


Abstract
Linopirdine [DuP 996, 3,3-bis(4-pyridinylmethyl)-1-phenylindolin-2-one], a putative cognition enhancing drug, increases acetylcholine release in rat brain tissue and improves performance in animal models of learning and memory. The mechanism whereby linopirdine enhances acetylcholine release has been proposed to involve inhibition of the M-type K+ current (IM). Our study examines the selectivity of linopirdine for IM by determining its effects on other ionic currents present in rat hippocampal CA1 neurons using patch clamp techniques. Linopirdine was found to block voltage-gated, calcium-activated and leak K+currents in a dose-dependent manner. Of the seven currents measured, linopirdine was most selective for IM with an IC50 of 2.4 ± 0.4 μM, followed by IC(measured as a medium afterhyperpolarization tail current, ImAHP) with an IC50 of 16.3 ± 2.4 μM. Both IM and IC were completely suppressed by linopirdine. At a concentration of 100 μM, linopirdine weakly inhibited the K+ leak current, IL, the transient outward current, IA, the delayed rectifier, IK, and the slow component of IAHP, by 28 ± 8, 37 ± 10, 36 ± 9 and 52 ± 10 percent, respectively. The mixed Na+/K+ inward rectifying current, IQ, was essentially unaffected by linopirdine (IC50 >300 μM). These results indicate that linopirdine selectively blocks IM at concentrations ≤ 3 μM, the approximate EC50 for acetylcholine release enhancement. Inhibition of other voltage-gated and calcium-activated K+ currents could also contribute to enhanced neurotransmitter release by linopirdine at intermediate (IC) and high (IL, IA, IK, IsAHP) concentrations.
Linopirdine is a putative cognitive enhancing drug that increases stimulus-evoked release of a number of neurotransmitters, including ACh (Nickolsonet al., 1990; Zaczek et al., 1995; Aiken et al., 1996). Although the exact mechanism by which linopirdine enhances ACh release is unknown, Maciag et al. (1994) showed that its effects were insensitive to 4-aminopyridine, atropine and Na+, Cl− and Ca++ channel antagonists. In addition, they found no apparent role for cholinergic autoreceptors. TEA was found to have actions similar to linopirdine on ACh release suggesting the involvement of a K+ channel in the action of linopirdine. Recently, Aiken et al. (1995) demonstrated that linopirdine reduced spike frequency adaptation and blocked IM in rat hippocampal CA1neurons in vitro. Because the concentration-response curves for IM block and ACh release have similar slopes and IC50/EC50 values, it has been proposed that M-current block may represent the mechanism underlying linopirdine-induced neurotransmitter release enhancement (Aiken et al., 1996).
Before ascribing pharmacological relevance to its M-current blocking activity, however, it is necessary to determine the effects of linopirdine on other K+ channels. Many K+ channel blockers have been shown to be non-selective. For example, TEA blocks IC, IK, IM, IK(IR) and IK(ATP), 4-AP blocks IA, ID, some types of IK and IK(ATP) and charybdotoxin blocks an intermediate-, as well as, the large-conductance calcium-activated K+ channel (Cook and Quast, 1990; Halliwell, 1990). Thus, there may be other K+ channels more sensitive than the M channel to the blocking action of linopirdine. In addition, a number of other potassium currents have been implicated in the control of neurotransmitter release. 4-AP and α-dendrotoxin, at concentrations that inhibit IA, increase the release of glutamate from guinea-pig cerebrocortical synaptosomes (Tibbs et al., 1989). Amoroso et al. (1990) demonstrated enhanced release of γ-aminobutyric acid by block of an adenosine triphosphate-sensitive K+ channel in substantia nigra, and Robitaille et al. (1993) reported an increase in transmitter release at the neuromuscular junction produced by block of Ca++-gated K+ channels.
Our study was undertaken to determine the selectivity of linopirdine for IM by determining its effects on the voltage-gated K+ currents IM, IA and IK, the afterhyperpolarization currents, ImAHP and IsAHP, the leak current, IL, and the inward rectifier, IQ, recorded from rat pyramidal CA1 neurons in the hippocampal slice. Portions of this work have previously been published in a preliminary form (Schnee and Brown, 1995).
Methods
Studies in this report were carried out in accordance with the Declaration of Helsinki and with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Rockville, MD).
