


Abstract
Two peptide agonists, eight nonpeptide agonists, and five nonpeptide antagonists were evaluated for their capacity to regulate FLAG (DYKDDDDK)-tagged human κ opioid receptors (hKORs) stably expressed in Chinese hamster ovary cells after incubation for 4 h with a ligand at a concentration ∼1000-fold of its EC50 (agonist) or Ki (antagonist) value. Dynorphins A and B decreased the fully glycosylated mature form (55-kDa) of FLAG-hKOR by 70%, whereas nonpeptide full agonists [2-(3,4-dichlorophenyl)-N-methyl-N-[(2R)-2-pyrrolidin-1-ylcyclohexyl-]acetamide (U50,488H), 17-cyclopropylmethyl-3,14-dihydroxy-4,5-epoxy-6-[N-methyl-trans-3-(3-furyl) acrylamido] morphinan hydrochloride (TRK-820), ethylketocyclazocine, bremazocine, asimadoline, and (RS)-[3-[1-[[(3,4-dichlorophenyl)acetyl]-methylamino]-2-(1-pyrrolidinyl)ethyl]phenoxy] acetic acid hydrochloride (ICI 204,448) caused 10–30% decreases. In contrast, pentazocine (partial agonist) and etorphine (full agonist) up-regulated by ∼15 and 25%, respectively. The antagonists naloxone and norbinaltorphimine also significantly increased the 55-kDa receptor, whereas selective μ, δ, and D1 receptor antagonists had no effect. Naloxone up-regulated the receptor concentration- and time-dependently and enhanced the receptor maturation extent, without affecting its turnover. Treatment with brefeldin A (BFA), which disrupts Golgi, resulted in generation of a 51-kDa form that resided intracellularly. Naloxone up-regulated the new species, indicating that its action site is in the endoplasmic reticulum as a pharmacological chaperone. After treatment with BFA, all nonpeptide agonists up-regulated the 51-kDa form, whereas dynorphins A and B did not, indicating that nonpeptide agonists act as pharmacological chaperones, but peptide agonists do not. BFA treatment enhanced down-regulation of the cell surface receptor induced by nonpeptide agonists, but not that by peptide agonists, and unmasked etorphine- and pentazocine-mediated receptor down-regulation. These results demonstrate that ligands have dual effects on receptor levels: enhancement by chaperone-like effects and agonist-promoted down-regulation, and the net effect reflects the algebraic sum of the two.
The κ opioid receptor (KOR) is one of the three major types (μ, δ, and κ) of opioid receptors that mediate physiological and pharmacological effects of opioids in vivo. Stimulation of the KOR generates many effects, such as antinociception (especially for visceral chemical pain), dysphoria, water diuresis, hypothermia, modulation of immune responses, and alleviation of craving for cocaine in addicts (reviewed in Liu-Chen, 2004). The cDNA clones of KORs have been isolated and characterized from several species including human, mouse, rat, guinea pig, zebra fish, frog, and Caenorhabditis elegans. The receptors are coupled preferentially to pertussis toxin-sensitive heterotrimeric Gi/o proteins. After receptor activation, both α and βγ subunits of G proteins recruit downstream signaling effectors to inhibit adenylyl cyclase and voltage-gated Ca2+ channels and to stimulate G protein-activated inwardly rectifying K+ channels, mitogen-activated protein kinase, and phospholipase Cβ (for a review, see Law et al., 2000b). In addition, activation of the KOR regulates Na+,H+-exchanger 3 via interaction with Ezrin-radixin-moesin-binding phosphoprotein-50/Na+,H+-exchanger regulatory factor-1 (EBP50/NHERF-1) (Huang et al., 2004).
Peptides derived from prodynorphin, including dynorphin A and dynorphin B, are endogenous ligands for the KOR. Many nonpeptide agonists and antagonists have been synthesized. U50,488H, an arylacetamide compound, is the prototypic nonpeptide selective κ opioid agonist (von Voigtlander et al., 1983). Several other arylacetamide compounds, including U69,593, ICI 204,448, and asimadoline, were subsequently found to be selective for the KOR (Szmuszkovicz, 1999). In addition, several nonarylacetamide compounds have been reported to be selective KOR agonists, including salvinorin A (Roth et al., 2002) and TRK-820 (Seki et al., 1999). Norbinaltorphimine (Nor-BNI) is the first selective κ antagonist (Portoghese et al., 1987). There are many nonselective opioid compounds, including bremazocine, etorphine, ethylketocyclazocine, pentazocine, naloxone, and naltrexone.
The number and activity of the receptor on the cell surface are important factors in determining its capacity to modulate downstream signaling molecules (Law et al., 2000b). The receptor number on the cell surface can be regulated through the biosynthesis pathway (including transcription, translation, protein folding, and transport) and degradation. Agonist-induced adaptative events of the KOR have been extensively investigated. Briefly, after activation by an agonist, the KOR is phosphorylated by G protein-coupled receptor kinases and then nonvisual arrestin is recruited, which reduces coupling between the receptor and G protein, causing receptor desensitization. Through the clathrin- and dynamin-dependent pathway, phosphorylated KOR is endocytosed (internalization) followed by either dephosphorylation and EBP50/NHERF-1-involved recycling (resensitization) or down-regulation via both lysosome and proteasome systems (for a review, see Liu-Chen, 2004). In the absence of an agonist, there is a low level of constitutive internalization (Li et al., 1999) and presumably down-regulation and resensitization. On the contrary, not much is known about regulation of the KOR along the protein biosynthesis pathway. We recently found that GEC1, a small tubulin-binding protein, bound to the KOR and enhanced cell surface expression of the receptor by facilitating trafficking from endoplasmic reticulum (ER) to Golgi to plasma membranes (Chen et al., 2006).
As the first intracellular compartment responsible for protein synthesis and processing, ER plays very prominent roles in controlling the fate of cellular proteins. It has been widely accepted that ER functions as the quality control system in cells (Ellgaard and Helenius, 2003). In particular, assuming a natively correct conformation is the prerequisite for proteins to avert the ER-associated degradation pathway and to exit this quality control system before reaching their action sites (Kleizen and Braakman, 2004).
A growing body of evidence has demonstrated that small-molecule pharmacological chaperones are able to stabilize native conformation and to promote ER-to-Golgi export and protein maturation. Generally, these small molecules are selective ligands, substrates, or inhibitors of the unstable receptors, enzymes, or transmembrane channels. Thus, the concentrations of the pharmacological chaperones required to facilitate the export of their target proteins from the ER are usually low enough to be therapeutically approachable and to minimize adverse effects (Bernier et al., 2004; Ulloa-Aguirre et al., 2004).
