ROSA)26Sortm9(CAG-tdTomato)Hze, JAX:007914), Atoh1Cre (Yang et al., 2001), En1Cre (En1tm2(cre)Wrst/J, JAX:007916):, Rosalsl-LacZ (Gt(ROSA)26Sortm1(CAG-lacZ,-EGFP)Glh, JAX:012429), Atoh1LacZ(Atoh1tm2Hzo, JAX:005970), Atoh1Flox(Atoh1tm3Hzo, MGI:4420944), Pcp2Cre (B6.129-Tg(Pcp2-cre)2Mpin/J, JAX: 004146), Slc32a1Flox (Slc32a1tm1Lowl/J, JAX: 012897), HoxA2::CreTG (Awatramani et al., 2003), Phox2b::CreTG (Tg(Phox2b-cre)3Jke, JAX:016223), Atoh1FlpO, Ai65 (Gt(ROSA)26Sortm65.1(CAG-tdTomato)Hze,JAX:021875), and Sox2::CreTG (Tg(Sox2-cre)1Amc, MGI:3801167). Ear tissue, collected from ear-marking, was used for PCR genotyping to minimalize stress on animals. For timed pregnancies, noon on the day of the vaginal plugging was set as embryonic day 0.5 (E0.5).
Generation of the Atoh1FlpO Mice
Request a detailed protocolThe FlpO sequence from pQUAST-FLPo was ligated to a PGK-Neo cassette flanked by lox2722 sites in a pUC vector. This FlpO-PGK-Neo cassette was then cloned into a pre-existing pBlueScript II KS+ plasmid that contained the Atoh1 5’ and 3’ targeting arms without disrupting the Atoh1 transcriptional start site, identical to the approach previously described (Rose et al., 2009b). This construct was then electroporated into B57/6J ES cells with an agouti mutation. These ES cells were expanded under neomycin selection and screened for correct recombination by Southern blot using external 3’ probes, and internal 5’ PCR. Six clones were further expanded, tested correct recombination using PCR and sequence validated. Three clones were chosen to be injected into albino B57/6J blastocyst to generate chimeras. Chimeras were backcrossed to albino B57/6J females to generate heterozygote Atoh1FlpO-Neo mice. These were crossed to Sox2Cre to remove the PGK-Neo cassette. See Figure 5—figure supplement 1 for genomic targeting diagram, and example PCR genotyping (forward primer: CTTCGTTGCACGCGAC, reverse primer: CACAATTTATCGTGTAGCCG. WT: 2.2 kb, FlpO-KI: 2.6 kb).
Immunofluorescence (IF) Assays
Request a detailed protocolIF and cryosectioning were performed using previously described protocols (Huang et al., 2012). Mice older than one week were perfused with 4% PFA, after which brains were dissected and post-fixed overnight in 4% PFA at 4°C. Embryonic and P0 brains were directly dissected from pups and dropped fixed in 4% PFA at 4°C. After fixation brains were cryopreserved in 30% sucrose solution in PBS until sunk and frozen in OCT. Frozen sections were cut at 40 µm and kept at 4°C in the dark until used for immunostaining. For immunolabeling sections were first blocked in 5% normal donkey serum, 0.5% Triton-X in PBS for one hour at room temperature. Next, sections were incubated in primary antibody overnight at 4°C in blocking solution, followed by three washes and secondary labeling for at least two hours at room temperature. Nuclei were labeled using DAPI (1:10,000) and slides were mounted in DAPI-free mounting solution (Vectashield). The following antibodies and their dilutions were used: rabbit anti-cFos (1:5000, Santa Cruz), rabbit anti-NK1R (1:2000, Advanced Targeting Systems), mouse anti-Synapsin (1:1000, Synaptic Systems), goat anti-TPH2 (1:1000, Santa Cruz), mouse anti-TH (1:200, ImmunoStar), and goat anti-ChAT (1:200, EMD Millipore). Secondary antibodies were conjugated with Alexa Fluor 488 (1:1000, Molecular Probes). We used a Leica TCS SP5 confocal microscope system to image fluorescent signal. Image brightness and contrast were normalized using ImageJ.