Tissue preparation.
Pathogen-free male CD rats from Charles River (Wilmington, MA) weighing 30 to 80 g (15–25 days old) were anaesthetized with halothane. After decapitation, the brain was rapidly excised (<1 min) and submerged in an ice-cold oxygenated physiological solution. The brain was bisected and transverse slices containing the hippocampus were prepared on a Vibratome tissue slicer. Slices were transferred to a Perspex holding chamber filled with chilled saline and allowed to reach room temperature (23°C). For recording, slices were placed on a nylon mesh in a submersion-type chamber (Medical Systems, Greenvale, NY), pinned to a Sylgard base and perfused with an oxygenated physiological saline solution at room temperature at a rate of 3 ml min−1. The physiological solution for both dissection and recording was (mM) NaCl (127.0), NaHCO3 (26.0), KCl (3.0), CaCl2 (2.5), NaH2PO4 (1.25), MgSO4 (1.0) and glucose (10.0), gassed with 5% CO2 in O2 (pH 7.35). TTX (0.1 μM) or Cd++ (0.3 mM) were added to the perfusion solution to block Na+ and Ca++ currents, respectively.
Electrophysiological recording.
Microelectrodes were pulled from borosilicate glass (1.5 mm OD/1.0 mm ID; World Precision Instruments, Sarasota, FL) using a Sutter P-80/PC electrode puller (Sutter Instruments, Novato, CA). Electrode resistances were 2–2.5 Mohms when filled with intracellular solution. Tight-seal (1–5 Gohm) whole-cell voltage-clamp recordings, with access resistances of ≤ 20 Mohms, were obtained from neurons in the CA1pyramidal cell body layer using the “blind” patch technique. Current recordings were obtained by means of an Axopatch 200A amplifier (Axon Instruments, Foster City, CA). Signals were filtered at 5 KHz and recorded with pClamp software (version 6.0.1, Axon Instruments). Series resistance compensation was not used in order to minimize noise and “ringing” of the amplifier. In preliminary studies, little difference in current amplitudes was noted between the presence and absence of series resistance compensation when using 2 to 2.5 Mohm resistance electrodes. Except where stated, application time of drugs was approximately 20 min. Internal solutions for whole cell recording were (in mM) Kgluconate (140), KCl (10), HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) (10), EGTA (ethylene glycol-bis(β-aminoethylether)-N,N,N′,N′-tetraacetic acid) (10), MgCl2 (2.0), CaCl2 (1.0) and MgATP (2.0), pH adjusted to 7.4 with KOH; later experiments used KMethylsulfate (140) or KAspartate (130), KCl (10), HEPES (10), BAPTA (1,2-bis(2-aminophenoxy)ethane- N,N,N′,N′-tetraacetic acid) (10), K2ATP (adenosine 5′-triphosphate, dipotassium salt) (5.0), MgCl2 (2.0) and CaCl2(1.0), pH adjusted to 6.7 (to avoid rundown; Brown et al., 1989; Cloues and Marrion, 1996) with KOH to record IM and pH 7.3 to record other currents.
Drugs.
Linopirdine (free base) was synthesized at the DuPont Merck Pharmaceutical Company (Wilmington, DE). A stock solution in 0.1 N HCl was prepared immediately before use and added to the superfusing solution. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Data analysis.
All calculated data are expressed as mean ± S.E.M. In cumulative dose-response experiments on the effect of linopirdine on IL amplitude over a range of voltage steps, a two-factor analysis of variance model (SuperANOVA, version 1.11, Abacus Concepts, Berkeley, CA) was used to test the hypothesis that mean control (pretreatment) values were unchanged with drug treatment. If there was evidence of a statistically significant treatment effect, Duncan’s New Multiple Range test (SuperANOVA) was used to identify the significant dose effects. Statistical significance level was set at P < .05.
Results
Voltage-Gated Currents
IM.
