


Abstract
LY354740 is a conformationally constrained analog of glutamate which is a potent agonist for group II cAMP coupled metabotropic glutamate receptors (mGluRs). The discovery of this novel pharmacological agent has allowed the exploration of the functional consequences of activating group II mGluRs in vivo. In an effort to evaluate the clinical utility of LY354740 as an anxiolytic, we examined its effects in the fear potentiated startle and elevated plus maze models of anxiety and compared the results with the clinically prescribed anxiolytic diazepam. In the fear potentiated startle and elevated plus maze models, both LY354740 and diazepam produced significant anxiolytic activity (ED50 values of 0.3 and 0.4 mg/kg p.o. for fear potentiated startle and 0.2 and 0.5 mg/kg for the elevated plus maze, respectively). The duration of pharmacological effect for LY354740 in the efficacy models was 4 to 8 hr. In contrast to diazepam, acute administration of LY354740 did not produce sedation, cause deficits in neuromuscular coordination, interact with central nervous system depressants, produce memory impairment or change convulsive thresholds at doses 100- to 1000-fold the efficacious doses in animal models of anxiety. Thus, LY354740 has anxiolytic activity in animal models that are sensitive to benzodiazepines such as diazepam. However, at anxiolytic doses in these models, LY354740 produced none of the unwanted secondary pharmacology associated with diazepam. These data indicate a functional role for group II mGluRs in fear/anxiety responses in animals and suggest that compounds in this class may be beneficial in the treatment of anxiety-related disorders in humans without the side effects seen with currently prescribed medications.
Glutamate is the major excitatory neurotransmitter in the mammalian CNS, contributing excitatory input to virtually all neurons of the brain and spinal cord. The ubiquitous nature of glutamate excitation supports a role for glutamatergic neurotransmission in most physiological processes of the CNS including sensory and motor functions, central regulation of respiratory and cardiovascular centers, cognition and regulation of perception and emotion. Similarly, altered glutamatergic neuronal transmission has also been hypothesized to have a central role in many neurological and psychiatric disorders, including neurodegenerative diseases, epilepsy, schizophrenia and anxiety (Danyszet al., 1995).
Until recently, the effects of glutamate were thought to be exclusively mediated by ion channel-mediated ionotropic receptors (iGluRs), which include NMDA, AMPA and kainate receptor subtypes (Hollmann and Heinemann, 1994). However, it is now known that glutamate can also couple to second messenger pathways through G-protein-dependent mechanisms (Sladeczek et al., 1985; Nicoletti et al., 1986; Sugiyama et al., 1987; Schoepp and Johnson, 1988). These metabotropic glutamate receptors (mGluRs) represent a heterogeneous family of proteins that are coupled to multiple second messenger systems (Schoepp and Conn, 1993; Nakanishi and Masu, 1994;Pin and Duvoisin, 1995) and are divided into three groups with similar pharmacology, molecular structure and second messenger coupling.
Group I mGluRs include mGluR1 and mGluR5, group II receptors include mGluR2 and mGluR3 and group III mGluRs consist of mGluR4, mGluR6, mGluR7 and mGluR8. When expressed in nonneuronal cells, group I mGluRs couple to the activation of phosphoinositide hydrolysis, although group II and group III mGluRs are negatively linked to cAMP formation. Each mGluR subtype is uniquely and differentially distributed in the CNS. Group II mGluRs (mGluR2 and mGluR3) are highly localized within forebrain regions in the rat suggesting specific roles in modulation of glutamatergic transmission in those regions (Ohishi et al., 1993a; Ohishi et al., 1993b; Schoepp, 1994; Pin and Duvoisin, 1995). Group II mGluRs are thought to exist at presynaptic sites where they function as negative feedback autoreceptors to inhibit the release of glutamate (possibly through effects on voltage-sensitive calcium channels), or postsynaptically where they can modulate cell function via alterations in A-kinase activity (ultimately by changing protein phosphorylation) and/or direct modulation of various ion channels (i.e., K+, Ca++) (Schoepp, 1994; Pin and Duvoisin, 1995). Specifically, forebrain areas that express group II mGluRs include limbic structures that participate in the control of emotionality and anxiety states. A compound acting on mGluRs in these brain areas would be hypothesized to modulate behavioral states in animals associated with these brain areas.
LY354740 [(1S,2S,5R,6S-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylate monohydrate], a conformationally constrained analog of glutamate, is a nanomolar potent, highly selective and orally active agonist at group II/cAMP coupled mGluRs (mGluR2 and mGluR3) (see fig. 1) (Monn et al., 1997;Schoepp et al., 1997). Until the recent discovery of LY354740, there were no known group II mGluR agonists that were nanomolar potent, highly selective and systemically active. Thus, the discovery of this novel pharmacological agent has allowed exploration of the functional consequences of activating group II mGluRs in vivo.

Chemical structure of LY354740 (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylate monohydrate.
The fear potentiated startle procedure, a model based on fear which is reportedly selective for the identification of anxiolytic drugs, has been described by Davis (1979, 1986). In this procedure, a neutral stimulus such as a light (conditioned stimulus) is repetitively paired with an aversive stimulus such as shock (unconditioned stimulus). After conditioning, when the animals are presented with loud acoustic stimuli, enhanced startle responses are elicited when the startle stimulus is preceded by the light. The augmentation of startle responses engendered in this model are reported to result from a classically conditioned increase in fear (Davis, 1979).
A second model of anxiety, the elevated plus maze, is a widely used test based on the natural aversion of rodents to heights and open spaces, which has been validated for both rats and mice (Lister, 1987;Dawson and Trinklebank, 1995; Helton et al., 1995), and, as with fear potentiated startle, is sensitive to both anxiolytic and anxiogenic drugs.
Clinically proven anxiolytics such as diazepam (Valium) and buspirone (Buspar) are effective in reducing the fear (increased startle responding) associated with the presentation of the light in the potentiated startle model (Davis, 1979; Kehne et al., 1988) and at increasing open-arm activity in the elevated plus maze (Heltonet al., 1995). However, all clinically available anxiolytics have limited clinical efficacy because of their adverse side effect profile, dependence producing properties (Dantzer, 1977; Greenblatt and Shader, 1978; Woods et al., 1987, 1992; Treit, 1991). Therefore, a nonaddictive, highly efficacious anxiolytic devoid of benzodiazepine-like side effects would be of great clinical value for the treatment of anxiety and related disorders.
In an effort to evaluate the role of group II mGluRs in anxiety, we examined the effects of LY354740 [or its racemic form, (±)LY314582] in two animal models that detect anxiolytic activity, the fear potentiated startle model and elevated plus maze. We also profiled the secondary (potential side-effect) pharmacology of LY354740 in comparison to diazepam, including the effects of both agents on spontaneous activity, neuromuscular coordination, interaction with a CNS depressant, electro-convulsant threshold and learning and memory.
Materials and Methods
Animals.