It has been demonstrated that some newly synthesized wild-type and mutant membrane-bound proteins, including cystic fibrosis transmembrane conductance regulator and several G protein-coupled receptors, cannot exit from the ER quality control system (Gelman and Kopito, 2002; Bernier et al., 2004; Ulloa-Aguirre et al., 2004). Thus, the wild-type receptors can be the targets of the pharmacological chaperones that can regulate their cell surface expression. In the present study, we explored how KOR ligands regulated cell surface expression of the receptor and whether they function as pharmacological chaperones. CHO cells stably expressing FLAG-tagged human-κ opioid receptor (FLAG-hKOR) were used as the model system.
Materials and Methods
Materials. Dynorphin A(1-17) and dynorphin B(1-13) were purchased from Phoenix Pharmaceuticals (Belmont, CA). U50,488H, ethylketocyclazocine, bremazocine, etorphine, norbinaltorphimine, pentazocine, and naltrindole were provided by the National Institute on Drug Abuse (Bethesda, MD). ICI 204,448 was purchased from Tocris Cookson Inc. (Ballwin, MO). Asimadoline and TRK-820 were generous gifts from Adolor Corporation (Exton, PA). Radioligands, l-[35S]methionine/cysteine (∼1175 Ci/mmol), were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Naloxone, naloxonazine, R(+)-SCH 23,390, brefeldin A, M1 mouse anti-FLAG monoclonal antibody, rabbit anti-FLAG polyclonal antibody, normal goat serum, CaCl2, EDTA, bovine serum albumin, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). SuperSignal West Pico Chemiluminescent substrate kit, EZ-Link Sulfo-NHS-SS-Biotin, and immobilized streptavidin were obtained from Pierce Chemical (Rockford, IL). Cell media (DMEM/F-12, 1:1 and DMEM without methionine/cysteine), Opti-MEM I reduced serum, and fetal bovine serum (FBS) were acquired from Invitrogen (Carlsbad, CA). Materials for cell counting, including Isoton II diluent, Accuvettes, and Coulter Clenz solution, were purchased from Beckman Coulter (Fullerton, CA). The following reagents were bought from the indicated companies: geneticin (G418) from Cellgro Mediatech (Herndon, VA); horseradish peroxidase-conjugated goat anti-rabbit IgG from New England Biolabs (Beverly, MA); complete EDTA-free protease inhibitor cocktail tablets from Roche Diagnostics (Indianapolis, IN); Immobilon-P PVDF 0.45-μm transfer membrane from Millipore (Bedford, MA); Alexa Fluor 488-conjugated goat anti-mouse IgG from Molecular Probes (Eugene, OR); PANSORBIN cells from Calbiochem (San Diego, CA); and Lab-Tek II Slide Chambers from Lab-Tek (Naperville, IL).
Cell Lines. A clonal CHO cell line stably expressing the FLAG-hKOR was generated previously (Li et al., 2002). Cells were cultured in 10-cm Petri dishes in DMEM/F-12 medium supplemented with 10% FBS and 0.2 mg/ml geneticin in a humidified atmosphere consisting of 5% CO2 and 95% air at 37°C.
SDS-PAGE, Western Blotting, and Ligand-Induced Regulation of FLAG-hKOR. CHO-FLAG-hKOR cells (∼90% confluence) were treated with ligands or vehicle at 37°C for 4 h, harvested with Versene buffer (0.54 mM EDTA, 140 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.46 mM KH2PO4, and 1 mM glucose, pH 7.4), and then cell number was determined by a Z1 cell and particle counter (Beckman Coulter). One million cells were solubilized in 200 μl of 2× Laemmli sample buffer [4% SDS, 100 mM dithiothreitol, 10% glycerol, 62.5 mM Tris-HCl (pH 6.8) and 0.1% bromphenol blue], and 2 × 105 cells (40 μl of sample buffer) per lane were subjected to Tricine-SDS-PAGE on 8% separating gel, along with SeeBlue-prestained protein molecular weight markers (Invitrogen). The separated protein bands were transferred to Immobilon-P PVDF transfer membranes, which were then incubated with blocking solution A [5% nonfat dry milk in TBS-T buffer (25 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween 20, pH 7.6)] for at least 1 h and then overnight with rabbit polyclonal anti-FLAG (F7425) antibody (0.8 mg/ml, 1:5000) in blocking solution A at 4°C on a head-over-head shaker. The PVDF membranes were washed three times with the TBS-T buffer and incubated at room temperature for 2 h with horseradish peroxidase-linked goat anti-rabbit IgG (1:1000) in blocking solution A. After the membrane was washed three times with TBS-T buffer, the protein bands were visualized by applying SuperSignal West Pico Chemiluminescent substrate and then digitalized with the Fuji LAS-1000 Plus Gel Documentation System (Fuji Film, Tokyo, Japan). The densitometric analyses of FLAG-hKOR bands were performed by using ImageGauge 4.1 software (Fuji Film). Quantitative comparison of optical densities of FLAG-hKOR between ligand-treated and vehicle-treated cells was carried out to assess ligand-promoted regulation of FLAG-hKOR. Percent change of FLAG-hKOR = 100 × (optical density in ligand-treated group – optical density in vehicle-treated group)/(optical density in vehicle-treated group).
Metabolic Labeling, Immunoprecipitation, and Autoradiography. Experiments were performed using a modified procedure as described by Petaja-Repo et al. (2002). For conducting [35S]Met/Cys labeling, 1.2 × 106 cells were subcultured into a 60-mm Petri dish, grown in complete medium for 24 h (to reach ∼90% confluence), and then preincubated with 2 ml of Met/Cys-free DMEM depletion medium at 37°C for 1.5 h and pulse-labeled with 150 μCi/ml l-[35S]methionine/cysteine in fresh depletion medium. After a 30-min labeling at 37°C, the pulse phase was terminated by washing the cells one time with the chase medium (complete medium supplemented with 5 mM l-methionine) and then incubating them with the chase medium for specified time periods. If BFA and/or ligands were used, BFA was present from the starting of the depletion phase until the end of chase phase, but ligands were added into the medium only in the chase phase. After the chase phase, cells were detached using Versene buffer, washed one time with phosphate-buffered saline (PBS) (140 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.46 mM KH2PO4, and 1 mM glucose, pH 7.4), pelleted by centrifugation at 2500g at 4°C for 5 min, and stored at –80°C until further studies.