M-current activates at potentials positive to −70 mV and is fully activated at −30 mV (Brown, 1988). To record IM, hippocampal CA1 neurons were stepped to −30 mV for 30 sec from a holding potential of −50 mV, repolarized by 20 mV for 2 sec and then stepped to −30 mV for 10 sec. Using this protocol, M-current deactivated in a mono-exponential manner during the 20 mV repolarization step. The amplitude of IM was measured by fitting a single exponential function (Clampfit, Axon Instruments) to the outward deactivation tail current and extrapolating to the onset of the repolarization step. Each measure of IM was the mean of nine repetitions of this protocol. Time constants for current deactivation from −30 mV were in the range of 110 to 220 msec, which were comparable to previously published reports (Brown, 1988). IM in this preparation was sensitive to 50 μM carbachol and 1 mM Ba++ (figs. 1A and B) and was stable for >60 min after breaking into whole cell mode (amplitude was 106 ± 5, 104 ± 6 and 104 ± 9% of control after 20, 40 and 60 min, respectively).
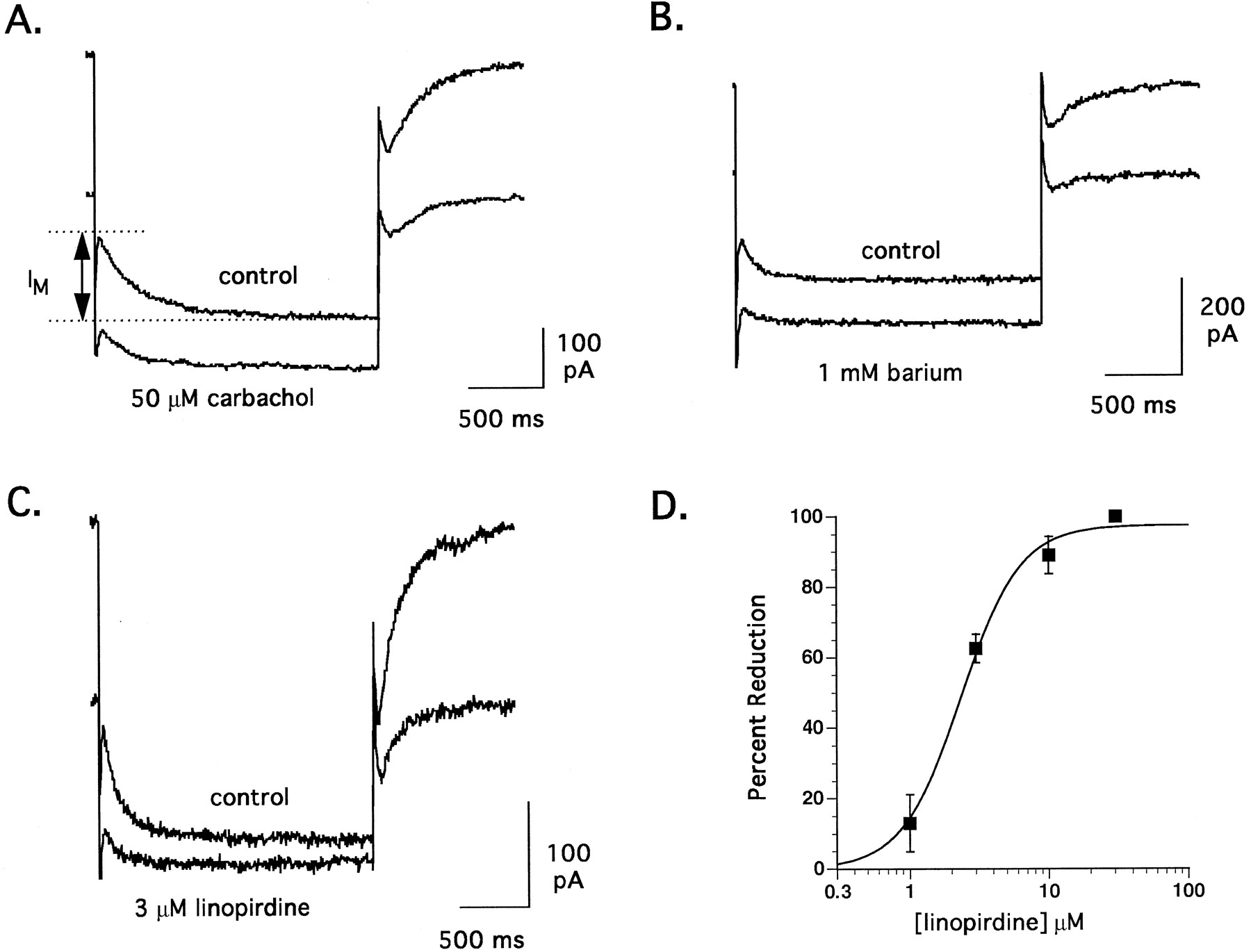
Representative traces of the effect of 50 μM carbachol (A), 1 mM barium (B) and 3 μM linopirdine (C) on M-current recorded from the soma of a CA1 neuron in a rat hippocampal slice. Each trace illustrates the deactivation tail current during a 2 sec voltage step from −30 to −50 mV; the amplitude of which is equivalent to the M-current. (D) Concentration-response curve for the inhibition of M-current by linopirdine (n = 5, 11, 5 and 2 at 1, 3, 10 and 30 μM linopirdine, respectively). Recordings were made in the presence of 0.1 μM TTX and 0.3 mM Cd++using a patch pipette with an internal pH of 6.7.