All experiments were performed in accordance with Eli Lilly and Company Animal Care and Use Policies. Male Long Evans rats (180–400 g) and male NIH Swiss mice (18–35 g) were obtained from Harlan Sprague-Dawley, Cumberland, IN. Male CD-1 mice (18–35 g) were obtained from Charles Rivers Laboratories, Portage, MI. All animals were acclimated at least 3 days before testing. Animals were housed at 23 ± 2°C (relative humidity 30–70%) and given Purina Certified Rodent Chow (PMI International, Inc., St. Louis, MO) and water ad libitum. The photo period was 12 hr of light and 12 hr of dark, with dark onset at approximately 1800 hr.
Drugs.
(+)LY354740 monohydrate [(1S,2S,5R,6S-2-aminobicyclo [3.1.0]hexane-2,6-dicarboxylate monohydrate], (−)LY366563 monohydrate or racemic (±)LY314582 (Monn et al., 1997) were dissolved in a vehicle of purified water and neutralized with 5 N NaOH to a pH of approximately 7 to 8. Diazepam (Sigma Chemical Co., St. Louis, MO) was suspended in purified water by the drop-wise addition of Tween 80. Hexobarbital (Sigma) was solubilized in purified water by the addition of 0.2 N sodium hydroxide. Hydrochloric acid (0.1 N) was subsequently added to adjust the pH prior to dilution to the final volume with purified water. NMDA (Research Biochemicals International, Natick, MA) was solubilized in 0.9% physiological saline. Control animals received the respective vehicle. Food was removed at least 1 hr before compound administration.
Fear potentiated startle.
SL-LAB (San Diego Instruments, San Diego, CA) chambers were used for conditioning sessions and for producing and recording startle responses. A respondent conditioning procedure was used to produce potentiation of startle responses. Briefly, on days 1 and 2, rats were placed into dark startle chambers in which shock grids were installed. After a 5-min acclimation period, each rat received a 1 mA electric shock (500 msec) preceeded by a 5-sec presentation of light (15 W) which remained on for the duration of the shock. Ten presentations of the light and shock were given in each conditioning session with an inter-trial interval of 10 sec. Startle responding was measured through transducers located in the center of a platform located under the animal. Recorded values represent a maximum change in voltage from baseline and are recorded and presented as a peak maximum voltage (Vmax). Forty-eight hours after the second conditioning session, rats were administered LY354740, diazepam, or water, and startle testing sessions were conducted. A block of 10 consecutive presentations of acoustic startle stimuli (110 dB, non-light-paired) was presented at the beginning of the session to minimize the influence of the initial rapid phase of habituation to the stimulus. This was followed by 20 alternating trials of the noise (110 dB white noise, 50 msec in duration) alone or noise preceeded by the light. All intertrial intervals were eight seconds in duration. Excluding the initial trial block, startle response amplitudes for each trial type (noise-alone vs. light + noise) were averaged for each rat across the entire test session. Data were presented as the difference between noise-alone and light + noise. Diazepam (0.03–1 mg/kg, i.p.) or LY354740 (0.01–10 mg/kg, oral) was administered 30 or 60 min, respectively, before evaluation in potentiated startle. For oral duration of action testing, LY354740 (3 mg/kg, oral) was administered 2, 4, 6 or 8 hr before testing.
Elevated plus maze.
Construction of the elevated plus maze was based on a design validated for mice by Lister (1987). The entire maze was made of clear Plexiglas. The maze was comprised of two open arms (30 × 5 × 0.25 cm) and two enclosed arms (30 × 5 × 15 cm). The floor of each maze arm was corrugated to provide texture. The arms extended from a central platform and angled at 90° from each other. The maze was elevated to a height of 45 cm above the table and illuminated by a red light. Individual infrared photocells were mounted along each arm of the maze. For closed and open arms three photocells were positioned at equal intervals along the axis of each arm. For nosepoke activity photobeams were 0.5 cm from the entry of the open and closed arms. NIH Swiss mice were individually placed in the closed arm of the maze and the number of closed, open and nosepoke counts were recorded and used as a measure of arm entries and time spent on various sections of the maze over a 5-min test period. LY354740 (0 or 3 mg/kg, oral) was administered to mice at 1, 4 or 6 hr before evaluation in the maze.
Spontaneous activity.
Spontaneous activity of individual rats was recorded using Multi-Varimax Activity Monitors (Columbus Instruments, Columbus, OH). Interruptions of three individual infrared photocells were recorded by computer. Activity counts were cumulated after administration of diazepam (1–10 mg/kg, oral) or LY354740 (10–300 mg/kg, oral) at 15-min intervals for 1 hr. Ambulatory (locomotor or horizontal movement) counts represent the sum of individual photocell beam breaks (n = 3 beams/cage) within a representative time interval. Nonambulatory (stereotyped or nonlocomotor movement) counts represent the sum of the repetitive breaking of individual photocells.
Sensorimotor reactivity—auditory startle.
The auditory startle response of each rat was recorded using San Diego Instruments startle chambers (San Diego, CA). Startle was assessed across 10 trials with a 5-min adaptation period at a background noise level of 70 ± 3 dBA, followed by 10 presentations of auditory (120 ± 2 dBA white noise, 50 msec in duration) stimuli presented at 8-sec intervals. Startle was measured through transducers located in center of a platform located under the animal. Recorded values represent a maximum change in voltage from baseline and are recorded and presented as a peak maximum voltage (Vmax). During testing, rats were administered LY354740 (1–10 mg/kg) orally and evaluated for sensorimotor responsiveness after a 60-min pretreatment period.
Neuromuscular coordination—rotarod.
Rats were placed on a rotarod (model V EE/85, Columbus Instruments) accelerating from 1 to 80 revolutions/4 min. All rats were given two control trials at least 12 hr apart before evaluation of diazepam or LY354740. Rats were tested on the rotarod 60 min after administration of diazepam (1–10 mg/kg, oral) or LY354740 (30–300 mg/kg, oral). The number of seconds each rat remained on the rotarod was recorded.
Interaction with CNS depressants—hexobarbital-induced sleep times.
Male CD-1 mice were given a single oral dose of diazepam (1–10 mg/kg) or LY354740 (1–10 mg/kg) 60 min before challenge with hexobarbital (100 mg/kg, i.p.). Observation of the mice began immediately after hexobarbital injection. Once the mouse was asleep, it was transferred to a prenumbered chamber and placed on its back. Mice were considered awake when they could successfully right themselves (all four feet in contact with the surface) or until 240 min had elapsed. Once a mouse righted itself, it was placed on its back once more and allowed to right a second time for confirmation. Sleep duration was recorded for each mouse.
Convulsive liability—electroshock-induced convulsions.