X-gal staining
Request a detailed protocolEmbryos were collected at E14.5, brains were dissected and fixed for 10 min in ice-cold formalin followed by brief washing in PBS on ice and in room-temperature wash buffer. β-galactosidase activity was assayed by embedding and sectioning tissue as previously described (Ben-Arie et al., 2000). X-gal stained whole brains were stored at 70% ethanol. X-gal stained sections were first counterstained with nuclear fast red (Vector laboratories) and then imaged using a bright-field Axio Imager M2 microscope, equipped with an Axio Cam MRc5 color camera (Carl Zeiss, Germany). Contrast and saturation were adjusted using ImageJ and Adobe Photoshop.
In situ hybridization (ISH) was performed on 25µm-thick sagittal sections cut from unfixed, fresh frozen E14.5 embryonic control and CKO mice covering the entire embryo. We generated digoxigenin (DIG)-labeled mRNA antisense probes against Atoh1 using reverse-transcribed mouse cDNA as a template and a RNA DIG-labeling kit from Roche. Primer and probe sequences for the probe is available on the Allen Brain Atlas website (www.brain-map.org) and were validated previously (Huang et al., 2012). ISH was performed by the RNA In Situ Hybridization Core at Baylor College of Medicine using an automated robotic platform as previously described (Yaylaoglu et al., 2005).
Assessing cFos expression
Request a detailed protocolTo analyze in vivo hypoxia or hypercapnia induced cFos expression, adult animals (6–8 weeks old) were habituated to the plethysmography chambers five hours the day prior to the experiment and one hour on the day of the experiment. Freely moving mice were placed in whole-body plethysmography chambers (Buxco) through which fresh air was pumped at a basal flow rate of 0.5 L/min. Next, animals were exposed to either room air, hypoxia (10% O2, balance N2), or hypercapnia (5% CO2, 21% O2, balance N2) for 1 hr. Animals were sacrificed within 30 min of exposure and tissue was processed as described in ‘Immunofluorescence (IF) Assays.’
P21 juvenile unrestrained whole-body plethysmography (UWBP)
Request a detailed protocolBreathing analysis was performed using previously reported protocols with minor modifications (Huang et al., 2012; Orengo et al., 2018; Yeh et al., 2017). To test respiratory parameters, weaning age mice (P19-21) were placed in the plethysmography chambers and habituated for at least one hour prior to the experiment. After this habituation period, we recorded twenty minutes of room air breathing to determine the baseline. Next, we exposed animals to nine minutes in either hypoxia (10% O2, balance N2) or hypercapnia (5% CO2, 21% O2, balance N2). After this chemochallenge, we recorded breathing behavior for another fifteen-minute recovery period in room air. To determine changes in breathing behavior in response to the chemochallenge, we normalized all breathing parameters to baseline values using the following formula: normalized y(t)=y(x)/(average(y(xbaseline)), where y is the average value of a parameter during any given minute (x) and baseline is minutes one to twenty.
We used Phonemah three software (DSI) to identify breath waveforms and used custom-written MATLAB (Mathworks) code to derive inspiratory time, expiratory time, and tidal volume (Source code 1). Breaths with an inspiration time shorter than 0.03 s or an expiration time longer than 10 s were excluded. Minute long segments in which the average breathing frequency (breaths/min) was higher than 500 were excluded from our analysis to prevent confounds from sniffing or exploring. Tidal volume was adjusted for body temperature as previously described (Ray et al., 2011). Apneas were defined as breaths longer than 0.5 s and at least twice the length of the average of the six surrounding breaths (three previous and three following). Apneas were divided in spontaneous apneas and apneas that were preceded by a sigh (or a sigh in the previous breath). Sighs were defined as breaths with an inspiratory tidal volume 2.5 as big and an inspiratory time 1.25 as long as the previous five breaths. Inter Breath Interval (IBI) irregularity was defined as: IBI irregularity = abs (breath length(n + 1)-breath length(n))/breath length(n). Minute ventilation was defined as the total amount of air breathed per minute (breathing frequency * tidal volume). Source data from the plethysmography analysis can be found in the source data files: Figure 2—source data 1, Figure 3—source data 1, Figure 3—figure supplement 1—source data 1, and Figure 6—source data 1.
Since we did not observe any difference in breathing parameters between male and female mice, or between Atoh1-heterozygote animals and any of the Cre-lines, we grouped these animals together in the analysis.