A comparison of the IM current relaxation before (control) and after a 20-min bath application of 3 μM linopirdine is illustrated in figure 1C. In this particular cell, linopirdine reduced the peak current by 64% without significantly affecting the rate of deactivation. In 11 neurons tested, linopirdine at 3 μM reduced IM by 63 ± 4%. The time constant of IM deactivation was unaffected by linopirdine (131 ± 7 msec in control, 151 ± 14 msec with 3 μM linopirdine). IM block was only weakly reversible, even with extended wash (>60 min). Figure 1D shows the concentration response for IM inhibition by linopirdine with a calculated IC50 of 2.4 ± 0.4 μM and a Hill slope of 1.9. Complete block of IM by linopirdine was observed in three of five cells at 10 μM and two of two at 30 μM. Thus, the potency of linopirdine in blocking IM in this preparation was comparable to that reported by Aiken et al. (1995) using single electrode voltage clamp in hippocampal CA1neurons and in cultured sympathetic cervical ganglia cells using the perforated patch technique (Lamas et al., 1997).
IA.
The transient outward potassium current, IA, observed in these studies was initially characterized by determining its electrophysiological and pharmacological properties. Steady-state inactivation was studied by applying, from a holding potential of −50 mV, 100 msec prepulses to potentials of −20 to −110 mV followed by a 300 msec voltage step to +50 mV. Steady state activation was examined by clamping to potentials between −40 and +50 mV from a −50 mV holding potential in 10 mV increments. These studies showed that >90% of IA inactivation was removed by prepulses to potentials more negative than −75 mV and that IAactivated positive to −30 mV (fig. 2A). These characteristics, along with the observed block of IA by 4-AP (fig. 2B), are consistent with known properties of this current.

A, Mean steady-state inactivation and activation curves for the peak of the transient outward current, IA, rat hippocampal CA1 neurons (n = 6). B and C, Representative traces of the effect of 4-AP (5 mM) and linopirdine (10, 100 μM) on IA in neurons held at −50 mV, clamped to −100 mV for 100 msec to remove inactivation, then stepped to +50 mV to activate IA. D, Concentration-response curve for the inhibition of IA amplitude and inactivation rate constant (τ) by linopirdine (n = 6, 5 and 5 at 10, 30 and 100 μM linopirdine, respectively). Recordings were made in the presence of 0.1 μM TTX, 10 mM TEA (except for B) using a patch pipette with an internal pH of 7.3.
To isolate IA for the examination of linopirdine effects, slices were pretreated with 10 mM TEA to reduce the large delayed rectifier component of outward current present in CA1 neurons (seen in fig. 2B). In the presence of TEA, IA amplitude was determined by measuring the difference between baseline current at the holding potential of −50 mV and the peak of the transient current component following step depolarization (fig. 2B). The effects of 10, 30 and 100 μM linopirdine on 1) the voltage dependence of steady state inactivation and activation, 2) peak current amplitude during activation (fig. 2C) and 3) rate of inactivation were determined. Linopirdine had no effect on either the voltage dependence of steady-state inactivation or the voltage dependence of activation at any concentration studied. Linopirdine did, however, significantly reduce peak IA amplitude and tau inactivation (fig. 2D) by 37 ± 10 and 49 ± 3%, respectively, at 100 μM. These results indicate that the effects of linopirdine on IA were weak in comparison to its block of IM.
