


Abstract
Chronic benzodiazepine treatment can produce tolerance and changes in γ-aminobutyric acid (GABA)A receptors. To study the effect of treatment on a selected population of receptors, assays were performed using [3H]RY-80, which is selective for GABAA receptors with an α5 subunit. Rats were given a flurazepam treatment known to produce tolerance and down-regulation of benzodiazepine binding, or a diazepam treatment shown to produce tolerance but not receptor down-regulation. Quantitative receptor autoradiography using sagittal brain sections bound with [3H]RY-80 showed binding in areas known to express α5 mRNA. Brains from flurazepam-treated rats showed significantly decreased 1 nM [3H]RY-80 binding in hippocampal formation (e.g., 32% decrease in CA1) and superior colliculus, but not other areas. Using 5 nM [3H]RY-80 showed similar decreases in hippocampus. A corresponding 29% decrease inBmax but no change inKd was found with a filtration binding assay using hippocampal homogenates. Down-regulation of [3H]RY-80 binding had returned to control by 2 days after withdrawing flurazepam treatment. The magnitude of down-regulation of [3H]RY-80 binding suggested that GABAAreceptors with an α5 subunit may play a prominent role in the adaptive responses associated with benzodiazepine tolerance. Chronic diazepam treatment also resulted in decreased [3H]RY-80 binding. However, the regional selectivity was even more pronounced than in flurazepam-treated rats, and only the hippocampal CA1 region showed decreased binding (27%). This localized down-regulation persisted for several days after the end of diazepam treatment. These data indicate that synapses in the hippocampal CA1 region are particularly involved in the adaptive response to chronic benzodiazepine treatments.
Benzodiazepines are psychoactive agents used for sleep disorders, sedation, anxiety, epilepsy, and other conditions. Tolerance to some benzodiazepine actions, particularly the antiepileptic action, can be pronounced. Several experimental correlates of tolerance have been reported (Hutchinson et al., 1996). Although many neurotransmitter systems may be affected during benzodiazepine treatment of intact animals, the γ-aminobutyric acid (GABA)A receptor has been the focus of most studies of tolerance.
The GABAA receptor is a ligand-gated Cl− channel with several modulatory sites, including the benzodiazepine site (Macdonald and Olsen, 1994). The receptor is a pentamer (Nayeem et al., 1994), and has a very complex stoichiometry with evidence of mammalian genes for at least 6α-, 3β-, and 3γ-subunits, as well as γ- and ε-subunits (Lüddens et al., 1995). The subunit composition of a GABAA receptor determines its benzodiazepine pharmacology. Only receptors incorporating a γ-subunit form benzodiazepine binding sites (Lüddens et al., 1995). GABAA receptors with an α4 or α6 subunit do not recognize benzodiazepines such as diazepam, but bind agents such as Ro15-4513 (“diazepam-insensitive” binding). Diazepam-sensitive receptors can be categorized based on their affinity for zolpidem: those with an α1 subunit have high affinity for zolpidem, those with an α2 or α3 subunit have a much lower affinity, and receptors with an α5 subunit are essentially insensitive to zolpidem (Pritchett and Seeburg, 1990; Hadingham et al., 1993; Lüddens et al., 1995). Benzodiazepine agonists act as positive modulators, increasing the anion channel opening frequency produced by the binding of GABA to its own recognition site (Macdonald and Olsen, 1994).
Decreased responsiveness of the GABAA receptor to benzodiazepines associated with chronic treatment has been demonstrated using such assays as electrophysiological studies in the in vitro hippocampal slice (Xie and Tietz, 1992; Zeng and Tietz, 1999) and GABA-mediated flux of36Cl− into partially purified synaptic elements (“microsacs”) (Yu et al., 1988; Li et al., 1993). These and other studies (Tietz et al., 1989; Roca et al., 1990; Primus et al., 1996) suggest that the functional “coupling” of benzodiazepine and GABA sites is altered so that benzodiazepine agonists are less able to potentiate GABA-gating of Cl− conductance, which would mean tolerance existing at the level of individual receptors. Such a change may reflect altered post-translational modification of the receptor, or may be related to receptor number or subunit assembly.
There is abundant evidence that chronic benzodiazepine exposure affects the dynamics of GABAA receptor expression. Many, although not all, chronic benzodiazepine treatments have been found to reduce the number of benzodiazepine binding sites (Wu et al., 1994a;Hutchinson et al., 1996). Although it is doubtful that down-regulation of benzodiazepine binding sites can fully explain tolerance, it does suggest changes in those processes involved in regulating receptor turnover. In fact, several studies have reported changes in GABAA receptor mRNA levels during chronic benzodiazepine exposure (Heninger et al., 1990; O'Donovan et al., 1992; Tietz et al., 1993; Zhao et al., 1994; Impagnatiello et al., 1996). Various patterns have been noted among brain regions, subunits affected, and according to treatment regimen. Although varied, most results suggest that decreases in GABAA receptor subunit mRNA levels might be a common response to chronic benzodiazepine treatment, but differing subunits may be affected, depending on the treatment regimen, and on the details of drug-receptor interactions among the many subtypes of GABAAreceptors in the various brain regions. Even a treatment that had no effect on [3H]benzodiazepine binding (Ramsey-Williams et al., 1994) can cause some decreases in GABAA receptor subunit mRNAs (Wu et al., 1994b). Such findings suggest changes in GABAA receptor turnover, and possibly in receptor subunit composition (“receptor remodeling”), during chronic benzodiazepine treatment.