P7 neonatal unrestrained whole-body plethysmography (UWBP)
Request a detailed protocolBreathing analysis for neonatal mice was performed using a DSI pup-plethysmography set-up designed especially for these experiments. Specialized, small pup plethysmography chambers (DSI) were connected to a FinePointe Whole Body Plethysmography Unit with gas switch capability (DSI). The unit provided the pup plethysmograph chambers with a constant 1 L/min airflow and amplified breathing waveforms. P7 (day of birth is P0) animals were placed in the plethysmography chambers and could acclimate for 10 min, followed by 5 min of room air recording and a 5 min gas-challenge (hypoxia (10% O2, balance N2) or hypercapnia (5% CO2, 21% O2, balance N2)).
FinePointe software was used to define breaths and calculate basic breathing parameters. Breaths were defined as followed: (1) peak inspiratory flow (PIF) must be before peak expiratory flow (PEF); (2) expiration or inspiration is less than a couple samples; (3) Rpef, EF50, and Tr can be computed; (4) conditioning coefficient can be computed; (5) MinimumBoxFlowThreshold is larger than 0.005; (6) Start of expiration can be identified on the AC flow waveform. Breaths were rejected if TV was smaller than 0.005 ml or larger than 1.0 ml; if TI was larger than 0.02 s or twice as large as TE; or if inspiratory volume was not within 50% to 150% of the couple expiratory volume. For each minute of breathing recording a rejection index (RINX) was calculated as the percentage of the trace from which individual breaths were defined (length breathing trace used for analysis/one minute * 100%). For our final analysis, we included only those minutes for which the RINX was less than 40%. All animals from which we recorded fewer than three reliable minutes of respiratory rhythms in both room air and during chemochallenge (RINX <40%) were excluded from our analysis. The percentage of animals of each genotype included in the final analysis were similar (23 control mice out of 33 (70%); 12 Atoh1::En1-CKO mice out of 16 (75%); Chi-Square = 0.133, p=0.7149). RINX of included animals was also similar between genotypes (control mice: 18%; Atoh1::En1-CKO mice: 19.8%; t-test, p=0.614). From the included minutes the following parameters were derived: inhalation time, exhalation time, breathing frequency, tidal volume, and minute ventilation. Apneas and sighs were determined by a trained observer who was blinded to the genotype of the mice.
Source data from the pup plethysmography analysis can be found in the source data file: Figure 4—source data 1.
Caffeine administration and ELISA
Request a detailed protocolCaffeine was administered to pups through the drinking water of lactating dams (0.3 gram/L) from postnatal day three until weaning. Caffeine was dissolved in water provided by the animal facility and stored in special dark bottles to prevent photodegradation. Plasma levels of caffeine in pups were determined using a commercially available Caffeine/Pentoxifylline ELISA kit (Neogen). We obtained blood and isolated plasma from P19 pups raised in cages with normal water or caffeinated water.
Quantification and statistical analysis
Request a detailed protocolAll quantification and statistical analyses were performed using MATLAB (Mathworks). Data in text and figures represent the mean ± standard error of the mean (SEM). Student’s t-test was used when comparing two independent groups. Chi-square test were performed to test Mendelian ratios of surviving offspring. We used two-way ANOVA to determine interaction effects between genotype (control vs. conditional knockout) and treatment (no caffeine vs. caffeine), followed by a Tukey-Kramer post-hoc analysis in case significance was reached. To determine time-specific significant differences in respiratory chemoresponses, serial t-tests or ANOVA’s were performed and the p-value was Bonferroni adjusted for multiple comparisons. Statistical significance was accepted at p<0.05 for all other tests.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files. Source data files have been provided for Figures 2, 3, 4 and 6.
References
-
Maturation of respiratory reflex responses in the fetus and neonateSeminars in Neonatology 9:169–180.https://doi.org/10.1016/j.siny.2003.09.003
-
Efficacy of caffeine in treatment of apnea in the low-birth-weight infantThe Journal of Pediatrics 90:467–472.https://doi.org/10.1016/S0022-3476(77)80718-X
-
Impaired response to hypoxia in the respiratory center is a major cause of neonatal death of the PACAP-knockout mouseEuropean Journal of Neuroscience 37:407–416.https://doi.org/10.1111/ejn.12054
-
Functional conservation of atonal and Math1 in the CNS and PNSDevelopment 127:1039–1048.