IK.
The delayed rectifier current, IK, observed in these studies was evoked by 2000 msec depolarizing steps to +60 mV from a holding potential of −50 mV in 10 mV increments without a hyperpolarizing prepulse. IK, measured as the steady-state current amplitude at the end of each depolarizing step (baseline values were 5416 ± 654 pA at +60 mV; n = 13), activated at potentials positive to −30 mV (fig. 3A) and only slowly inactivated during the command pulse (fig. 3B). These characteristics, along with the blocking effects of TEA (fig. 3B), are established properties of IK.

A, Mean steady-state activation curve for the peak of the delayed rectifier outward current, IK, rat hippocampal CA1 neurons. B and C, Representative traces of the effect of TEA (10 mM) and linopirdine (10, 100 μM) on IK in neurons held at −50 mV and stepped to +50 mV to activate IK. D, Concentration-response curve for the inhibition of IK amplitude by linopirdine (n = 9, 7 and 5 at 10, 30 and 100 μM linopirdine, respectively) Recordings were made in the presence of 0.1 μM TTX using a patch pipette with an internal pH of 7.3.
For the examination of linopirdine effects, cells were recorded with an intracellular pipette solution that allowed the rundown of IM within 5 min (pH = 7.3). Under these conditions, linopirdine, at 10 to 100 μM, reduced the amplitude of IK in a dose-dependent manner (figs. 3C and D) with the concentration that inhibited IK by 50% exceeding 100 μM. Linopirdine-induced block of IK was voltage-independent as activation V1/2 values were 14.4 ± 0.4, 15.0 ± 0.6, 15.3 ± 0.7 and 17.4 ± 0.7 mV under control and 10, 30 and 100 μM linopirdine conditions, respectively. In contrast to IA, linopirdine had no apparent effect on the rate of IK inactivation at any concentration studied (fig. 3C).
Afterhyperpolarization Currents
The outward tail current recorded from hippocampal CA1 neurons after 10 mV incremental depolarizing steps from a holding potential of −50 mV to +60 mV in the absence of calcium channel blockers (figs. 4A and 6B) appeared to be a composite of two currents which could be differentiated kinetically (medium and slow) as well as pharmacologically. Voltage steps of 3.5 to 108 msec duration to 10 to +60 mV elicited tail currents which decayed in a biexponential manner (figs. 4, B, C and D). The first component was not well resolved under the experimental conditions used. Results of the second exponential fit (as performed by the Clampex portion of pCLAMP; mixed method) were used to measure ImAHP parameters. Maximum ImAHP amplitude (744 ± 129 pA;n = 7) with a deactivation rate constant of 33 ± 7 msec (n = 7) was evoked by a 108 msec step to +60 mV. The slowest tail current component (referred to as IsAHP) was activated by long (1.5 sec) depolarizing steps to −30 mV and above (fig. 6A). Typically, IsAHP peaked 3 to 5 sec after termination of the depolarizing step (at an amplitude of 228 ± 45 pA;n = 14) and deactivated over the next several seconds (fig. 6B and C). The effect of linopirdine on ImAHP and IsAHP was determined using a step duration and amplitude at or near maximum for each current component; i.e., a step to +60 mV for 54 ms for ImAHP and 1.5 sec for IsAHP.

A, Representative response of a rat hippocampal CA1 neuron held at −50 mV, stepped to + 50 mV for 54 msec and returned to −50 mV. Upon repolarization to −50 mV, a biexponentially decaying tail current (an expansion of which is shown in B) can be visualized. The amplitude of the second component, measured at the time indicated by the solid triangle, was recorded as ImAHP/IC. The effect of step potential and duration of the preceding depolarizing pulse on the mean activation of ImAHP/IC (n = 8) is shown in C and D, respectively. Recordings were made in the presence of 0.1 μM TTX using a patch pipette with an internal pH of 7.3.

A, Mean steady-state activation curve for the peak of the slow afterhyperpolarization tail current, IsAHP, which occurs in rat hippocampal CA1 neurons after a 1.5-sec depolarization step from a holding potential of −50 mV (n = 8). B and C, Representative traces of the effect of norepinephrine (10 μM) and linopirdine (3, 30 μM) on the slow afterhyperpolarization tail current. D, Concentration-response curve for the inhibition of IsAHP by linopirdine (n = 12, 13, 11 and 7 at 3, 10, 30 and 100 μM linopirdine, respectively). Recordings were made in the presence of 0.1 μM TTX using a patch pipette with an internal pH of 7.3.