Protein solubilization was accomplished by thawing the cells in 400 μl of TTSEC buffer [2% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA and protease inhibitor cocktail tablet] with shaking for 1 h at 4°C. The supernatant was obtained by centrifugation at 13,500g for 15 min. For a better signal/noise ratio, two-time antibody-PANSORBIN precipitations were performed in tandem to purify the [35S]-labeled FLAG-hKOR. First, 400 μl of supernatant was incubated with 2 μg of polyclonal anti-FLAG antibody at 4°C for 1 h and mixed with 20 μl of PANSORBIN at 4°C for 1 more h. The receptors adsorbed on PANSORBIN cells were washed three times by repeated centrifugation and resuspension in TBS-T buffer and then eluted by incubating the pellets in 20 μl of 2× Laemmli sample buffer at room temperature for 15 min. The 20 μl of supernatant was added into 380 μl of TTSEC buffer for the second round of precipitation experiments. Adsorbed receptors were eluted with 50 μl of 2× Laemmli sample buffer for Tricine-SDS-PAGE on 8% separating gel. The gel was dried using a gel dryer (Bio-Rad, Hercules, CA) and then exposed to prebleached storage phosphor screen for 2 days. The autoradiogram was captured by a Cyclone Storage Phosphor System (PerkinElmer Life and Analytical Sciences). The intensities of radiolabeled receptor bands were quantitated with the OptiQuant software. The background signal of each lane was subtracted before quantitative kinetic data analysis was performed.
Transformation of the immature receptor to the mature receptor and turnover of both receptor forms seem to follow the first-order kinetics (see Fig. 3). All analyses were performed using Prism 3.0 (GraphPad Software, San Diego, CA) to fit the data to the following equations (Tallarida and Murray, 1987): 
 where X is the amount of FLAG-hKOR, A is a constant for each equation, ka is the transformation rate constant of the immature to mature receptor, ke is the turnover rate constant of the mature or immature receptor, and t is the time of chase. Based on the amount of mature receptor versus time and the calculated rate constants, the maturation extent of the FLAG-hKOR, peak time of mature receptor, and half-lives of the mature and immature receptors were computed. The maturation extent is defined as the total amount of the mature FLAG-hKOR transformed from the immature receptor, which is determined by the area under the curve in the plot of the amount of the mature receptor against time (Fig. 3B). The turnover rate constant (ke) means the fraction of the receptor degraded per unit of time. Half-life (t1/2) is the time for the receptor to reduce by 50%, and it is equal to 0.693/ke. Peak time (tmax) is the time point at which the amount of the mature receptor is maximal and is calculated according to the following equation (Tallarida and Murray, 1987):
where X is the amount of FLAG-hKOR, A is a constant for each equation, ka is the transformation rate constant of the immature to mature receptor, ke is the turnover rate constant of the mature or immature receptor, and t is the time of chase. Based on the amount of mature receptor versus time and the calculated rate constants, the maturation extent of the FLAG-hKOR, peak time of mature receptor, and half-lives of the mature and immature receptors were computed. The maturation extent is defined as the total amount of the mature FLAG-hKOR transformed from the immature receptor, which is determined by the area under the curve in the plot of the amount of the mature receptor against time (Fig. 3B). The turnover rate constant (ke) means the fraction of the receptor degraded per unit of time. Half-life (t1/2) is the time for the receptor to reduce by 50%, and it is equal to 0.693/ke. Peak time (tmax) is the time point at which the amount of the mature receptor is maximal and is calculated according to the following equation (Tallarida and Murray, 1987): 
Immunofluorescence Staining of FLAG-hKOR on the Cell Surface. Surface FLAG-tagged hKOR was visualized using an “antibody feeding” method described by Wang et al. (2005). Briefly, CHO-FLAG-hKOR cells were grown on Lab-Tek II Slide Chambers for 24 h to reach ∼90% confluence, and the surface receptors were specifically labeled by incubating cells with monoclonal M1 anti-FLAG antibody (1:1000) in complete medium at 37°C for 30 min. Labeled cells were then fixed with 4% paraformaldehyde in TBS buffer for 15 min at room temperature. After washing 3 times with TBS buffer, cells were blocked with blocking solution B (4% normal goat serum in TBS buffer) for 15 min, and then treated with Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (1:1000) in TBS buffer containing 1% normal goat serum for 45 min at room temperature. Because of the requirement of Ca2+ for binding of M1 anti-FLAG antibody to the FLAG epitope, CaCl2 (final concentration of 1 mM) was added into all solutions in steps from incubation with M1 antibody. Immunostained cells were mounted with Slow Fade mounting medium, and coverslips were sealed with nail polish. An ELIPSE TE300 fluorescence microscope (Nikon, Tokyo, Japan) equipped with a 60× NA 1.4 objective and fluorescein filter sets was used to examine receptor distribution. Cells treated in the absence of M1 anti-FLAG antibody were employed as the control group, which did not show any staining.
Cell Surface Biotinylation and Separation of FLAG-hKOR. Experiments were carried out using a protocol recommended by the manufacturer (Pierce). For biotinylation of cell surface proteins, 10 mM stock solution of Sulfo-NHS-SS-Biotin (cell-impermeable and cleavable) was prepared immediately before use. Cells (∼90% confluence in a 60-mm Petri dish) were washed three times with ice-cold PBS buffer containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/Ca2+/Mg2+ buffer, pH7.4) and incubated with gentle agitation for 30 min at 4°C in PBS/Ca2+/Mg2+ buffer containing 1 mM Sulfo-NHS-SS-Biotin. Excess biotin reagent was quenched by treating the cells with 50 mM Tris-HCl buffer (pH 7.4) for 10 min at 4°C. The biotin-labeled cells were washed twice, detached with Versene buffer, and stored at –80°C until the protein solubilization step.
Cells were solubilized after thawed cells were treated in 1% TTSEC buffer (TTSEC buffer containing 1% Triton X-100) for 1 h at 4°C with head-over-head shaking and then precipitated with 30 μlof immobilized streptavidin-agarose beads. After cells were washed three times with 1% TTSEC buffer, the streptavidin-bound biotinylated proteins were eluted by incubating the beads in 50 μl of 2× Laemmli sample buffer for 1 h at room temperature. Biotinylated proteins were incubated with polyclonal anti-FLAG antibody and PANSORBIN for immunoprecipitation of FLAG-hKOR as described above.