-
Developmental changes in the hypoxic ventilatory response in C57BL/6 miceRespiration Physiology 128:179–186.https://doi.org/10.1016/S0034-5687(01)00271-7
-
Hindbrain interneurons and axon guidance signaling critical for breathingNature Neuroscience 13:1066–1074.https://doi.org/10.1038/nn.2622
-
A SIDS-like phenotype is associated with reduced respiratory chemoresponses in PACAP deficient neonatal miceAdvances in Experimental Medicine and Biology 551:77–83.https://doi.org/10.1007/0-387-27023-X_13
-
Postnatal emergence of synaptic plasticity associated with dynamic adaptation of the respiratory motor patternRespiratory Physiology & Neurobiology 164:72–79.https://doi.org/10.1016/j.resp.2008.06.013
-
Subnuclear organization of the efferent connections of the parabrachial nucleus in the ratBrain Research Reviews 319:229–259.https://doi.org/10.1016/0165-0173(84)90012-2
-
Transcription factors and the genetic organization of brain stem respiratory neuronsJournal of Applied Physiology 104:1513–1521.https://doi.org/10.1152/japplphysiol.01383.2007
-
Central respiratory chemoreceptionThe Journal of Comparative Neurology 518:3883–3906.https://doi.org/10.1002/cne.22435
-
Hypoxia and respiratory control in early lifeAnnual Review of Physiology 46:629–643.https://doi.org/10.1146/annurev.ph.46.030184.003213
-
Fetal cardiovascular and breathing responses to an Adenosine A2a receptor agonist in sheepAmerican Journal of Physiology-Regulatory, Integrative and Comparative Physiology 274:R152–R159.https://doi.org/10.1152/ajpregu.1998.274.1.R152
-
Adenosine A(1) and A(2A) receptors modulate sleep state and breathing in fetal sheepJournal of Applied Physiology 91:343–350.https://doi.org/10.1152/jappl.2001.91.1.343
-
Adenosine A₂a receptors and O₂ sensing in developmentAmerican Journal of Physiology. Regulatory, Integrative and Comparative Physiology 301:R601–622.https://doi.org/10.1152/ajpregu.00664.2010
-
Developmental neuropathology of brainstem respiratory centers in unexplained stillbirth: What's the meaning?International Journal of Developmental Neuroscience 53:99–106.https://doi.org/10.1016/j.ijdevneu.2016.06.007
-
Apnoea of prematurityPaediatric Respiratory Reviews 5:S377–S382.https://doi.org/10.1016/S1526-0542(04)90067-X
-
Changes in carotid body and nTS neuronal excitability following neonatal sustained and chronic intermittent hypoxia exposureRespiratory Physiology & Neurobiology 205:28–36.https://doi.org/10.1016/j.resp.2014.09.015
-
Role of the parabrachial nucleus in ventilatory responses of awake ratsThe Journal of Physiology 489:877–884.https://doi.org/10.1113/jphysiol.1995.sp021100
-
CO2, brainstem chemoreceptors and breathingProgress in Neurobiology 59:299–331.https://doi.org/10.1016/S0301-0082(99)00008-8
-
A novel functional neuron group for respiratory rhythm generation in the ventral medullaThe Journal of Neuroscience 23:1478–1486.https://doi.org/10.1523/JNEUROSCI.23-04-01478.2003
-
Motor neuron degeneration correlates with respiratory dysfunction in SCA1Disease Models & Mechanisms 11:dmm032623.https://doi.org/10.1242/dmm.032623
-
Central respiratory rhythmogenesis is abnormal in lbx1- deficient miceJournal of Neuroscience 28:11030–11041.https://doi.org/10.1523/JNEUROSCI.1648-08.2008
-
Modulation of reflex and sleep related apnea by pedunculopontine tegmental and intertrigeminal neuronsRespiratory Physiology & Neurobiology 143:293–306.https://doi.org/10.1016/j.resp.2004.02.012
-
Genetically and functionally defined NTS to PBN brain circuits mediating anorexiaNature Communications 7:11905.https://doi.org/10.1038/ncomms11905
-
Caffeine reversal of opioid-evoked and endogenous inspiratory depression in perinatal rat en bloc medullas and slicesAdvances in Experimental Medicine and Biology 669:123–127.https://doi.org/10.1007/978-1-4419-5692-7_25
-