Electroshock was conducted in male CD-1 mice according to the method ofSwinyard et al. (1952) with the following modifications. Twenty min after i.p. administration of diazepam (0.3–3 mg/kg), NMDA (25 mg/kg) or LY354740 (administered as the racemate (±)LY314852) (50–200 mg/kg), saline was placed on each cornea immediately before placement of the eyes in corneal electrodes. Current was then delivered with shock intensities ranging from 4 to 9 mA (0.2-sec shock duration). The types of convulsions (clonic, tonic or tonic-extensor) observed within 10 sec were recorded.
Learning and memory—conditioned avoidance responding.
Rats were anesthetized with isoflurane gas and surgically implanted with Alzet osmotic pumps (Alza Corporation, Vacaville, CA) filled with water or LY354740 delivering 3 mg/kg/day. All rats were implanted on the same day and testing was conducted in three different groups according to the following schedule: rats receiving 3 mg/kg/day LY354740 or vehicle were tested on days 2 through 6. Separate rats receiving 3 mg/kg/day LY354740 or vehicle were tested on days 7 through 11. Separate rats with 3 mg/kg/day LY354740 or vehicle exposure for 12 days were tested on days 1 through 5 after pump removal. An Omnitech Shuttle System (Columbus, OH) was used to collect behavioral data. The avoidance procedure was carried out in a two-compartment shuttle box with a grid floor through which electric current was delivered. Each test session consisted of 20 trials and each animal was tested for 5 consecutive days. The intertrial interval for each test session was variable (30 ± 10 sec). A combination of light (6 W) and acoustic (90 dB tone) stimuli was used, with a signal duration of 5 sec. Shock intensity was set at 0.5 mA and the shock interval was 5 sec. To avoid the shock, rats learned to move from one side of the chamber to the other side at the cue (avoidance) or move when the shock was administered (escape). Data were tabulated as mean percent escape or mean percent avoidance responses.
Learning and memory—passive avoidance responding.
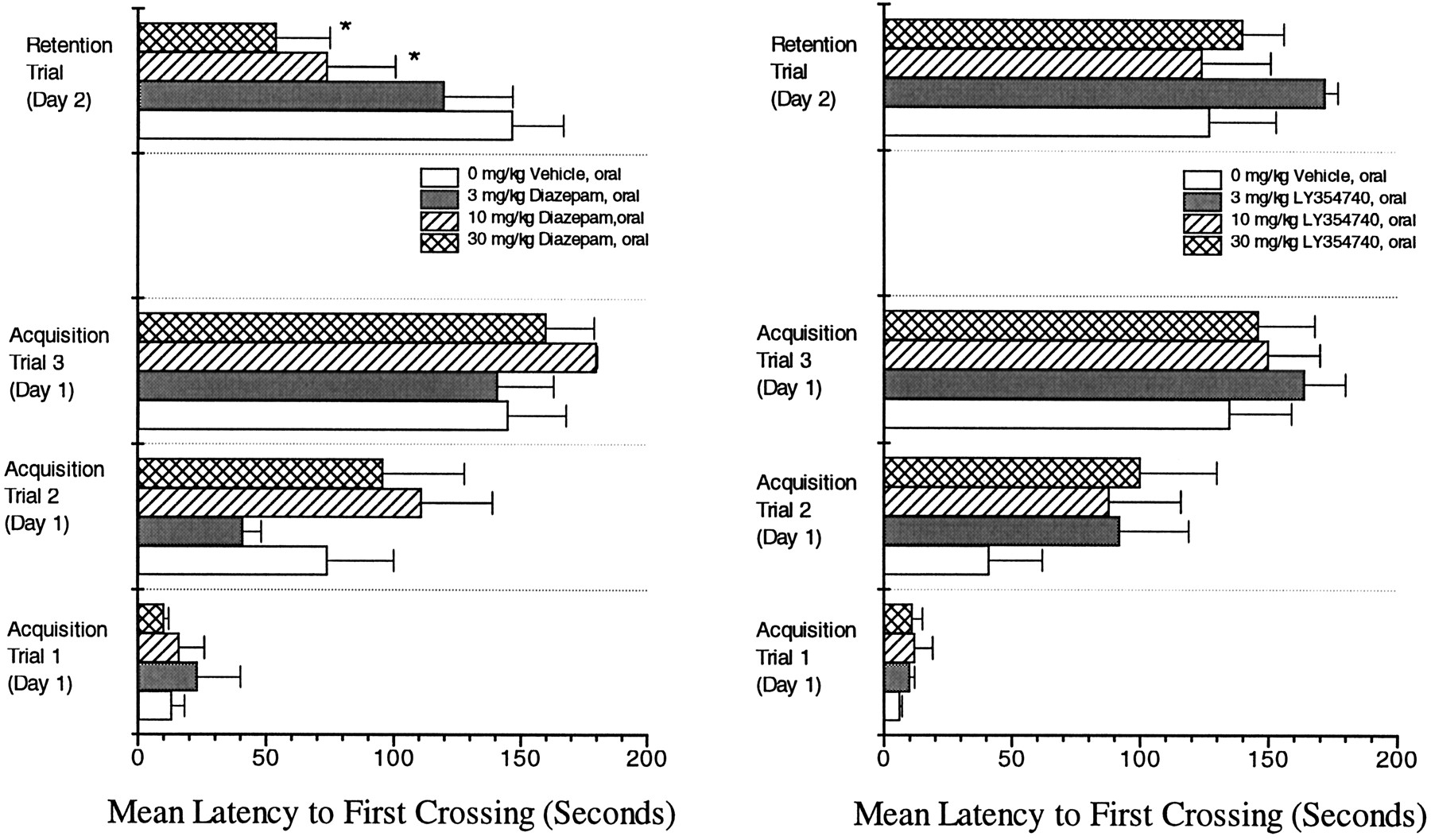
Male CD-1 mice were tested in a 2-day passive avoidance task. Diazepam (3–30 mg/kg, oral) or LY354740 (3–30 mg/kg, oral) was administered on test day one 60 min before acquisition training. Passive avoidance performance was evaluated using the Omnitech Shuttle Control System (Omnitech Electronics), which consisted of eight, two-compartment shuttle chambers and a constant current shock generator. Mice were individually placed in the shuttle chamber, and the session was initiated with a one min acclimation period. Day 1 acquisition testing consisted of three, 180-sec training trials which were signaled by illumination of a light (6 W) on the side the animal was on in the chamber. During each trial, a foot shock of 0.5 mA was delivered through the grid floor if the mouse crossed to the other compartment which was kept dark. When the animal crossed into the dark compartment a footshock was administered continuously until the animal returned to the illuminated compartment. Trials were separated by 1-min intervals of total darkness. On day 2, 24-hr (approximately) retention testing consisted of a 1-min acclimation period followed by a single, 180-sec retention trial in which no foot shock to mice was delivered upon crossing in any group. Computer-recorded data for each animal for each trial are reported as the mean latency to first crossing.
Statistical evaluations.