Benzodiazepine site ligands selective for GABAAreceptors of particular subunit composition may be useful tools for studying changes in GABAA receptor expression. One such ligand, zolpidem, was used to study benzodiazepine receptor regulation in tolerant rats. [3H]Zolpidem binding decreased after shorter treatments, and in more areas, than the binding of the nonselective ligand, [3H]flunitrazepam (Wu et al., 1994a, 1995). The findings indicated a particular involvement of [3H]zolpidem binding sites in down-regulation, and suggested an increase in zolpidem-insensitive [3H]flunitrazepam binding sites. To further evaluate the effects of benzodiazepine tolerance on binding, [3H]RY-80, a ligand selective for receptors that include an α5 subunit (Skolnick et al., 1997), was used after flurazepam or diazepam treatments that have been used previously to study regulation of benzodiazepine binding (Rosenberg and Chiu, 1981a), tolerance (Rosenberg, 1995), changes in GABAAreceptor function (Zeng et al., 1995), and mRNA levels (Wu et al., 1994b; Zhao et al., 1994).
Materials and Methods
Chronic Benzodiazepine Treatment.
Male Sprague-Dawley rats (Harlan, Indianapolis, IN) were used for these studies. Rats were housed in climate-controlled rooms with a 12-h light/dark cycle, and allowed free access to standard rat food. Rats of varying sizes were used for the different treatments so that they would be of similar size and age at the end of all treatments. For flurazepam treatment, flurazepam was administered in a 0.02% saccharin solution as drinking water, according to the procedure described previously (Rosenberg and Chiu, 1981a,b; Wu et al., 1994a). For 1-week treatment, rats initially weighed 160 to 175 g. The flurazepam concentration was adjusted daily to provide a daily dose of up to 100 mg/kg over 24 h for the first 3 days, and 150 mg/kg for the next 4 days (but subject to a maximum concentration of 1.0, and then 1.5 mg/ml). For the 4-week treatment, rats were initially 75 to 99 g. The drug concentration was adjusted to provide up to 100 mg/kg daily for the first week, and 150 mg/kg/day thereafter. Controls were handled identically, but received drug-free saccharin solution. To be included, rats must have consumed a minimum average dose of 100 mg/kg flurazepam daily. Although these doses would certainly cause severe motor impairment if ingested as a single dose, the brain levels are much lower because the drug is consumed over the 24-h period, and rats metabolize flurazepam and its active metabolites very rapidly, with corresponding plasma half-lives of less than 2 h (Lau et al., 1987). This treatment does not cause overt ataxia or sedation, nor are spontaneous withdrawal signs noted after treatment (Rosenberg and Chiu, 1981a). Drug treatment was stopped by replacing drug solution with saccharin solution for various times (12 h, 2 days, or 1 week) before collecting the brains. Because of the rapid metabolism of flurazepam and its active metabolites in rats (Lau et al., 1987), almost all active drug would be eliminated even after only 12 h of withdrawal. As an additional control, an acute treatment group (250–274 g) was treated with desalkyl-flurazepam, 2.5 mg/kg p.o., 12 h before sacrifice (Xie and Tietz, 1992). (Desalky-flurazepam is a much more potent metabolite of flurazepam, and probably accounts for most of the drug actions during chronic oral flurazepam treatment.) In a preliminary experiment, 4-week flurazepam-treated rats were used with no drug-free interval. A corresponding acute treatment group received desalkyl-flurazepam only 30 min before tissue collection.
For diazepam treatment, the drug was administered by implanting diazepam-filled silastic reservoirs (Gallager et al., 1985). In a previous study, this produced brain diazepam levels of 250 to 275 ng/g, and resulted in anticonvulsant tolerance (Ramsey-Williams et al., 1994). Rats (initial weight 225–249 g for 1-week treatment and 100–124 g for 3-week treatment) were anesthetized with methoxyflurane, and two silastic reservoirs, each containing 90 mg of diazepam, were inserted s.c. Control animals were handled identically, but received empty, sealed silastic tubes. For the 3-week treatment, an additional tube was implanted, while the rat was anesthetized with methoxyflurane, on day 10 of treatment. To stop the treatment, all tubes are removed, during methoxyflurane anesthesia, at various times (12 h, 2 days, or 1 week) before collecting the brains. Because rats metabolize diazepam and desmethyldiazepam very rapidly, with elimination half-lives of about 1 h (Friedman et al., 1986), almost all active drug would be eliminated even after only 12 h of withdrawal.
[3H]Benzodiazepine Binding by Quantitative Receptor Autoradiography.
After decapitation, the brains were quickly removed and immersed in isopentane cooled in an acetone-dry ice bath, and then stored at −70°C in air-tight vials. Parasagittal brain slices, 10 μm in thickness, were prepared at −14°C using a microtome, and thaw-mounted onto slides that had been coated with 0.5% gelatin and 0.05% chrome alum. The slides were then transferred to ice-cold slide boxes and stored at −70°C until the time of the binding assay.