ImAHP.
To characterize ImAHP pharmacologically, the effects of TEA and cobalt, inhibitors of hippocampal IC, were evaluated. TEA (1.5 mM) was effective in reducing the amplitude of ImAHP (by 57 ± 4.5%; n = 6) (fig. 5A) and had no effect on IsAHP. Cobalt (2 mM) also reduced ImAHP amplitude (72%, n = 2), whereas cholinergic agonists (carbachol and muscarine, 5–50 μM) and norepinephrine (3–10 μM) had no consistent effect on ImAHP. Because ImAHP was blocked by TEA and cobalt, not blocked by carbachol and had a deactivation time constant of approximately 30 msec, it was assumed that ImAHP is largely comprised of what is commonly referred to as IC (Storm, 1990).

A and B, Representative traces of the effect of TEA (1.5 mM) and linopirdine (3, 30 μM) on ImAHP/IC (measured as described in the legend to fig. 4) in neurons held at −50 mV, stepped to + 50 mV for 54 msec and returned to −50 mV. C, Concentration-response curve for the inhibition of ImAHP by linopirdine after a 54 msec depolarization to +50 mV. (n values for ImAHP were 5, 8, 8, 5, respectively, at 3, 10, 30 and 100 μM linopirdine.) Recordings were made in the presence of 0.1 μM TTX using a patch pipette with an internal pH of 7.3.
Linopirdine also inhibited ImAHP (fig. 5B and C). At 3 μM, a concentration more than the IC50 for inhibition of IM, linopirdine had no significant effect on ImAHP. At 10 to 30 μM linopirdine, however, ImAHP was significantly depressed and essentially completely blocked at 100 μM. Accordingly, the IC50 for linopirdine-induced inhibition of ImAHP was 16.3 ± 2.4 μM (fig. 5C). As observed for IM, the effect of linopirdine on ImAHP was not readily reversible. However, this was not due to rundown of the current since, in control experiments, the amplitude of ImAHP remained stable over the time period of linopirdine exposure (122 ± 10% of control at 60 min). In addition to IM, the amplitude of ImAHP is the only K+current in our study to be completely suppressed by linopirdine. The deactivation rate constant of ImAHP was not significantly affected by linopirdine (data not shown).
IsAHP.
The current underlying the slow afterhyperpolarization in hippocampal pyramidal neurons can be blocked by several neurotransmitters, including norepinephrine and acetylcholine (Storm, 1990). To verify the identity of IsAHP as the current commonly referred to in the literature as IAHP, the effects of norepinephrine, muscarine and TEA were evaluated. In agreement with published reports, 10 μM norepinephrine (fig.6B) and 10 μM muscarine markedly attenuated IsAHP, whereas 1.5 mM TEA had no effect on IsAHP in the same preparations in which it blocked ImAHP by an average of 57 percent (data not shown).
Linopirdine exerted a concentration-dependent inhibition of the amplitude of IsAHP while having little or no effect on the rate of deactivation (fig. 6C and D). Its effects on IsAHP were, however, weak and similar to those on IA and IK in that a concentration of 30 μM was required to inhibit IsAHP by approximately 50%, with no additional effect at 100 μM. These results indicate that, of the two components of depolarization-induced outward tail currents measured, linopirdine most potently blocked ImAHP.
Inward Rectifier and Leak Current
IQ.
IQ, a mixed Na+/K+ current, was recorded in CA1 neurons in response to 10 mV incremental hyperpolarizing voltage steps from a holding potential of −30 mV (fig. 7A). IQ slowly activates (τ = 241 ± 7.5 msec at −100 mV) at potentials negative to −60 mV, is noninactivating and is sensitive to 5 mM cesium (fig. 7B). Linopirdine, at 30 μM, had no effect on IQ (fig. 7C) and, at concentrations as high as 300 μM, only slightly reduced IQ (by 26%). Thus, the inwardly rectifying current examined in this study was even less sensitive to linopirdine than were the weakly inhibited IA, IK and IsAHP.