Potentiated startle, elevated plus maze, spontaneous activity, sensorimotor reactivity, neuromuscular coordination, hexobarbital-induced sleep time and learning and memory assessments were evaluated by an analysis of variance. When significant treatment effects were obtained, post hoc comparisons were made using a Tukey’s studentized range (HSD) test. In all analyses, two-tailed statistical tests were used and the level of significance was set at P ≤ .05. Respective SI50 values [shock intensity (mA) inducing tonic-extensor convulsions in 50% of the mice] for electroshock were calculated by probit analysis (log10) and compared to respective SI50 values obtained in the presence of LY354740 at the 95% confidence limits. Statistical analyses were performed using Statistical Analysis Systems (SAS Institute, Inc., Cary, NC).
Results
Fear potentiated startle.
Administration of diazepam (fig.2) resulted in a dose-related attenuation of fear potentiated startle responding with significant attenuation at 0.1, 0.3 and 1 mg/kg diazepam (ED50 = 0.4 mg/kg) (Tukey’s HSD, P ≤ .05). Oral administration of LY354740 (60-min pretreatment) produced a dose-dependent attenuation of fear potentiated startle responding in rats with significant reductions at 0.1, 1, 3 and 10 mg/kg (ED50 = 0.3 mg/kg LY354740) (fig.3) (Tukey’s HSD, P ≤ .05). Baseline startle responding during fear potentiated startle was not altered at any dose of diazepam or LY354740 examined (data not shown; see fig. 7 for LY354740 acute startle results). The inactive isomer of LY354740, (−)LY366563 (Monn et al., 1997), did not attenuate the fear potentiated startle response at oral doses up to 10 mg/kg (fig. 3). Oral administration of 3 mg/kg LY354740, a dose that produced a significant reduction in fear potentiated startle, produced a significant attenuation of fear potentiated startle responding for at least 6 hr after administration (fig. 4) (Tukey’s HSD, P ≤ .05).

Effect of intraperitoneally administered diazepam on fear potentiated startle responding in male Long Evans rats (n = 8 rats/treatment group). Values represent the mean (±1 S.E.) startle amplitude (Vmax) expressed as the difference between noise-alone and light + noise. Diazepam was administered 30 min before evaluation of startle responding. * Significantly different from control P ≤ .05.

Effect of orally administered (+) LY354740 (above left) or orally administered (−)LY366563 (the inactive isomer) (above right) on fear potentiated startle responding in male Long Evans rats. Values represent the mean (±S.E.) startle amplitude (Vmax) expressed as the difference between noise-alone and light + noise. LY354740 or LY366563 was administered orally 60 min before evaluation of startle responding. * Significantly different from control P ≤ .05.

Effect of orally administered LY354740 on sensorimotor reactivity as evaluated using auditory startle responding in Long Evans rats. LY354740 was administered by oral gavage 60 min before testing. Values represent the mean (±S.E.) peak response (maximum startle amplitude) averaged over 10 trials (n= 10 rats/treatment group).
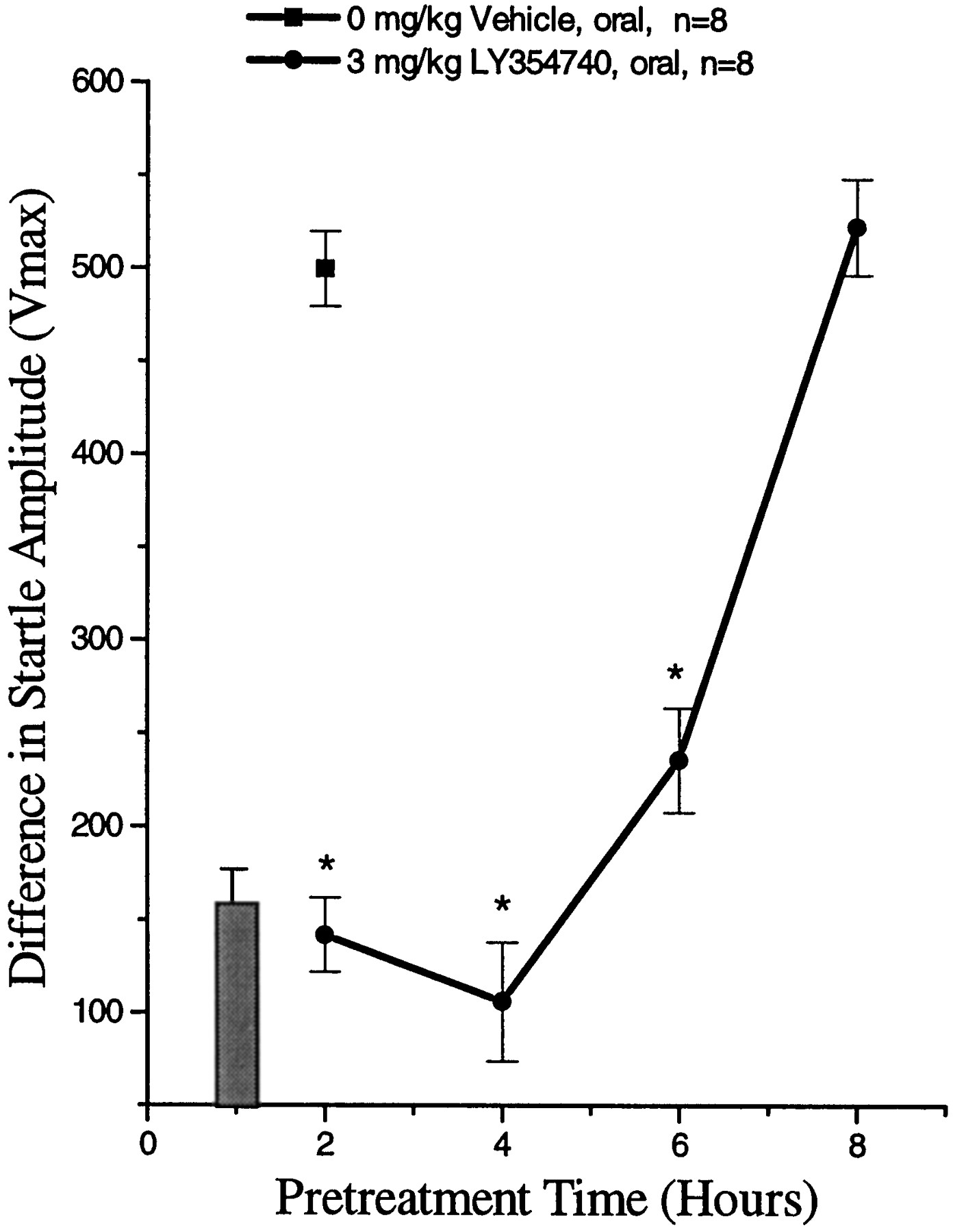
Effect of LY354740 (3 mg/kg) on fear potentiated startle responding in male Long Evans rats after oral pretreatment times of 2, 4, 6 or 8 hr (circles). The black bar represents the effect of LY354740 at 1 hr (data taken from fig. 3). Values represent the mean (±S.E.) startle amplitude (Vmax) expressed as the difference between noise-alone and light + noise. * Significantly different from water control P ≤ .05 (square).