After removing the slide-mounted brain slices from the freezer, they were rapidly dried with a stream of cold air, and then prewashed four times for a total of 30 min at 0°C in 50 mM Tris-citrate, pH 7.8, in 0.2 M NaCl. For most of the [3H]RY-80 binding assays, slides were incubated with 1 nM [3H]RY-80 (55 Ci/mmol; New England Nuclear, Boston, MA) at 0°C for 1 h. The concentration was selected to be near the reported Kd in tissue homogenates (Skolnick et al., 1997). Nonspecific binding to adjacent tissue slices was determined in the presence of 1 μM flumazenil. The incubation was terminated by rinsing twice for 30 s in ice-cold 50 mM Tris-citrate, pH 7.8, in 0.2 M NaCl. The slides were then dipped briefly in cold distilled water and dried with a stream of cool air. Slide-mounted tissue sections were fixed with paraformaldehyde vapor at 80°C, and exposed to tritium-sensitive film for 4 to 5 weeks. In preliminary studies, slices had been incubated 10 min to 2 h, and then scraped and counted by scintillation spectroscopy. The results showed that equilibrium was essentially complete by 30 min, and the binding remained stable for at least 2 h.
An additional preliminary autoradiographic binding study was also done, focusing on the hippocampal formation. These tissues were collected at the end of the 4-week flurazepam treatment with no drug-free interval, and from the corresponding control group and acute treatment group. For this experiment, 5 nM [3H]RY-80, a near-saturating concentration was used. This would better compete with any residual drug remaining from chronic treatment, and help ensure that the observation of reduced 1 nM [3H]RY-80 binding described below was not an artifact of residual drug in the tissue. It would also indicate whether a similar degree of down-regulation was present before a drug-free withdrawal period.
For [3H]flunitrazepam binding, the brain slices were prewashed as described above, and then preincubated in 0.17 M Tris-HCl buffer (pH 7.4) at 0°C for 30 min. Slices were then incubated in Tris-HCl buffer containing 2 nM [3H]flunitrazepam (85 Ci/mmol; New England Nuclear) for 1 h at 0°C. Nonspecific binding was determined in the presence of 2 μM clonazepam. Incubation was terminated by washing the slices twice (30 s each) in ice-cold 0.17 M Tris-HCl buffer, followed by a brief rinse in ice-cold distilled water. Finally, the slides were dried with a stream of cold air, fixed with paraformaldehyde vapor at 80°C, and then exposed to tritium-sensitive film for 2 weeks.
After developing the exposed film, the images were digitized, and ligand binding was quantified with computer-assisted densitometry using the NIH Image software. To quantify optical density over particular brain regions, the optical density of coexposed standards (Amersham, Arlington Heights, IL) were determined, and a standard curve was generated. The standards had been calibrated using rat brain paste standards containing known amounts of [3H]thymidine. The different brain regions were identified according a standard atlas (Paxinos and Watson, 1984). Specific binding was determined by subtracting nonspecific binding from total binding, and by converting optical density measurement to picomoles per milligram of protein.
Homogenate Binding Assay.
Membrane homogenates were prepared as described previously (Wu et al., 1994a). Separate saturation assays were performed using hippocampal tissue from each of five treated and five control rats. After decapitation, tissue was collected and homogenized in 0.32 M sucrose. The homogenates were centrifuged at 1000g for 10 min at 4°C. The supernatant was recentrifuged at 20,000g for 20 min. The resulting pellet was washed two more times with 50 mM Tris-citrate, pH 7.8, by resuspension followed by centrifugation, and then finally resuspended in 0.2 M NaCl containing 50 mM Tris-citrate, pH 7.8.
Binding was performed by incubating hippocampal membrane homogenates (0.1–0.2 mg of protein/ml) with [3H]RY-80 in 50 mM Tris-citrate, pH 7.8, in 0.2 M NaCl for 60 min at 4°C. Hippocampal membranes were incubated, in duplicate, with 0.2 to 8 nM [3H]RY-80. Additional tubes also containing 1000-fold excess flumazenil were used to determine nonspecific binding. The reaction was terminated by addition of 5 ml of ice-cold 50 mM Tris-citrate, pH 7.8, in 0.2 M NaCl, and rapid filtration under controlled suction through glass fiber filters (no. 32; Schleicher & Schuell, Keene, NH). The filters were washed two more times with 5 ml of ice-cold 50 mM Tris-citrate, pH 7.8, in 0.2 M NaCl, and were allowed to equilibrate overnight in CytoScint scintillation fluid (ICN, Costa Mesa, CA) before counting. The binding data were evaluated using GraphPad Prism software (GraphPad Software, San Diego, CA).
Data Analysis.
For the results of [3H]RY-80 binding studied by quantitative autoradiography, data were collected from those brain regions that showed a clear signal compared with film background. For each brain, the value for ligand binding over each area of interest was taken as the average of the values determined from three slices prepared from the same rat. The results were analyzed (SigmaStat software; SPSS Inc., Chicago, IL) by ANOVA, with the data grouped according to two independent variables, treatment group (chronic treatment or control) and brain region. In the case of a significant treatment effect, post hoc analysis by the method of Tukey was used to determine which brain regions had been affected by the treatment. In two analyses of the effect of 4-week flurazepam treatment, the data from treated and control rats were also compared with results from acutely treated rats. For the results of the homogenate binding assay, theKd and Bmaxvalues of treated and control tissue were compared by ttest. In all cases, P < .05 was required for statistical significance.