Multiple rhythmic states in a model of the respiratory central pattern generatorJournal of Neurophysiology 101:2146–2165.https://doi.org/10.1152/jn.90958.2008
-
Rnx deficiency results in congenital central hypoventilationNature Genetics 24:287–290.https://doi.org/10.1038/73516
-
Structural and functional architecture of respiratory networks in the mammalian brainstemPhilosophical Transactions of the Royal Society B: Biological Sciences 364:2577–2587.https://doi.org/10.1098/rstb.2009.0081
-
Lateral parabrachial nucleus mediates shortening of expiration and increase of inspiratory drive during hypercapniaRespiratory Physiology & Neurobiology 165:9–12.https://doi.org/10.1016/j.resp.2008.10.009
-
Lateral parabrachial nucleus mediates shortening of expiration during hypoxiaRespiratory Physiology & Neurobiology 165:1–8.https://doi.org/10.1016/j.resp.2008.10.007
-
Responses to hypoxia during early developmentRespiratory Physiology & Neurobiology 136:115–129.https://doi.org/10.1016/S1569-9048(03)00076-4
-
Cerebellar zonal patterning relies on Purkinje cell neurotransmissionJournal of Neuroscience 34:8231–8245.https://doi.org/10.1523/JNEUROSCI.0122-14.2014
-
Development of the cerebellum: from gene expression patterns to circuit mapsWiley Interdisciplinary Reviews: Developmental Biology 2:149–164.https://doi.org/10.1002/wdev.65
-
A V0 core neuronal circuit for inspirationNature Communications 8:544.https://doi.org/10.1038/s41467-017-00589-2
-
Multiple developmental defects in Engrailed-1 mutant mice: an early mid-hindbrain deletion and patterning defects in forelimbs and sternumDevelopment 120:2065–2075.
-
Respiratory-related outputs of glutamatergic, hypercapnia-responsive parabrachial neurons in miceJournal of Comparative Neurology 523:907–920.https://doi.org/10.1002/cne.23720
Article and author information
Author details
Meike E van der Heijden

Funding
American Heart Association (Predoctoral fellowship award number 17PRE33660616)
- Meike E van der Heijden
Howard Hughes Medical Institute
- Huda Y Zoghbi
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Dr. T Klisch for expertise and guidance generating the Atoh1FlpO knock-in mouse line, Dr. R Ray for continuous advice on project design, and V Brandt for her insightful comments and edits on the manuscript. We also thank Dr. S Dymecki for editorial and scientific comments to the manuscript and for sharing the HoxA2::CreTG mice. We thank Drs. J Elmquist, L Gan, A Joyner, and R Sillitoe for sharing the Phox2b::CreTG, Atoh1Cre, En1Cre mice, and Pcp2Cre/+;Slc32a1Flox/Flox respectively. This project was supported by the Neurobehavioral Core facility (U54HD083092), the Neuroconnectivity Core facility (U54HD083092), and the RNA In Situ Hybridization Core facility (with the expert assistance of Cecilia Ljungberg, Ph.D., 1S10 OD016167 and IDDRC grant 1U54 HD083092, Eunice Kennedy Shriver National Institute Of Child Health and Human Development). This work was supported by American Heart Association AWRP Predoctoral Fellowship to MEH (17PRE33660616). HYZ is a Howard Hughes Medical Institute investigator.
Ethics
Animal experimentation: All animals were housed in a Level 3, AALAS-certified facility on a 14hr light cycle. Husbandry, housing, euthanasia, and experimental guidelines were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine (protocol number: AN1013).
Version history
- Received: May 17, 2018
- Accepted: July 1, 2018
- Accepted Manuscript published: July 4, 2018 (version 1)
- Version of Record published: July 31, 2018 (version 2)
Copyright
© 2018, van der Heijden et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,231
- views
-
- 403
- downloads
-
- 25
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Loss of Atoh1 from neurons regulating hypoxic and hypercapnic chemoresponses causes neonatal respiratory failure in mice
eLife 7:e38455.
https://doi.org/10.7554/eLife.38455




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}