A, Representative traces of the voltage-dependent activation of IQ in rat hippocampal CA1 neurons upon incremental 10 mV hyperpolarization steps to −100 mV from a holding potential of −30 mV. B and C, Effect of cesium (5 mM) or linopirdine (30 μM) on IQ and IL in neurons stepped to −100 from −30 mV. D, Concentration-dependent effect of linopirdine on IL from −40 to −100 mV. ▪ Baseline, ⋄ 10 μM, ▴ 30 μM and ○ 100 μM linopirdine. For the purpose of clarity, standard error bars were not included on the 10 and 30 μM linopirdine lines. Recordings were made in the presence of 0.1 μM TTX using a patch pipette with an internal pH of 7.3.
IL.
IL, a leak potassium current, was measured in CA1 neurons as the instantaneous current component evoked by the voltage protocol utilized to activate IQ (fig. 7C). In agreement with Benson et al. (1988), the instantaneous current was nonrectifying over the range of −40 to −100 mV (fig. 7D). In four cells, linopirdine induced a small, concentration-dependent reduction of IL which was statistically significant at both 30 and 100 μM. Under control conditions, a voltage step from −30 to −100 mV induced an instantaneous current of −892 ± 108 pA which was reduced by 9 ± 5, 17 ± 8 and 28 ± 8% in the presence of 10, 30 and 100 μM linopirdine, respectively. These results indicate that, like IA, IK, IsAHP and IQ, IL is weakly inhibited by linopirdine.
Discussion
Our objective was to determine the selectivity of linopirdine for IM by measuring its effects on other voltage-dependent and calcium-activated K+currents. The order of sensitivity to linopirdine among the seven currents recorded from hippocampal CA1 neurons was IM > ImAHP ≫ IK = IsAHP > IL = IA ≫ IQ. Thus, linopirdine showed approximately seven times more selectivity for IM than the next most sensitive current, ImAHP, and a more than 50-fold selectivity for IM over the other five measured currents. In addition, IM and ImAHP were the only K+currents that could be completely blocked by linopirdine.
The inhibition of IM by linopirdine observed in our study confirms previous reports on its effects in rat hippocampal CA1 neurons (Aiken et al., 1995) and rat superior cervical ganglia (Lamas et al., 1997; Costa and Brown, 1997). The IC50 of 2.4 μM determined in this study agrees well with reported values in the range of 3.4 to 8.5 μM. In addition, in all four studies, linopirdine, in a concentration range of 30 to 100 μM, inhibited IM by 100%. This, together with the lack of effect of internal GTPγS, GDPβS or BAPTA on IM inhibition (Costa and Brown, 1997) and the block of M channels in outside-out membrane patches (Lamaset al., 1997), strongly indicate that linopirdine is a direct M channel blocker as opposed to working through a G-protein- or calcium-coupled second messenger system. The ability of linopirdine to block M channels is, as previously discussed (Aiken et al., 1995), likely to account for its voltage-dependent depolarization of resting membrane potential and reduction of spike frequency adaptation.
Given the putative role of IC in mediating spike repolarization (Storm, 1990), the block of ImAHP(IC) observed in the present study by linopirdine may account for its effects on action potential duration in hippocampal CA1 neurons. In studies performed at room temperature, a single concentration of linopirdine (10 μM) had no effect on action potential duration (Aiken et al., 1995). Under these conditions, action potential duration was already prolonged relative to physiologic temperature and, based on the IC50 of 16.3 μM determined in this study, IC inhibition was likely to be less than 50%. However, in earlier studies performed at 37°C using a concentration range of 5 to 100 μM (Lampe and Brown, 1991), linopirdine exerted a concentration-dependent prolongation of action potential duration, with small effects (20–30% increase) noted at 10 μM and a more than 100% increase seen at 30 μM. In addition, the weak inhibition of IA and IK observed in this study (and by Lamas et al., 1997 in rat superior cervical ganglia) may also contribute to the prolongation of action potential duration exerted by ≥ 30 μM linopirdine.