Results
In the autoradiographic binding assays, nonspecific binding of [3H]RY-80 and of [3H]flunitrazepam was very low, similar to film background. Both [3H]RY-80 and [3H]flunitrazepam showed binding over brain areas, such as the hippocampus, known to express α5 subunits. [3H]RY-80 binding was similar to that reported for another α5 subunit-selective ligand (Sur et al., 1999). There was an obvious regional heterogeneity of binding, and many areas densely labeled by [3H]flunitrazepam but which do not express α5, such as the substantia nigra and cerebellum, had very low binding with the α5-selective ligand (Fig.1).

Representative autoradiographs of 2 nM [3H]flunitrazepam (top) and 1 nM [3H]RY-80 binding (bottom) to parasagittal slices of rat brain.
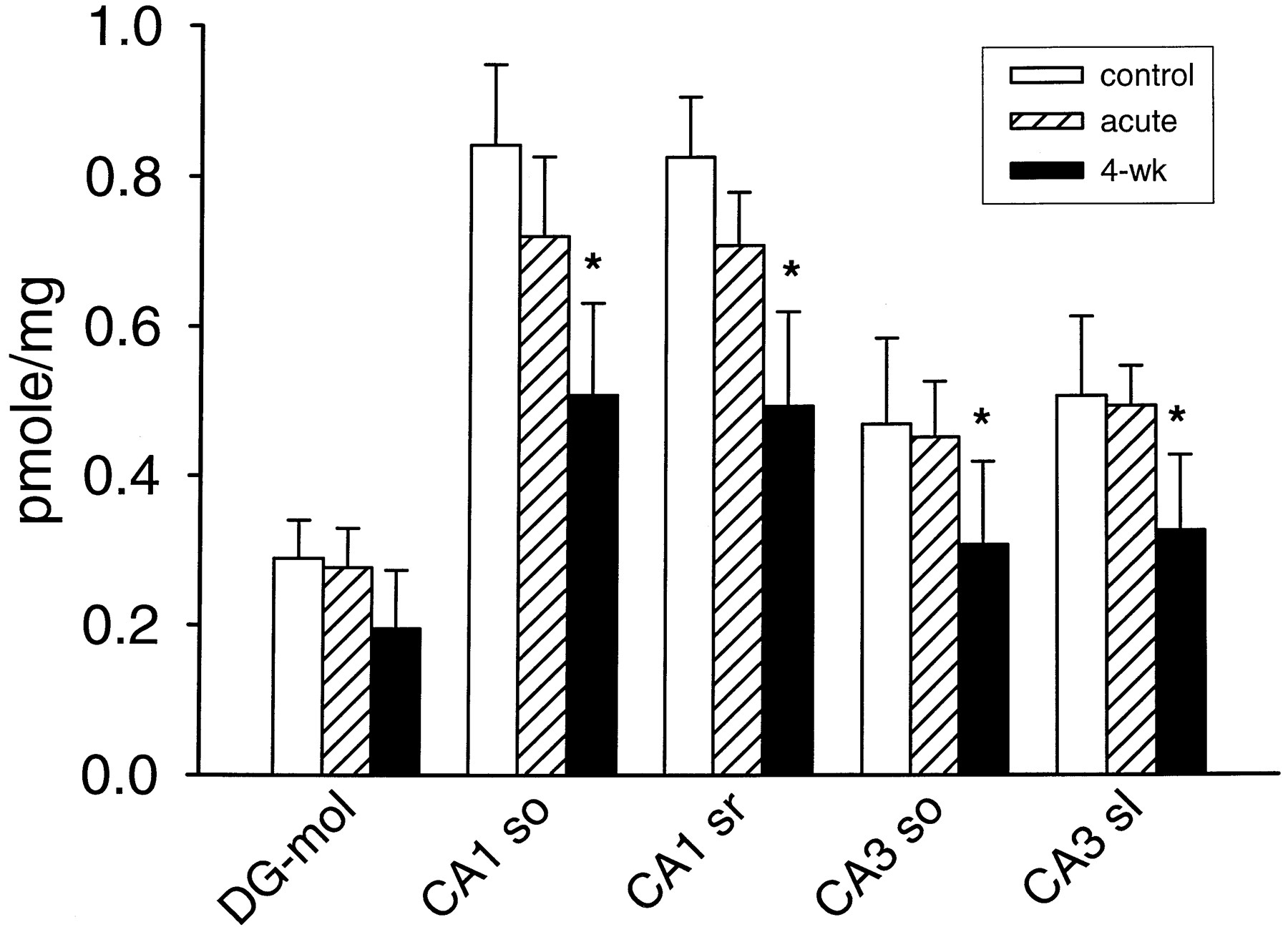
The 4-week flurazepam treatment used in this study is known to produce down-regulation of [3H]flunitrazepam and [3H]zolpidem binding (Wu et al., 1994a, 1995). [3H]RY-80 binding was also reduced by the 4-week flurazepam treatment. In the preliminary study, 5 nM [3H]RY-80 binding to hippocampal formation was studied in 4-week flurazepam-treated, control, and acutely treated animals (n = 5 in each group). The results (Fig.2) showed that the flurazepam treatment had significantly reduced [3H]RY-80 binding. As expected, there was also a significant difference in the density of [3H]RY-80 binding among brain areas. Further evaluation of these results (Tukey test) showed that the 4-week treatment group had significantly less binding compared with both the control group and the acute treatment group. Moreover, the acute treatment did not affect [3H]RY-80 binding; there was no significant difference between the control and the acute desalkyl-flurazepam treatment group. Looking at individual hippocampal formation regions, it was found that the small apparent binding decrease in dentate gyrus was not significant, but all of the other regions examined showed a significant decrease after 4-week flurazepam treatment (Fig. 2).