Elevated plus maze.
Oral administration of 3 mg/kg LY354740 produced a significant increase in nosepoke activity at 1, 4 and 6 hr after administration (fig. 5) (Tukey’s HSD, P ≤ .05). Significant increases in open-arm activity was found at 1 and 4, but not 6, hr postdosing (fig. 5). Closed-arm activity counts were not significantly altered at any timepoint (fig.5). The inactive isomer, (−)LY366563, did not alter open, nosepoke or closed-arm activity at oral doses up to 10 mg/kg (data not shown).

Effect of LY354740 (3 mg/kg) on automated elevated plus maze in male NIH Swiss mice after oral pretreatment times of 1, 4 and 6 hr. Nosepoke counts represent incomplete movements (head only) into the open arm from the closed arm. Open counts represent activity in the open (sideless) arms of the maze. Closed counts represent activity in the enclosed arms of the maze. Scores represent mean ± S.E. of activity counts in each zone during a 5-min test period. * Significantly different from control P ≤ .05.
Spontaneous activity.
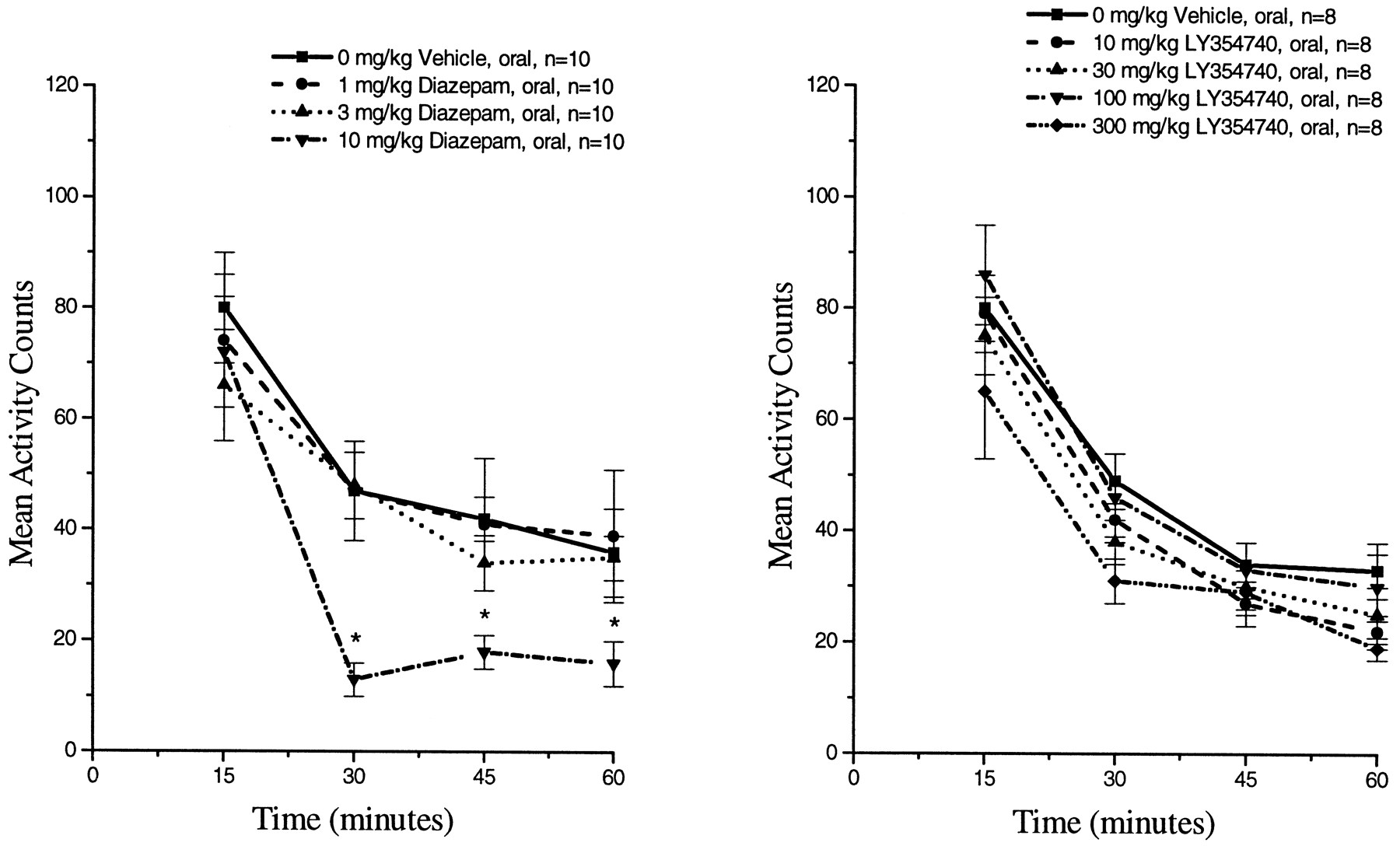
Diazepam administration significantly reduced spontaneous ambulatory (fig. 6) and nonambulatory (data not shown) activity levels in rats at 10 mg/kg (oral) from 30 through 60 min postdosing (Tukey’s HSD, P ≤ .05). LY354740 did not significantly alter spontaneous ambulatory (fig. 6) or nonambulatory (data not shown) activity levels at oral doses up to 300 mg/kg.

Effect of orally administered diazepam (left panel) or orally administered LY354740 (right panel) on spontaneous ambulatory activity levels in male Long Evans rats accumulated at 15-min intervals for 60 min. Activity counts represent the sum of individual photocell beam breaks averaged at 15 min intervals (mean ± S.E.). * Significantly different from control P ≤ .05.
Sensorimotor reactivity—auditory startle.
There were no significant changes in auditory startle responding in rats at oral doses of LY354740 as high as 10 mg/kg (fig.7), a dose that completely suppresses fear potentiated startle responding (see fig. 5). In addition, no significant changes were seen in startle habituation at doses up to 10 mg/kg (data not shown).
Neuromuscular coordination—rotarod.
Orally administered diazepam significantly reduced the number of sec rats spent on the rotarod at doses of 3 and 10 mg/kg given 60 min before testing (fig.8) (Tukey’s HSD, P ≤ .05). LY354740 administration (30 min pretreatment) did not produce any significant changes in neuromuscular coordination as evaluated on the rotarod at oral doses up to 300 mg/kg (fig. 8).

Effect of orally administered diazepam (left panel) or orally administered LY354740 (right panel) on neuromuscular coordination as evaluated on the rotarod in male Long Evans rats (n = 10 rats/dose group). Diazepam or LY354740 was administered by oral gavage 30 min before testing. Values represent the mean (±S.E.) number of sec rats remained on the rotarod. * Significantly different from control P ≤ .05.