Fluorescence Flow Cytometry and Ligand-Induced Regulation of Cell Surface FLAG-hKOR in the Presence of BFA. A fluorescence flow cytometry assay was conducted according to a modified protocol described by Li et al. (2003). CHO-FLAG-hKOR cells (∼90% confluence) grown on 100-mm Petri dishes were first incubated with 5 μg/ml BFA for 2 h and then treated with ligands or vehicle in the presence of BFA at 37°C for 4 h. After harvesting, 1.5 × 106 cells were used for a further flow cytometric assay without permeabilization of cell membranes. Because calcium ion is necessary for M1 monoclonal anti-FLAG antibody binding with the antigen, all solutions used in the subsequent steps contain 1 mM CaCl2. Cells were washed once with Opti-MEM I and then incubated with M1 antibody (1 μg/ml, 1:2000) in 1 ml of Opti-MEM I for 1 h. After another three washes with PBS buffer, cells were incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG (1 μg/ml, 1:2000) in 1 ml of Opti-MEM I for 1 h, washed three times and then resuspended with 1 ml of PBS buffer. All solutions used were ice-cold and all of the above procedures were performed in a cold room unless specified otherwise. Surface FLAG-hKOR immunofluorescence of 1 × 104 cells was quantitated using a FACScan (BD Biosciences, San Jose, CA), and the mean fluorescence intensity of a single cell was calculated. The mean fluorescence intensity of cells stained only with the second antibody was also measured and subtracted from the mean intensity. Ligand-induced percent change of cell surface FLAG-hKOR = 100 × (fluorescence intensity in ligand group – fluorescence intensity in vehicle group)/(fluorescence intensity in vehicle group).
Data Analysis. All quantitative data are presented as means ± S.E. if they were acquired from at least three independent experiments. Student's t test was used for comparing two sets of independent samples. The difference was defined to be significant if the p value was < 0.05. All statistical analyses were performed using Prism 3.0.
Results
Different Regulatory Effects of KOR Ligands on the Steady-State Level of the Mature Form of FLAG-hKOR. Immunoblotting data showed that FLAG-hKOR stably expressed in CHO cells migrated as two bands of Mr 55,000 and 45,000 (Fig. 1A). We have previously demonstrated that the 45-kDa band represents N-linked glycosylated high-mannose intermediates, which mainly distribute in ER and/or cis-Golgi, whereas the 55-kDa band represents the fully glycosylated mature forms of FLAG-hKORs, most of which locate in plasma membranes (Chen et al., 2006). In addition, our receptor binding results showed that cell surface receptors accounted for ∼85% of the total receptor (Xu et al., 2005). Cell surface receptors are the ones responding to agonists, leading to signal transduction. Therefore, to evaluate how different ligands regulated the FLAG-hKOR, we focused on the 55-kDa mature form. The 55-kDa band seems to be a doublet, which may be due to heterogeneity of glycosylation. The heterogeneity may be attributed to different chain lengths of carbohydrate. In addition, there are two N-linked glycosylation sites (Asn25 and Asn39) in the N-terminal domain and the observed heterogeneity may result from glycosylation at one versus two Asn residues. Because they both represent fully glycosylated receptors, we did not examine their regulation separately.

Differential regulation by prolonged treatment (4 h) with KOR ligands of the steady-state level of the mature form (doublet at ∼55 kDa) of FLAG-hKOR stably expressed in CHO cells. A, immunoblotting pattern of FLAG-hKOR in CHO cells. Cells were solubilized with 2× Laemmli sample buffer, and proteins were separated by SDS-PAGE followed by immunoblotting with polyclonal anti-FLAG antibody. This represents one of the three experiments performed with similar results. B and C, immunoblotting and quantitation of ligand-induced changes in the mature receptor level. Cells were incubated with each drug for 4 h at a concentration ∼1000-fold of its EC50 (agonists) or Ki (antagonists). Thereafter, 2 × 105 cells were solubilized and subjected to SDS-PAGE. FLAG-hKOR was detected with immunoblotting with anti-FLAG antibody and quantitated (mean ± S.E., n = 4) using ImageGauge software. B represents one of the four experiments performed with similar results. Dyn A, dynorphin A; Dyn B, dynorphin B; U50, U50,488H; TRK, TRK-820; Brema, bremazocine; EKC, ethylketocyclazocine; Asim, asimadoline; ICI, ICI 204,488; Etor, etorphine; PTZ, pentazocine; Nal, naloxone; Nor-BNI, norbinaltorphimine; NalAz, naloxonazine; NTI, naltrindole; SCH, R(+)-SCH 23,390.
Fifteen ligands were examined for their abilities to change the expression level of the receptor (Fig. 1). Unless indicated otherwise, the concentration of each drug was ∼1000-fold of its EC50 (agonists) or Ki (antagonists) (Henry et al., 1995; Zhu et al., 1997; Roth et al., 2000; Wang et al., 2005; our unpublished data) and the incubation duration was 4 h (Li et al., 2000; Zhang et al., 2002).
Dynorphin A and dynorphin B, endogenous peptides and full agonists for the KOR, reduced the mature FLAG-hKOR by ∼70%. By comparison, the nonpeptide full agonists U50,488H, TRK-820, bremazocine, ethylketocyclazocine, asimadoline, and ICI 204,448, caused much less reduction, ranging from 10 to 30%. In contrast, the nonpeptide full agonist etorphine and the nonpeptide partial agonist pentazocine up-regulated the mature receptor by ∼25 and ∼15%, respectively.
Four nonpeptide opioid antagonists were also examined. After a 4-h incubation, the nonselective opioid antagonist naloxone and the KOR-selective antagonist Nor-BNI significantly enhanced the mature form by ∼25 and ∼15%, respectively. However, naloxonazine and naltrindole, selective for μ and δ opioid receptors, respectively, did not have significant effects on the receptor level, nor did the selective D1 dopamine antagonist R(+)-SCH 23,390. These results indicate that receptor binding is required for the regulatory effects of ligands.