Specific binding of 5 nM [3H]RY-80 to brain slices from vehicle-treated controls; brain slices from rats treated for 4 weeks with flurazepam and not allowed any drug-free interval (4-wk); and tissue from rats pretreated with 2.5 mg/kg desalkyl-flurazepam, 30 min before collecting brain tissue (acute). Columns indicate mean + S.E. (n = 5 in each group). Abbreviations for brain regions, after regional definition according to Paxinos and Watson (1984): DG-mol, molecular layer of dentate gyrus; CA1 so, hippocampal CA1 region, stratum oriens; CA1 sr, CA1 stratum radiatum; CA3 so, CA3 stratum oriens; CA3 sl, CA3 stratum lucidum. ∗, significantly different from corresponding value in control rat brain.
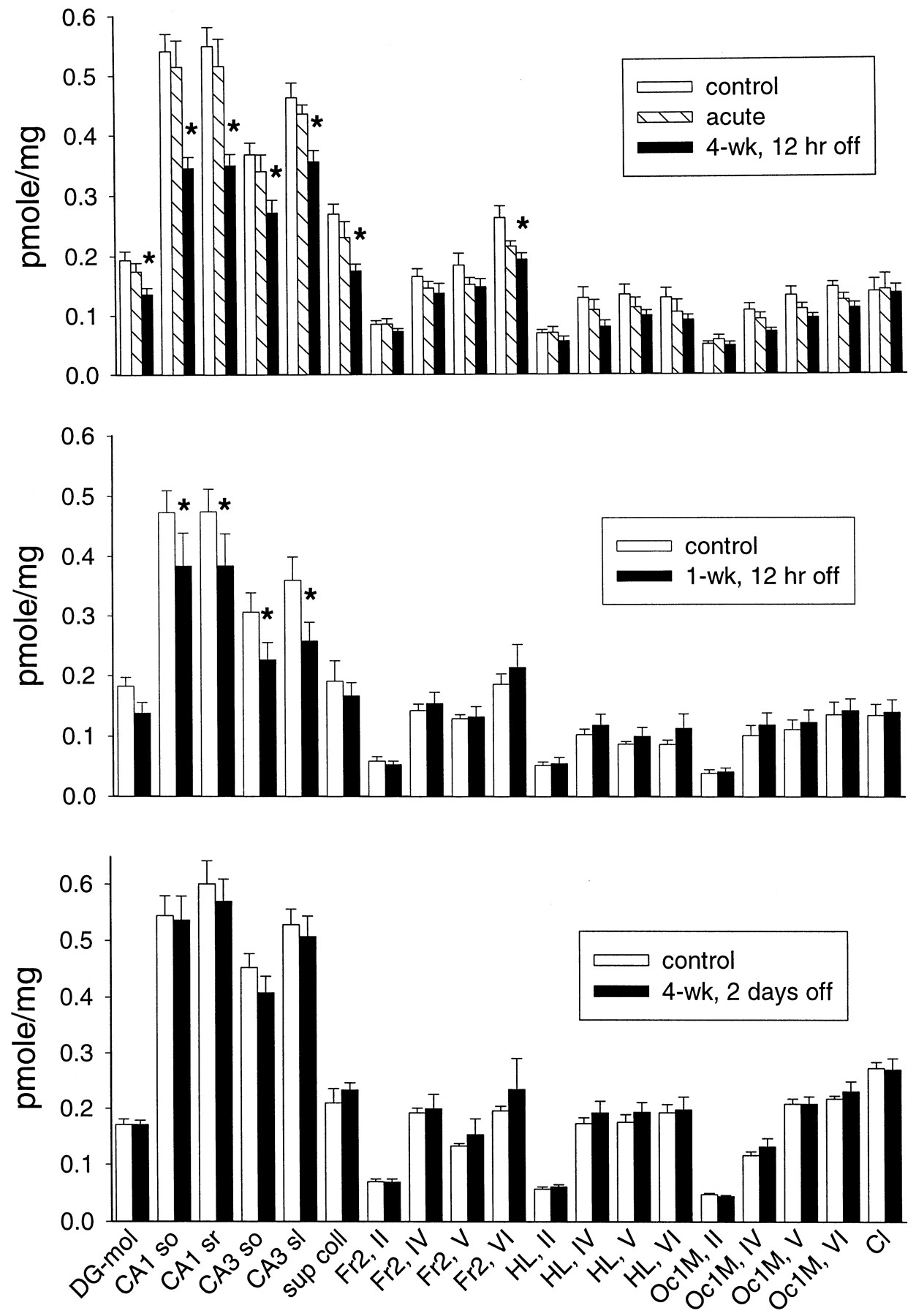
In the other experiments, more brain regions were examined using 1 nM [3H]RY-80 binding. As shown in Fig.3, there was again a decrease in [3H]RY-80 binding 12 h after flurazepam treatment. After the 4-week flurazepam treatment, [3H]RY-80 binding was significantly different from both control and 12-h pretreated rats. There was also a small effect of the 12-h desalkyl-flurazepam pretreatment. However, further analysis failed to show a significant effect of the 12-h pretreatment in any of the brain regions studied. In contrast, the 4-week flurazepam treatment produced a significant decrease in [3H]RY-80 binding in all hippocampal formation regions examined (both laminae in hippocampal CA1 and CA3 regions and the dentate gyrus), and in superior colliculus. However, the flurazepam treatment did not affect all brain regions that showed [3H]RY-80 binding. There was no treatment effect on [3H]RY-80 binding in the claustrum or, with only one exception, the many regions of the cerebral cortex examined (Fig. 3). This down-regulation of [3H]RY-80 binding reversed fairly rapidly after the end of the 4-week flurazepam treatment, and there was no significant treatment effect in brain tissue taken from animals 2 days after the end of flurazepam treatment (Fig. 3). Similarly, [3H]RY-80 binding a week after the end of flurazepam treatment showed no residual treatment effect (data not shown).