Interaction with CNS depressants—hexobarbital-induced sleep times.
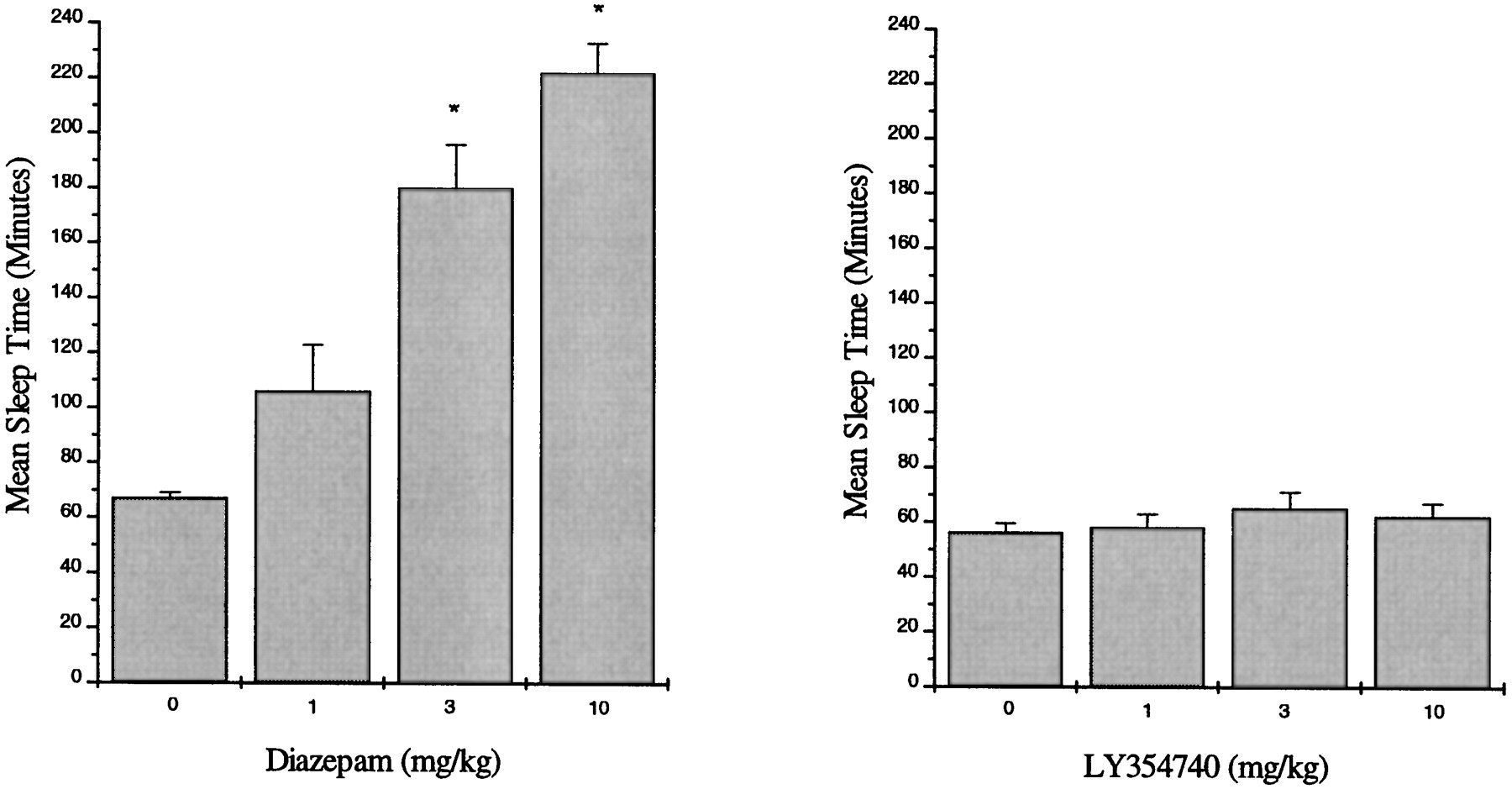
Orally administered diazepam produced dose-dependent increases in hexobarbital-induced sleep times in CD-1 mice with significant effects at 3 and 10 mg/kg (169 and 231% increases with respect to control, respectively) (fig.9) (Tukey’s HSD, P ≤ .05). There were no significant increases in sleep time after oral administration of LY354740 (60 min pretreatment) at doses up to 10 mg/kg (fig. 9).

Effect of orally administered diazepam (left panel) or orally administered LY354740 (right panel) on hexobarbital-induced sleep time in male CD-1 mice. Diazepam or LY354740 was administered by oral gavage 60 min before administration of hexobarbital (10 mg/kg, i.p.). Values represent mean (±S.E.) sleep time in min (n = 10 mice/treatment group). * Significantly different from control P ≤ .05.
Convulsive liability—electroconvulsive shock.
Diazepam (i.p.) effectively attenuated all electroshock-induced tonic convulsions at doses of 1 and 3 mg/kg (fig. 10). In contrast, the ionotropic glutamate receptor agonist, NMDA, produced a significant increase in the number of electroshock-induced convulsions when tested at a single i.p. dose of 25 mg/kg (fig. 10). No significant changes were observed in the current intensity producing 50% tonic convulsions following administration of LY354740 (administered as the racemate (±)LY314582, i.p.) at doses up to 200 mg/kg (fig. 10).

Effect of i.p. administered diazepam (left panel) or i.p. administered NMDA or LY354740 (administered as the racemate (±)314582) (right panel) on electroshock-induced convulsive patterns in male CD-1 mice. Diazepam, NMDA or LY354740 was administered i.p. 20 min before testing. Values represent the number of animals (expressed as percent) displaying tonic convulsions at each current level (n = 40 mice/dose-response).
Learning and memory—conditioned avoidance responding.