In current clamp studies using sharp microelectrodes, we observed that 10 μM linopirdine had no significant effect on: 1) normal resting membrane potential, 2) the voltage sag during a prolonged hyperpolarizing pulse from resting potential (Lampe and Brown, 1991) and 3) the slow afterhyperpolarization after a train of action potentials evoked by a prolonged depolarizing pulse (Aiken et al., 1995). Because these potentials can, at least in part, be accounted for by IL, IQ and the IsAHP, respectively, our findings of a weak or absent effect of 10 μM linopirdine on these currents are in agreement with the current clamp results. At ≥30 μM, we have seen a depolarization of normal resting potential induced by linopirdine (B.J. Lampe, P.A. Murphy and B.S. Brown, unpublished observation) which may be due to its small but significant inhibition of IL.
Using cultured rat sympathetic ganglia, Lamas et al.(1997) also studied the effects of linopirdine on a variety of ionic currents. In general, there is good agreement between their results and those of our study in that linopirdine potently inhibited IM, weakly inhibited IA and IK and, at 10 μM, had no significant effect on IAHP or IQ. One notable difference between the two studies, however, is the effect of linopirdine on IC. In sympathetic ganglia, 10 μM linopirdine had no effect on IC whereas in hippocampal CA1 neurons, ImAHP/IC was inhibited at an IC50 of 16.3 μM and was one of two currents completely suppressed by linopirdine. The difference in the pharmacology of IC between ganglia and CA1 neurons may represent a difference in the expression of BK channel subtypes, because the activation of BK channels is believed to correspond to the ICmacroscopic current (Sah, 1996). A similar explanation has been proposed to account for the well known difference in the pharmacology of the small-conductance, calcium-activated K+ current in sympathetic ganglia and hippocampus (Lancaster and Adams, 1986; Storm, 1989; Sah, 1996) where the ganglionic current is apamin-sensitive but the hippocampal current is apamin-insensitive. This pharmacological difference may be related to the expression of different SK channel subtypes in the two tissues (Kohler et al., 1996).Similarly, because there are at least two major subtypes of BK channels (for review, see Gribkoff et al., 1997), a difference in BK expression between sympathetic ganglia and hippocampal CA1 neurons could explain the apparent difference in sensitivity of IC to linopirdine in the two cell types.
Previous studies into the mechanism whereby linopirdine enhances neurotransmitter release indicated an inhibition of M-current as the most likely site of action (Aiken et al., 1995, 1996). Our results support this conclusion and, in addition, suggest that the inhibition of IC may also play an important role in the action of linopirdine because there was only a 7-fold separation between the IC50 values for the two currents. Accordingly, both an inhibition of IC/BK channels and the resultant action potential broadening have been associated with enhancement of transmitter release (Robitaille et al., 1993;Jackson et al., 1991). Furthermore, although the mechanism of M-current block has been studied in some detail, with results indicating a direct channel interaction (Lamas et al., 1997;Costa and Brown, 1998), similar studies have not been performed with BK channels. However, because most, if not all, known activators and blockers of BK channels are direct channel blockers (Gribkoff et al., 1997), the same may also be true of linopirdine.
We have shown that linopirdine is a selective blocker of M-current at concentrations below 10 μM in hippocampal CA1neurons. At higher concentrations, it will first block IC, then IK, IsAHP, IL, IA and IQ. This progression of ion channel effects, as well as its interaction with peripheral ligand-gated channels (Lamas et al., 1997), must be considered when interpreting the functional and toxicological properties of linopirdine.
Footnotes
-
Send reprint requests to: Dr. Barry S. Brown, DuPont Pharmaceuticals Research Laboratories, P.O. Box 80400, Wilmington, DE 19880-0400.
- Abbreviations:
- IM
- M-current
- IA
- transient K+-current
- IK
- delayed rectifier K+ current
- ImAHP
- medium calcium-activated K+ current
- IC
- TEA-sensitive, large conductance calcium-activated K+ current
- IsAHP
- slow calcium-activated K+ current
- IL
- leak potassium current
- IQ
- slow inward rectifier Na+/K+ current
- ID
- slowly inactivating, highly 4-aminopyridine-sensitive, delay K+ current
- IK(ATP)
- ATP-sensitive K+ current
- ACh
- acetylcholine
- TEA
- tetraethylammonium
- 4-AP
- 4-aminopyridine
- TTX
- tetrodotoxin
- Received November 17, 1997.
- Accepted April 10, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}