Specific binding of 1 nM [3H]RY-80 to brain slices from rats receiving flurazepam treatment (n = 6 for all groups). Top, results from rats receiving the 4-week flurazepam treatment and withdrawn for 12 h (4-wk, 12 h off), their corresponding controls, and rats pretreated with 2.5 mg/kg desalkyl-flurazepam 12 h before (acute). Center, results from rats treated for only 1 week with flurazepam, and then withdrawn 12 h (1-wk, 12 h off), and their controls. Bottom, results from rats treated for 4 weeks with flurazepam, and then withdrawn for 48 h (4-wk, 2 days off), and their controls. Abbreviations for brain regions, after regional definitions according to Paxinos and Watson (1984) (Roman numerals refer to the laminae of cerebral cortical regions): sup coll, superior colliculus; Fr2, frontal cortex, area 2; HL, hindlimb area of cerebral cortex; Oc1, occipital cortex, area 1; Cl, claustrum. Other abbreviations as in Fig. 2. ∗, significantly different from corresponding value in control rat brain.
A shorter, 1-week flurazepam treatment can produce smaller decreases in [3H]flunitrazepam binding (Tietz et al., 1986), fairly pronounced decreases in [3H]zolpidem binding (Wu et al., 1995), and also produce tolerance (Rosenberg, 1995). This 1-week flurazepam treatment produced a significant decrease of [3H]RY-80 binding, which was localized to the CA1 and CA3 regions of the hippocampus, with no changes noted in other regions (Fig. 3).
[3H]RY-80 binding was also studied in hippocampal homogenates from five 4-week flurazepam-treated rats and five controls (Fig. 4). Flurazepam treatment produced a significant decrease of 29% (t test,P < .01) in the mean Bmax(control = 613 ± 33 fmol/mg, treated = 433 ± 32 fmol/mg of protein). There was no significant effect (ttest, P = .5) on the Kd(control = 2.16 ± 0.08 nM, treated = 2.28 ± 0.14 nM).

A, representative Scatchard analysis of specific [3H]RY-80 binding to well-washed homogenates of hippocampal tissue taken from a control rat (▪) and a rat 12 h after the end of the 4-week flurazepam treatment (▴). B, summary of results of saturation analysis of the binding to hippocampal homogenates. Columns indicate mean + S.E. of five control (▨) and five flurazepam-treated rat brains (▪). There was a significant decrease in Bmax, but no significant change in Kd.
In contrast to the down-regulation of benzodiazepine binding produced by some chronic benzodiazepine treatments, the diazepam treatment used in this study, although capable of producing tolerance (Gallager et al., 1985; Ramsey-Williams et al., 1994; Rosenberg, 1995), did not produce measurable effects in homogenate binding assays using [3H]flunitrazepam (Gallager et al., 1985;Ramsey-Williams et al., 1994) or [3H]zolpidem (Wu et al., 1994a). However, using the α5-selective ligand and the more sensitive autoradiographic binding technique revealed significantly decreased binding 12 h after 3-week diazepam treatment, but not after the shorter 1-week treatment (Fig.5). This down-regulation was highly localized to the hippocampal CA1 region, with no effect in CA3 or any other region studied (Fig. 5). When the withdrawal period was extended to 48 h, there was a smaller decrease, which also appeared localized to the CA1 region (Fig. 6). Although there was no overall significant effect 48 h after stopping the diazepam treatment, specific comparisons (Tukey test) were made for only the stratum oriens and stratum radiatum of CA1. Both showed a significant drug effect, suggesting that there was a localized decrease in binding still present at this time point. Very similar results were found 1 week after the 3-week diazepam treatment (Fig. 6).