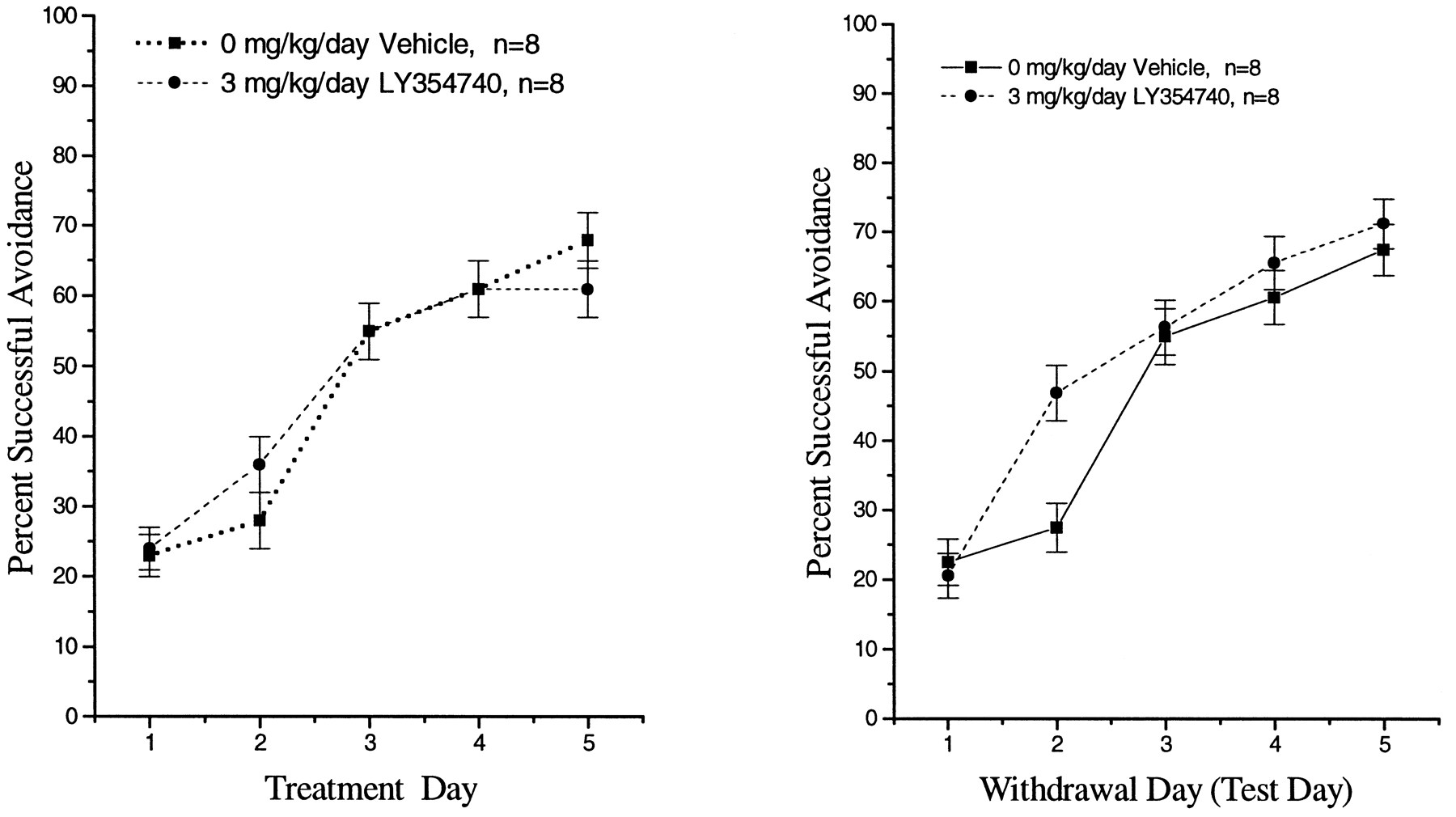
Continuous administration of LY354740 (3 mg/kg/day, s.c. infusion) to rats did not impair the acquisition of avoidance responding on days 2 to 6 (fig. 11) or days 7 to 11 of exposure (data not shown). Cessation of LY354740 exposure did not disrupt the acquisition of avoidance responding during the first 5 days of withdrawal (fig. 11).

Effect of s.c. administered LY354740 (3 mg/kg/day) on the acquisition of avoidance responding during treatment (left panel) or after the withdrawal of LY354740 (right panel) in male Long Evans Rats. LY354740 was administered for 12 consecutive days in osmotic pumps. Values represent mean (±S.E.) percent successful avoidance (n = 8 rats/treatment group).
Learning and memory—passive avoidance.
Oral diazepam (0, 3, 10 or 30 mg/kg) did not alter the acquisition phase (acquisition trials 1, 2, 3—learning day 1) of passive avoidance responding in CD-1 mice at any dose examined. However, diazepam produced a dose-dependent disruption of the retention phase (retention trial—memory day 2) of passive avoidance with significant deficits in responding at 10 and 30 mg/kg (fig. 12) (Tukey’s HSD, P ≤ .05). LY354740, at oral doses up to 30 mg/kg, did not disrupt the acquisition phase or the retention phase of passive avoidance responding (fig. 12).

Effect of p.o. administered diazepam (left panel) or p.o. administered LY354740 (right panel) on the acquisition (acquisition trials 1, 2, 3—learning day 1) and retention (retention trial—memory day 2) of passive avoidance responding in male CD-1 mice. Diazepam or LY354740 was administered 60 min before testing during the acquisition phase. Values represent mean (±S.E.) of latency to first crossing (n = 8 mice/treatment group). * Significantly different from control P ≤ .05.
Discussion
LY354740, a conformationally constrained analog of glutamate, is a highly potent and selective group II (mGluR2 and mGluR3) receptor agonist with no appreciable agonist/antagonist activity at metabotropic group I and group III mGluRs or ionotropic (NMDA, AMPA or kainate) glutamate receptors (Monnet al., 1997; Schoepp et al., 1997). The high potency and selectivity of LY354740 for group II mGluRs make LY354740 a novel pharmacological tool for investigating the role of group II mGluRs in physiological processes and exploring the potential therapeutic applications of such agents.
Benzodiazepines such as diazepam (Valium) are the most widely prescribed medications for the treatment of anxiety and related disorders. The 5HT1a agonist, buspirone (Buspar), is also available, but is limited in use due to marginal efficacy and delayed onset of action (Riblet et al., 1982). Benzodiazepines act at a specific site on the GABAA receptor to facilitate fast-inhibitory synaptic transmission throughout the CNS. This likely accounts for their widespread therapeutic applications including use as anticonvulsants, anxiolytics, muscle relaxants, sedative-hypnotics and anesthetic agents. However, the widespread distribution of benzodiazepine receptors also produces side-effects and other liabilities such as CNS depression, abuse potential, physical dependence and withdrawal reactions (Woods et al., 1992). In most synapses, the inhibitory effects of GABA are opposed by glutamate excitation and theoretically, agents that block the postsynaptic excitatory effects of glutamate produce pharmacology similar to benzodiazepine pharmacology (Coyle, 1987). Consistent with this theory, antagonists for NMDA and/or AMPA receptors have been shown to produce anxiolytic effects in the fear-potentiated startle (Kim et al., 1993; Anthony and Nevins, 1993) or elevated plus maze models (Wiley et al., 1995) of anxiety. However, in contrast to NMDA or AMPA receptors that are postsynaptic and participate directly in fast-excitatory synaptic transmission, group II mGluRs have unique pre- and postsynaptic localizations (Petralia et al., 1996) which can alter second messengers to indirectly modulate fast-synaptic transmission (Schoepp and Conn, 1993). To evaluate the potential clinical utility of LY354740 as an anxiolytic, we examined its effects in the fear potentiated startle and elevated plus maze animal models of anxiety.