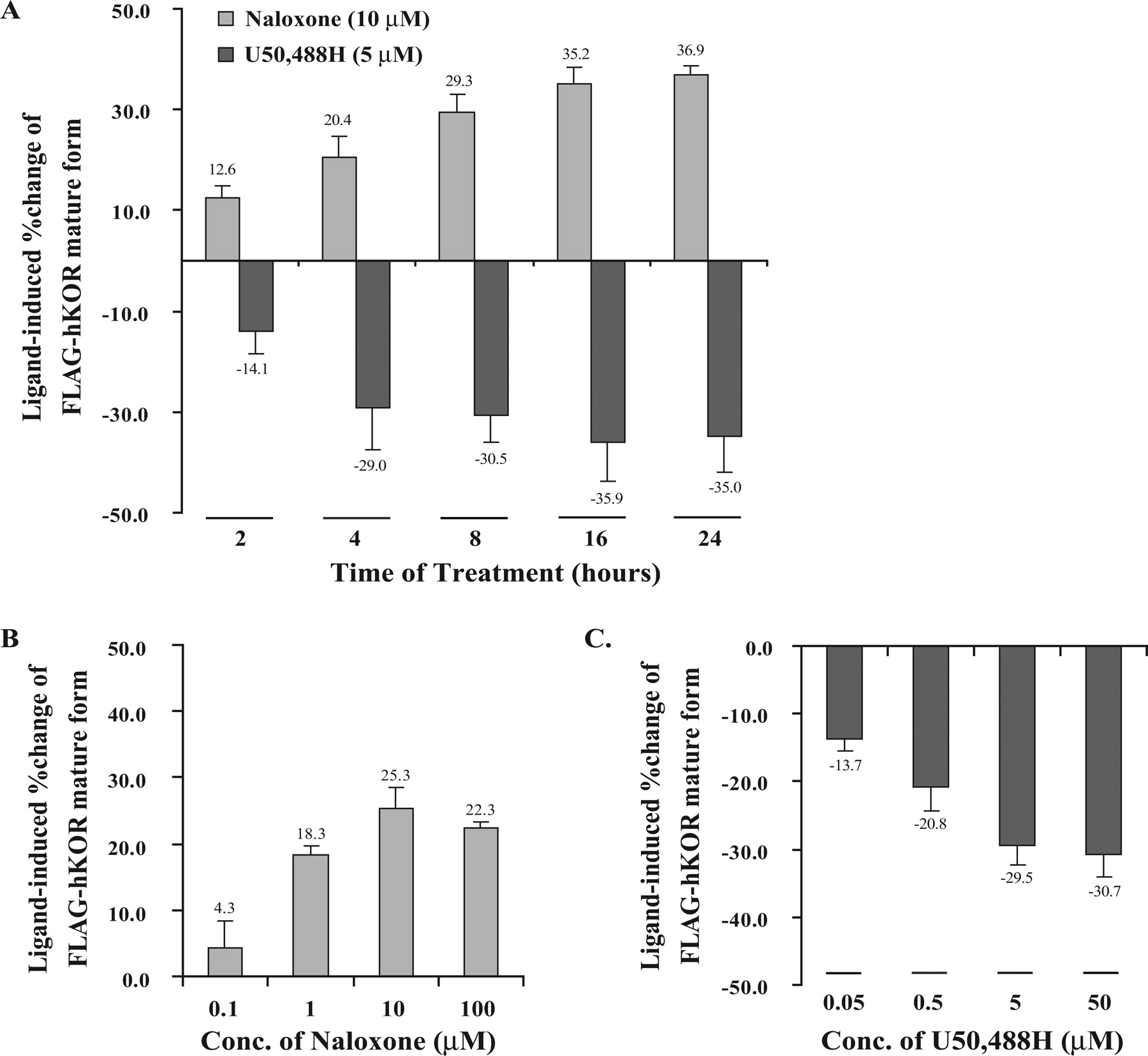
Time and concentration dependence of ligand-mediated regulation of the steady-state level of mature FLAG-hKOR. Cells were treated with (A) naloxone (10 μM) or U50,488H (5 μM) for indicated time periods or (B and C) different concentrations of naloxone or U50,488H for 4 h. FLAG-hKOR was detected by immunoblotting and quantitated (mean ± S.E., n = 3) by densitometry as described under Materials and Methods.
Time and Concentration Dependence of Ligand-Mediated Receptor Regulation. We then examined whether the effects of the agonist U50,488H and the antagonist naloxone on receptor level were dependent on incubation duration and concentration. As shown in Fig. 2, U50,488H (5 μM) induced KOR down-regulation and naloxone (10 μM) promoted up-regulation in time-dependent manners, reaching the respective plateaus after the 16-h treatment. In addition, when cells were treated for 4 h, both naloxone-promoted increases and U50,488H-induced decreases were concentration-dependent, attaining maximal effects at 10 and 5 μM, respectively. Thus, these results demonstrate that their effects on FLAG-hKOR levels are time- and concentration-dependent, indicating that this process observes the law of mass action and requires receptor occupancy.
Naloxone-Enhanced Conversion of 45- to 55-kDa Form of hKOR. Naloxone treatment does not cause activation-dependent receptor internalization and down-regulation (Li et al., 1999, 2000). Therefore, it is a good tool to study how a ligand up-regulates cell surface receptor. The pulse-chase technique was used to determine the rates and extents of maturation and turnover of the newly synthesized receptors. As shown in Fig. 3A, after metabolic labeling with [35S]Met/Cys (pulse) for 30 min, most of the labeled FLAG-hKOR existed as the intracellular immature form (45 kDa). After removal of [35S]Met/Cys and incubation with chase medium, the 45-kDa protein band gradually decreased and, concomitantly, the fully glycosylated 55-kDa protein band gradually increased, reaching the highest level around 2 h. Incubation with 10 μM naloxone expedited turnover of the immature receptors and significantly enhanced maturation extent of FLAG-hKOR (Fig. 3B; Table 1). In addition, this ligand treatment did not alter the turnover rate of the mature receptors. Therefore, naloxone-induced enhanced mature receptor expression is due to increased transformation from immature species to mature ones but not increased stability of the mature receptor.
Kinetic parameters of both newly generated receptor species (55-kDa mature form and 45-kDa immature form) in control and naloxone-treated cells
All parameters were determined from the data in Fig. 3B as described under Materials and Methods.
Naloxone Acted Intracellularly in the ER as a Pharmacological Chaperone. We next examined whether naloxone acted intracellularly along the biosynthesis pathway (from ER to Golgi apparatus to plasma membrane). After a 6-h treatment with BFA, which disrupts the Golgi apparatus, there was a new receptor form with a Mr of 51,000 that migrated between the immature and the mature receptors (Fig. 4A). The incubation time was identical to that used by Petaja-Repo et al. (2002). Pulse-chase experiments showed that this new form was generated from the 45-kDa species (Fig. 4B). Incubation of cells with naloxone enhanced the 51-kDa form in BFA-treated cells and the 55-kDa form in control cells (Fig. 4C).
We next investigated whether the 51-kDa form was located intracellularly or on cell surface by immunofluorescence staining, flow cytometry, and cell surface biotinylation. Immunofluorescence staining with anti-FLAG antibody without permeabilization of the plasma membranes showed that incubation with BFA resulted in an apparent reduction of cell surface FLAG-hKOR (Fig. 5A). Flow cytometric assay revealed that, after BFA treatment, there was a ∼40% decrease in cell surface receptors (Fig. 5B). For cell surface receptor biotinylation (Fig. 5C), cells were incubated with [35S]Met/Cys for 30 min to label newly synthesized proteins followed by the 4-h chase in the absence or presence of naloxone and/or BFA; cell surface proteins were labeled with biotin and then precipitated with immobilized streptavidin agarose beads followed by anti-FLAG antibody and PANSORBIN to immunoprecipitate biotinylated FLAG-hKOR. Although in control cells a broad radiolabeled 55-kDa band was detected, in BFA-treated cells no radioactive protein band was observed. Taken together, these results demonstrate that the 51-kDa species is located intracellularly. Thus, by disrupting the Golgi apparatus, BFA interrupts maturation and membrane targeting of the receptor. In addition, in control cells, naloxone treatment up-regulated the mature form (55 kDa); however, in BFA-treated cells, naloxone did not promote trafficking of the 51-kDa species to the cell surface or induce generation of the 55-kDa form (Fig. 5C).