Specific binding of benzodiazepines to brain slices from rats receiving diazepam treatment (n = 7 for all groups). Top, specific 1 nM [3H]RY-80 binding to brain sections from rats receiving 1 week of diazepam treatment, and then withdrawn for 12 h (1-wk DZP, 12 h off), and the corresponding controls. Center, specific 1 nM [3H]RY-80 binding to brain sections from rats receiving the 3-week diazepam treatment and withdrawn 12 h (3-wk DZP, 12 h off), and their controls. Bottom, specific 2 nM [3H]flunitrazepam (FNP) binding to brain sections from rats receiving the 3-week diazepam treatment and withdrawn 12 h, and their controls. Abbreviations as in Figs. 2and 3. ∗, significantly different from corresponding value in control rat brain.

Specific binding of 1 nM [3H]RY-80 to brain slices from rats receiving 3-week diazepam treatment (n = 7 for all groups). Top, results from rats receiving the 3-week diazepam treatment and withdrawn for 2 days (3-wk DZP, 2 days off), and their corresponding controls. Bottom, results from rats receiving the 3-week diazepam treatment and withdrawn for 7 days (3-wk DZP, 1 wk off), and their controls. Abbreviations as in Figs. 2 and 3. ∗, significantly different from corresponding value in control rat brain.
[3H]Flunitrazepam binding was also evaluated in tissue from 3-week diazepam-treated rats. In keeping with the lack of change previously reported using tissue homogenates (Gallager et al., 1985; Ramsey-Williams et al., 1994), there was no overall significant effect on [3H]flunitrazepam binding. However, inspection of the results (Fig. 5) suggested a small, localized decrease in CA1. Specific comparison of only the CA1 region indicated a significant decrease (Tukey test) of [3H]flunitrazepam binding in this region.
Discussion
Using benzodiazepine site ligands that have selectivity for GABAA receptors containing particular subunits can provide insights into regulation of particular subpopulations of receptors. During some chronic benzodiazepine treatment regimens, there is a down-regulation of receptors. By using selective ligands, it was shown that GABAA receptors containing α1 subunits are particularly affected (Galpern et al., 1990; Wu et al., 1994a, 1995). The present study used [3H]RY-80, a ligand that is selective for the benzodiazepine recognition site of GABAA receptors containing an α5 subunit (Skolnick et al., 1997). The results showed that this receptor subpopulation also plays a role in receptor down-regulation, and possibly in tolerance. Moreover, the results indicate that neurons in the hippocampal CA1 region play a prominent role in these processes.
The effects of flurazepam treatment on [3H]RY-80 binding can be compared with the changes noted with a nonselective benzodiazepine site ligand, [3H]flunitrazepam. Flurazepam treatment has been shown to be associated with decreased [3H]flunitrazepam and [3H]zolpidem binding of varying degrees, involving large regions of the brain (Rosenberg and Chiu, 1981b; Tietz et al., 1986; Wu et al., 1994a, 1995). [3H]Zolpidem binding (Wu et al., 1994a) had shown larger, more rapid, and more regionally widespread down-regulation than could be detected with a nonselective ligand (Wu et al., 1995). In contrast, [3H]zolpidem binding was not affected by diazepam treatment (Wu et al., 1994a). It was suggested that, in large parts of the brain, GABAA receptors that include an α1 subunit are particularly affected by chronic flurazepam. In particular, the data suggested that there must have been an increase in zolpidem-insensitive receptors in those situations wherein there was a decrease in [3H]zolpidem binding, but not in [3H]flunitrazepam binding (Wu et al., 1994a,1995), which was similar to the conclusion drawn in a previous study (Galpern et al., 1990). One possibility was that an increase in expression of α5 subunits, as was reported for diazepam treatment (Impagnatiello et al., 1996; Longone et al., 1996), might result in zolpidem-insensitive receptors. The results of this study do not support such a possibility.
[3H]RY-80 binding revealed that GABA receptors containing an α5 subunit display a substantial amount of down-regulation. Unlike the results with [3H]zolpidem, which suggested a regionally widespread involvement of α1 subunit-containing receptors, the decrease in [3H]RY-80 binding was limited anatomically. Compared with a previous study (Tietz et al., 1986) in which regional down-regulation of [3H]flunitrazepam binding was studied after the same flurazepam treatment used in the present study, the decrease in [3H]RY-80 binding was seen in fewer areas, primarily in the hippocampus. However, the fractional change of [3H]RY-80 binding was somewhat greater in the present study. For example, in CA1, there was a 32% decrease in [3H]RY-80 binding compared with a 15% decrease in [3H]flunitrazepam binding after 4-week flurazepam treatment. Although these data resulting from only a single concentration of each ligand cannot be strictly compared, it is suggested that down-regulation of benzodiazepine recognition sites on α5 subunit-containing GABAA receptors plays a disproportionate role. Saturation assay using tissue homogenates allows a more quantitative evaluation. In the binding assay using homogenates of hippocampal tissue, there was a similar decrease of 29% (Fig. 4) in the Bmax of specific [3H]RY-80 binding. This may be compared with the 14 to 18% decrease in hippocampal [3H]flunitrazepamBmax previously found following 4-week flurazepam treatment (Rosenberg and Chiu, 1981b; Wu et al., 1994a). Comparing the Bmax noted above for controls with that previously found for [3H]flunitrazepam (Rosenberg and Chiu, 1981b;Wu et al., 1994a), and by the direct comparison of [3H]RY-80 and [3H]flunitrazepam binding (Skolnick et al., 1997), it appears that binding to α5 subunit-containing receptors in hippocampus accounts for roughly 18 to 25% of total benzodiazepine binding. Thus, down-regulation of binding to α5 subunit-containing receptors in hippocampus appears to account for over half of the decrease in hippocampal benzodiazepine binding after flurazepam treatment. The 40% decrease in 5 nM [3H]RY-80 binding in the CA1 region (Fig. 2) supports this idea.