The potentiated startle procedure is a model based on fear which is selective for the identification of anxiolytic drugs (Davis, 1979;1986; see above). The amygdala has been implicated as an important brain region for acquisition, retention and expression of conditioned fear and the actions of anxiolytic drugs (Davis, 1992; Davis et al., 1993, 1994). The suppression of excitatory glutamatergic synaptic transmission within the amygdaloid complex has also been linked to anxiolytic responses in animals (Campeau and Davis, 1995). For example, intraamygdala injections of NMDA receptor antagonists prevent the acquisition of fear-potentiated startle responses in rats, although having no effects on the expression of fear responses in conditioned animals (Miserendino et al., 1990; Anthony and Nevins, 1993). However, the intraamygdala injection of the AMPA/kainate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione blocks the expression of fear-potentiated startle responding in animals (Kimet al., 1993). Within the amygdaloid complex there is evidence that tonic excitation through glutamate pathways and evoked excitatory postsynaptic potentials can be blocked by antagonists for NMDA and AMPA/kainate ionotropic receptors that act at postsynaptic sites (Rainnie et al., 1991), or metabotropic agonists (group II and III) that act presynaptically to diminish postsynaptic AMPA receptor mediated fast-excitatory postsynaptic potentials (Rainnie and Shinnick-Gallagher, 1992). Interestingly, the oral administration of LY354740 produced a dose-dependent attenuation of fear potentiated startle responding with significant reductions at 0.1, 1, 3 and 10 mg/kg (ED50 = 0.3 mg/kg LY354740) (fig. 3). Baseline startle responding was not altered at any dose of LY354740 examined (fig. 7). The effects observed were stereoselective because the inactive enantiomer of (+)LY354740, (−)LY366563, was without effect in these models at oral doses up to 10 mg/kg (fig. 3). Additionally, oral administration of 3 mg/kg LY354740, a dose that produced a significant reduction in fear potentiated startle, produced a significant attenuation of fear potentiated startle responding during the first 6 hr after administration (fig. 4). Thus, in the fear potentiated startle model, LY354740 and diazepam produced significant anxiolytic activities with very similar potencies (ED50 values of 0.3 and 0.4, respectively, see table 1).
Comparative doses (ED50) of diazepam (DIAZ) or LY354740 in efficacy and secondary pharmacology models
We have previously shown that in the elevated plus maze, oral administration of LY354740 produces dose-dependent increases in open arm activity with no effect on closed-arm activity (Monn et al., 1997). In this study, we further examined the time course of activity for LY354740 after oral administration of 3 mg/kg, a dose that produced maximal increases in open-arm activity when administered 30 min before testing. LY354740 produced significant increases in open-arm and nosepoke activity through the first 4 hr after administration. Again, closed-arm activity counts were not significantly altered at any timepoint (data not shown).
In addition to anxiolytic activity, administration of diazepam and other benzodiazepines can result in sedative, ataxic and cognitive side-effects (Greenblatt and Shader, 1978; Dantzer, 1977) and with repeated exposure, tolerance and dependence. In our series of experiments, diazepam produced sedation with therapeutic ratios (based on efficacy ED50/Rotarod ED50) of only 19.8- and 36-fold higher than those producing anxiolytic effects in fear potentiated startle and the elevated plus maze, respectively (table 1). In contrast, LY354740 did not produce changes in neuromuscular function at doses as high as 300 mg/kg providing therapeutic ratios of >1000 and >600, respectively (table 1).
The difference in side-effect profile between diazepam and LY354740 is also reflected in overt clinical signs and spontaneous activity tests. In these tests, diazepam, but not LY354740, produced marked changes in coordination, and activity (table 1). Additionally, LY354740 did not produce cognitive impairment during the first 11 days of continuous administration or during the first 5 days after withdrawal. Furthermore, the acquisition (learning) of a passive avoidance task was not disrupted by either diazepam or LY354740. However, although memory for the task was not altered by LY354740, a significant disruption of retention was noted for diazepam treated animals (fig. 12).
As a class of pharmaceutical compounds, benzodiazepines consistently enhance the effects of other CNS depressants. Potentiation of hexobarbital-induced sleep time was used to evaluate potential interactions of LY354740 with CNS depressants. Hexobarbital is a short-to-intermediate acting barbiturate that exerts its primary pharmacological effect on the CNS by enhancing inhibition of GABA-mediated neurotransmission (Olsen, 1982). Test compounds may shorten or prolong hexobarbital-induced sleep time by acting directly on the CNS (excitation or depression) or indirectly via inhibition of hepatic enzymes involved in hexobarbital metabolism. As expected, hexobarbital-induced sleep times were dose-dependently increased by diazepam supporting its known interactive activity with sedative-hypnotics. However, LY354740 did not increase sleep time indicating that LY354740 may not produce CNS depression or interact with other CNS depressants at clinically relevant doses (fig. 9).
Benzodiazepines are also used clinically as anticonvulsants. Changes in electroconvulsive shock thresholds after administration of diazepam or LY354740 were evaluated. Although diazepam increased the threshold for electroshock-induced convulsions, no significant increases in convulsive thresholds were seen with LY354740 (fig. 10). In addition, the proconvulsive ionotropic receptor agonist, NMDA, decreased convulsive thresholds in electroshock (fig. 10). These data suggest that LY354740 may not produce CNS depression or proconvulsive activities in humans.
In summary, LY354740 is a novel, nanomolar potent, highly selective and orally active agonist at group II/cAMP coupled metabotropic glutamate receptors. These data indicate that activation of in vivogroup II mGluRs with LY354740 produces benzodiazepine-like efficacy in the fear-potentiated startle and elevated plus maze models of anxiety. However, unlike benzodiazepines, LY354740 produces no CNS depression and other unwanted benzodiazepine-like activities in animals. This atypical profile of anxiolytic activity and large safety margin in animals for LY354740 may reflect the novel mechanism and regional selectivity produced by this agent. These data support the functional role of group II mGluRs in anxiety. Additionally, LY354740 may be a useful pharmacological tool for evaluating the therapeutic relevance of group II receptor activation in anxiety and other CNS-related disorders in humans.
Acknowledgments
The authors thank Vanessa Benagh, Angie Boring, Kenneth Firman, Kelly Griffey, Allen Johnson and Deah Modlin for their fine technical assistance in performing the behavioral studies.
Footnotes
-
Send reprint requests to: Dr. Darryle D. Schoepp, Neuroscience Research Division, Lilly Corporate Center, drop 0510, Eli Lilly and Company, Indianapolis, IN 46285.
-
↵1 Current address: Quintiles BRL, 1300 N. 17th Street, Suite 300, Arlington, VA 22209.
- Abbreviations:
- CNS
- central nervous system
- iGluR
- ionotropic glutamate receptor
- NMDA
- N-methyl-d-aspartic acid
- AMPA
- 2-amino-3(3-hydroxy-5 methylisoxazol-4-yl)propionic acid
- mGluR
- metabotropic glutamate receptor
- HSD
- Tukey’s studentized range
- Received May 23, 1997.
- Accepted October 27, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}