Effect of naloxone (10 μM) on the kinetics of newly synthesized FLAG-hKOR: pulse-chase study. A, time courses of generation and decline of the immature and mature receptors in the absence or presence of 10 μM naloxone. After a 90-min incubation in Met/Cys-free DMEM depletion medium, cells were pulse-labeled with 150 μCi/ml [35S]Met/Cys at 37°C for 30 min and then incubated with complete medium containing 5 mM methionine (chase) for specified time periods. Cells were treated with naloxone (Nal) or vehicle (Veh) during the chase phase. After chase, cells were harvested and solubilized with TTSEC buffer, and proteins were immunoprecipitated with anti-FLAG antibodies and PANSORBIN twice. Immunoprecipitated materials were resolved with SDS-PAGE followed by gel drying and autoradiography. The figure represents one of the four experiments performed with similar results. B, quantitative autoradiography results were acquired by densitometric analysis using the OptiQuant software (mean ± S.E., n = 4).

Naloxone enhanced the 51-kDa form in cells treated with BFA. A, immunoblotting pattern of FLAG-hKOR in control and BFA-treated CHO cells. After incubation with vehicle (Veh) or 5 μg/ml BFA for 6 h, cells were solubilized for SDS-PAGE and immunoblotting as described under Materials and Methods. B, BFA treatment resulted in production of a new receptor intermediate (Mr 51,000) from the immature form (Mr 45,000) as demonstrated with the pulse-chase study. All experiments were conducted using the same protocol as described in the legend to Fig. 3 except that cells were treated with BFA during the 1.5-h depletion phase, the 0.5-h pulse phase, and the subsequent chase phase for the indicated time periods. C, naloxone (Nal) promoted production of the mature (55-kDa form) FLAG-hKOR in control cells and the 51-kDa form in BFA-treated cells. The pulse-chase assay was performed as described in the legend to Fig. 3. In BFA-treated cells, BFA was added into the medium during the entire depletion-pulse-chase phases lasting for 6 h. Naloxone treatment was only carried out during the 4-h chase phase. Each figure represents one of three experiments performed with similar results.
The results in Figs. 4 and 5 indicate that naloxone acts intracellularly, most likely at the ER, to enhance the level of the 55-kDa form in control cells and the 51-kDa form in BFA-treated cells. Thus, naloxone acts as a pharmacological chaperone in the ER to stabilize the receptor and thus to facilitate maturation of the receptor.
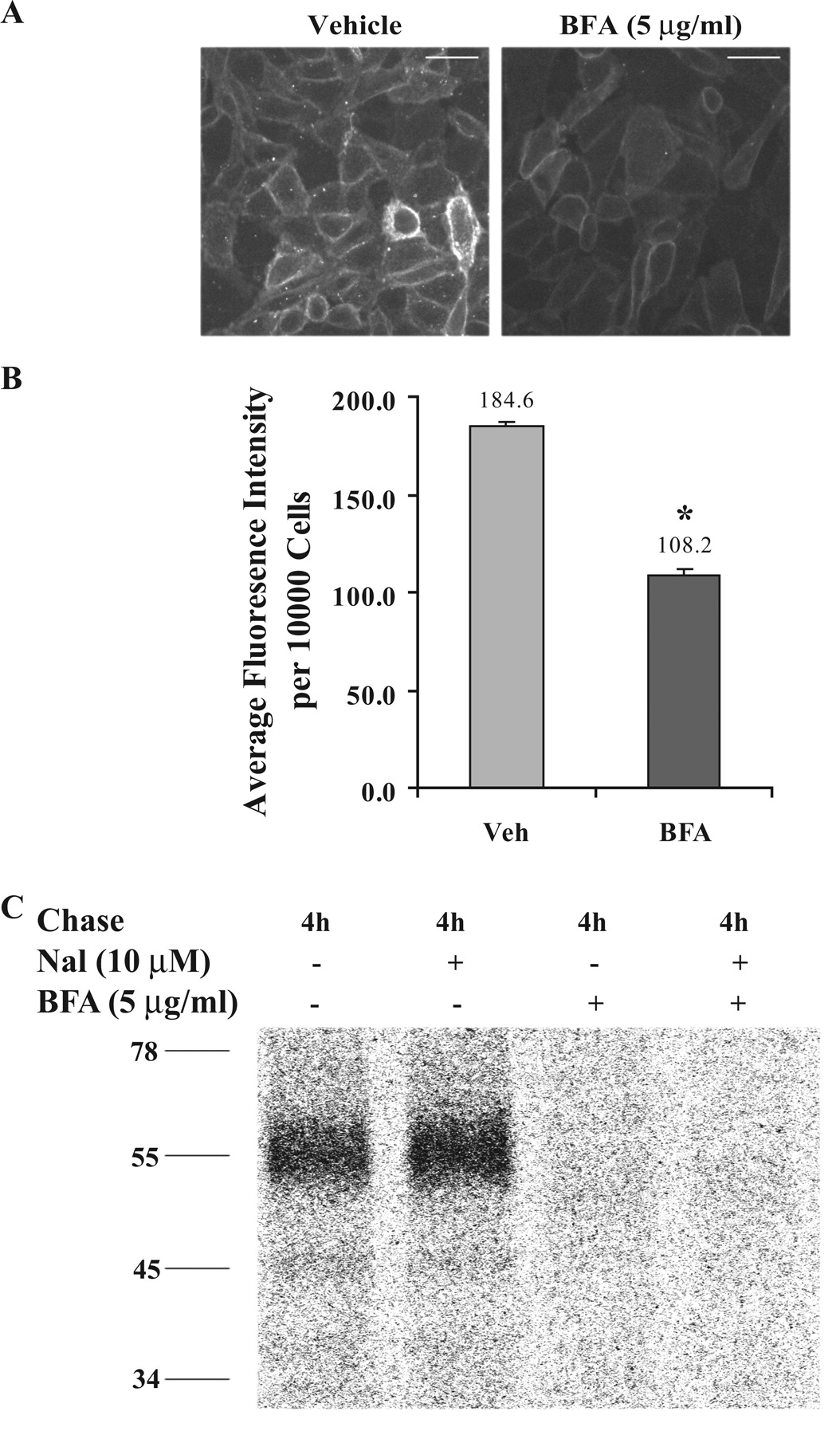
BFA-induced 51-kDa FLAG-hKOR is located intracellularly. A, BFA treatment reduced cell surface (mature) FLAG-hKOR immunofluorescence. After treatment with vehicle or BFA (5 μg/ml) for 6 h, cells were incubated with monoclonal M1 anti-FLAG antibody without membrane permeabilization and then fixed by 4% paraformaldehyde. The surface receptors were further stained with Alexa Fluor 488-conjugated goat anti-mouse IgG antibody and visualized using a fluorescence microscope. This figure represents one of four experiments performed with similar results. Scale bar, 30 μm. B, BFA treatment significantly decreased cell surface FLAG-hKOR. Cells were incubated with BFA or vehicle (Veh) for 6 h, and cell surface receptors were labeled by monoclonal M1 anti-FLAG antibody and then Alexa Fluor 488-conjugated goat anti-mouse IgG antibody. Immunofluorescence intensity was determined (mean ± S.E., n = 4) using a fluorescence activated cell sorter. *, P < 0.05 compared with the control using a two-tailed Student's t test. C, the BFA-induced 51-kDa form was located intracellularly. After pulse-chase labeling as described in the legend to Fig. 4C, cell surface receptors were biotinylated with water-soluble EZ-link Sulfo-NHS-SS-Biotin. Cells were solubilized and incubated with immobilized streptavidin to precipitate biotinylated proteins, which were then incubated with polyclonal anti-FLAG antibody and PANSORBIN for immunoprecipitation of biotinylated receptors. Immunoprecipitated materials were resolved with SDS-polyacrylamide gel electrophoresis, and radioactivity was detected with autoradiography. This figure represents one of three experiments performed with similar results.