Down-regulation of benzodiazepine binding has not been observed after all treatments, even those for which tolerance has been found (Ramsey-Williams et al., 1994). Thus, although down-regulation of benzodiazepine binding has been noted frequently after flurazepam treatment, and may be produced by some diazepam treatments (Zanotti et al., 1996), it had not been observed in rats given the diazepam treatment used in the present study (Gallager et al., 1985; Wu et al., 1994a). However, using the selective ligand, and the more sensitive autoradiographic binding technique, it can now be seen that there is, indeed, down-regulation of benzodiazepine binding after the 3-week diazepam treatment. The regional specificity of this down-regulation was pronounced, involving only the CA1 region, where there was a decrease of 26 and 29% in stratum oriens and stratum radiatum, respectively (Fig. 5). Relatively smaller decreases of [3H]flunitrazepam binding (16 and 10%) also appeared to be present in these laminae (Fig. 5). Although quantitative comparison is difficult without further experiments, the data do suggest that this highly localized down-regulation is largely a result of decreased binding to receptors that include the α5 subunit.
Decreased benzodiazepine binding, or other GABAAreceptor changes associated with tolerance, might be correlated with changes in α5 protein and mRNA levels. Several studies have addressed this question. In rats given the same 4-week flurazepam treatment as in the present study, levels of α5 mRNA in hippocampal homogenates were found to be reduced after the second week of treatment, but had returned to control values by the conclusion of week 4 (Zhao et al., 1994). A similar transient decrease of α5 mRNA was also reported to occur during a very different chronic flurazepam treatment (O'Donovan et al., 1992). Using in situ hybridization, no change in α5 mRNA was found in hippocampal regions of rats given a 1-week flurazepam treatment (Tietz et al., 1999a). These data may indicate dynamic changes in α5 mRNA turnover, but they do not suggest any obvious correlation between changes in mRNA levels and changes in binding to benzodiazepine sites containing α5 receptors. Additional information using subunit-selective antibodies would be helpful in understanding the regulation of the GABAA receptor in hippocampus of flurazepam-tolerant rat brain. Studies have also looked at diazepam treatment. One study, using a different diazepam treatment than the one used in this study, reported increases in α5 mRNA in some cerebral cortical regions and in hippocampus, an increase in α5 immunoreactivity in cortex, but no change in benzodiazepine binding (Impagnatiello et al., 1996). Another study, using the same 3-week diazepam treatment as the present work, reported small but significant decreases in α5 mRNA in hippocampus and cerebral cortex (Wu et al., 1994b). Because [3H]RY-80 binding was decreased in only the former area, there again does not seem to be any clear link between the mRNA level and the down-regulation of benzodiazepine binding, although changes in turnover of mRNA certainly may be occurring.
The pattern of [3H]RY-80 down-regulation indicated the importance of the hippocampus in the changes in GABAA receptors that occur during chronic benzodiazepine exposure. In particular, using the selective ligand allowed a new recognition that the 3-week diazepam treatment does cause down-regulation, especially of benzodiazepine sites that include an α5 subunit, and this is very specific to the hippocampal CA1 region. Several studies have reported functional changes in the hippocampal CA1 region after chronic flurazepam (Xie and Tietz, 1992; Poisbeau et al., 1997; Tietz et al., 1999b; Zeng and Tietz, 1999), and clonazepam (Davies et al., 1988). The findings reported here indicate that this region may be particularly involved in the production of benzodiazepine tolerance and dependence.
The differing pattern of down-regulation is of interest considering the differing patterns of tolerance that can be produced by various chronic treatment regimens. For example, the 1-week flurazepam and 3-week diazepam treatments have been used to study tolerance to the anticonvulsant effect of several benzodiazepines (Rosenberg, 1995). It was found that the patterns of cross-tolerance to various benzodiazepines differed according to the chronic treatment drug used. Although the down-regulation found in the present study does not correlate with the presence of tolerance (e.g., tolerance to several drugs 48 h after ending the flurazepam treatment), it does show that the pattern of change in GABAA receptors differs with differing chronic treatments, and could thus provide a basis for differing patterns of tolerance.
Acknowledgments
We are indebted to Elizabeth I. Tietz, Ph.D., for valuable discussion, and to Eugene Orlowski for excellent technical assistance.
Footnotes
-
Send reprint requests to: Dr. Howard C. Rosenberg, Department of Pharmacology and Therapeutics, Medical College of Ohio, 3035 Arlington Ave., Toledo, OH 43614-5804. E-mail: hrosenberg{at}mco.edu
-
↵1 This work was supported by research Grant RO1-DA02194 from the National Institute on Drug Abuse.
- Abbreviation:
- GABA
- γ-aminobutyric acid
- Received April 27, 2000.
- Accepted July 27, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}