Pharmacological Chaperone Effects of the Agonists. BFA has been widely used as a tool to examine whether a ligand has a chaperone-like effect on wild-type and mutant receptors (Petaja-Repo et al., 2002; Chaipatikul et al., 2003; Van Craenenbroeck et al., 2005). We then used BFA-treated cells to assess whether the agonists tested in Fig. 1 had chaperone-like effects similar to those of naloxone. Figure 6 shows that both hydrophilic peptide agonists (Dyn A and Dyn B) did not up-regulate the 51-kDa species but that all hydrophobic nonpeptide agonists did enhance its level. Therefore, nonpeptide, but not peptide, ligands behave as pharmacological chaperones to facilitate anterograde trafficking of the receptor along the biosynthesis pathway, indicating that membrane permeability is required for the chaperone roles.
Ligand-Induced Regulation of Surface FLAG-hKOR after Treatment with BFA. Next we examined how the ligands regulated the cell surface receptor if membrane targeting of proteins was blocked by BFA. Because both the 55- and 51-kDa bands are broad and diffuse, there was overlap of the two bands, making it difficult to quantify the 55-kDa band. Fluorescence flow cytometry, instead of immunoblotting, was used to quantify cell surface receptors for evaluating ligand-mediated receptor regulation. It is reasonable to use cell surface receptor to represent the mature form because most of fully glycosylated proteins are located on the cell surface. With BFA treatment, Dyn A and Dyn B (Fig. 7) still caused the highest degrees of down-regulation; the down-regulation extent (65%) was comparable with that (70%) in the absence of BFA determined by immunoblotting (see Fig. 1). On the other hand, in the presence of BFA, all nonpeptide full agonists mediated higher levels of down-regulation than they did in the absence of BFA (see Fig. 1). In particular, etorphine and pentazocine, which up-regulated the receptor in the absence of BFA (see Fig. 1), down-regulated cell surface receptors in the presence of BFA (Fig. 7). In addition, with BFA treatment, naloxone had no influence on the expression of FLAG-hKOR on plasma membranes, although it up-regulated the receptor without BFA. These results demonstrate that the effects of ligands on cell surface expression are a combination of activation-dependent down-regulation and pharmacological chaperone effects.

Nonpeptide KOR ligands enhanced the BFA-induced intracellular 51-kDa form of FLAG-hKOR, but peptide ligands did not. Pulse-chase experiments were conducted as described in the legend to Fig. 4C. Cells were only treated with each ligand during the 4-h chase phase at the same concentration as indicated in Fig. 1. This figure represents one of three experiments performed with similar results.

Effects of BFA on the ligand-induced regulation of cell surface FLAG-hKOR. CHO-FLAG-hKOR cells were preincubated with BFA for 2 h and vehicle or ligands were added for 4 h in the presence of BFA. After these treatments, cell surface FLAG-hKOR was labeled and quantitated (mean ± S.E., n = 4) as described in the legend to Fig. 5B.
Discussion
To the best of our knowledge, this is the first report showing that membrane-permeable nonpeptide full agonists caused less down-regulation of cell surface receptors than membrane-impermeable peptide full agonists due to their pharmacological chaperone effects counteracting activation-dependent degradation.
Cell-Permeable Ligands Act in the ER to Produce Chaperone Effects. We have shown previously that dynorphin A, U50,488H, U69,593, ethylketocyclazocine, and tifluadom promote similar levels of hKOR internalization (Li et al., 1999, 2003). Therefore, the observed differential down-regulation of cell surface hKOR by peptide and nonpeptide agonists is probably due to differences in trafficking in the biosynthesis pathway and in receptor degradation after internalization.
BFA causes rapid disintegration of the Golgi apparatus and blockade of ER-to-Golgi vesicular transport and retrograde transport of Golgi resident proteins, such as glycosyl-transferases, back into the ER (Rios et al., 1994). For G protein-coupled receptors (GPCRs) of which the sorting of internalized receptor is Golgi-dependent (e.g., Tawfeek and Abou-Samra, 2004), it is conceivable that BFA may have a negative impact on postendocytic recycling of the receptor. However, our results demonstrated that BFA did not affect the extents of Dyn A- and Dyn B-induced reduction of cell surface hKOR (Figs. 1 and 7). In addition, Law et al. (2000a) reported that inhibition of receptor recycling with monensin had no effect on reduction of the cell surface μ opioid receptor after prolonged agonist treatment (≥4 h). Thus, under our experimental conditions, the effects of BFA are primarily attributed to blockade of the ER to Golgi transport.
After BFA treatment, a new 51-kDa form of FLAG-hKOR was generated, which is localized intracellularly and represents the receptor formed in the ER by retrogradely transported Golgi enzymes. We found that nonpeptide KOR ligands, regardless of efficacy, up-regulated the 51-kDa form, whereas peptide ligands did not, indicating that nonpeptide, but not peptide, ligands act as pharmacological chaperones. Exit from the ER to the Golgi apparatus seems to be a rate-limiting step for many GPCRs. The membrane-permeable KOR ligands most likely act in the ER to bind to the newly synthesized receptors in various states of the folding pathway. The interaction of nonpeptide ligands with amino acid side chains within the transmembrane hydrophobic core most likely promotes energy-favorable conformational states of the receptor, leading to increased exit of the receptor from the ER to the Golgi (Bernier et al., 2004; Ulloa-Aguirre et al., 2004).
Mechanisms of Differential Effects of Ligands. With BFA, all nonpeptide agonists decreased cell surface receptors to greater degrees than without BFA, whereas peptide agonists caused receptor down-regulation to similar extents. In addition, after BFA incubation, etorphine and pentazocine caused down-regulation, whereas in the absence of BFA they up-regulated the mature receptor. Moreover, BFA abrogated naloxone-induced up-regulation.
The results are interpreted as follows. Chronic ligand treatment potentially has dual effects on receptor levels: activation-induced receptor degradation and chaperone-mediated enhancement. At the two ends of the spectrum are the peptide agonists and nonpeptide antagonists. Membrane-impermeable peptide agonists cause activation-induced receptor degradation without any chaperone effect. Membrane-permeable antagonists exert chaperone-like actions without activation-promoted degradation. In between the two ends, cell-permeable nonpeptide agonists have both effects, and their net effects are the algebraic sum of the two. Thus, all nonpeptide full agonists, except etorphine, led to smaller reductions of the steady-state mature receptor than peptide full agonists. Etorphine or pentazocine induced receptor up-regulation, because of their chaperone effects and low efficacy to induce receptor degradation.
Down-Regulation and Chaperone Effects Seem to Have Similar Time Courses in Vitro. For down-regulation, ligand binding to cell surface receptors causes internalization of the receptor via clathrin-coated pits, and internalized receptors are trafficked through endocytic vesicles, early endosomes, late endosomes, and finally lysosomes. For a chaperone effect, the ligand has to penetrate plasma membranes to reach the ER to stabilize favorable receptor conformations that allow the receptor to exit the ER to the Golgi and then to plasma membranes. Thus, both ligand-induced down-regulation and chaperone effects involve a series of vesicle fusion events; therefore, their time courses may be similar.
Nonpeptide Agonists Have Differential Activities in Down-Regulating the KOR and as Pharmacological Chaperones. Nonpeptide full agonists produced higher degrees of down-regulation with BFA than without BFA. However, BFA did not cause these nonpeptide agonists to down-regulate the hKOR to the same levels. These results suggest that nonpeptide agonists have different activities in regulating the KOR. This notion is supported by our finding that although etorphine is a full agonist, it does not cause hKOR internalization (Li et al., 1999, 2003), which is required for down-regulation (Li et al., 2000). In addition, internalized receptors may be sorted differently depending on the agonist used. Marie et al. (2003) have reported that compared with [d-Pen2,5]enkephalin and deltorphin I, etorphine results in less lysosome-mediated degradation, but more recycling, of the human δ-opioid receptor. This possibility remains to be investigated for the hKOR. Moreover, among nonpeptide agonists, there may be differences in their abilities to pass through membranes. Asimadoline and ICI 204,448, two peripherally acting KOR agonists (Shaw et al., 1989; Barber et al., 1994), caused higher degrees of down-regulation (Fig. 1), probably because they are less able to penetrate membrane. Furthermore, this discrepancy may be due to differential stability of these ligands in the incubation medium, which includes 10% FBS that contains numerous enzymes.
Other Mechanisms Seem Not to Play Important Roles in Ligand-Induced KOR Up-Regulation. To date, several other mechanisms have been demonstrated for ligand-induced receptor up-regulation, including enhanced transcription and translation and reduced internalization and degradation. In the present study, we used the hKOR cDNA construct, which does not contain the promoter region, and expression of the receptor is driven by the constitutively active cytomegalovirus promoter. Thus, regulation at the level of DNA transcription or mRNA translation does not play a role.
Chronic antagonist-induced decreases in lysosomal enzyme (β-glucuronidase and β-hexoseaminidase) activities and in trafficking proteins (G protein-coupled receptor kinase 2 and dynamin2) were reported to be important in facilitating receptor expression (Belcheva et al., 1992; Rajashekara et al., 2003). However, our results do not support this explanation because naloxone treatment did not affect the turnover rate of the mature receptor (Fig. 3; Table 1).
Antagonist-Promoted KOR Up-Regulation in Vivo. After chronic administration of naloxone, naltrexone, or buprenorphine, the KOR in brain regions was up-regulated (Morris et al., 1988; Belcheva et al., 1993; Lesscher et al., 2003). After chronic antagonists, functional supersensitivity to κ agonist-mediated antinociception was demonstrated (Millan et al., 1988), indicating that the up-regulated KOR is functional. It is likely that the mechanisms uncovered here are applicable in vivo.
Nonpeptide Ligands Act as Pharmacological Chaperones of Other GPCRs. μ and δ Opioid receptors. Nonpeptide agonists and antagonists, but not peptide ligands, have been reported to act as chaperones to stabilize the ER forms and enhance cell surface levels of the wild-type and D95A mutant of the δ opioid receptor and two deletion mutants and several constitutively active mutants of the μ opioid receptor (Li et al., 2001a,b; Petaja-Repo et al., 2002; Chaipatikul et al., 2003).
Nonopioid receptors. Ligand-induced enhancement in cell surface receptors has been demonstrated for several other wild-type and mutant GPCRs, for example, vasopressin V2, dopamine D4, and β2-adrenergic receptors (Samama et al., 1997; Morello et al., 2000; Van Craenenbroeck et al., 2005). Cell-permeable antagonists/inverse agonists and agonists up-regulated cell surface receptors. Infusion with the β2-antagonist ICI 118,551 for 7 days resulted in a 50-fold increase in density of constitutively active mutated β2-adrenergic receptor in the myocardium (Samama et al., 1997), indicating that pharmacological chaperone effects occur in vivo.
Differential Effects of Nonpeptide and Peptide Ligands. In contrast to the nonpeptide ligands examined, the two peptide ligands, dynorphin A and B, did not have pharmacological chaperone actions. Similar differential effects of nonpeptide versus peptide ligands have been observed for other GPCRs, including μ and δ opioid, kinin B1, and gonadotropin-releasing hormone receptors and a V2 vasopressin receptor mutant (Morello et al., 2000; Janovick et al., 2002; Petaja-Repo et al., 2002; Chaipatikul et al., 2003; Fortin et al., 2006).
Membrane Permeability of Peptide Ligands. Most peptide ligands are generally accepted to be cell impermeable, whereas some water-soluble short peptides (≤35 residues) that contain the so-called membrane translocation sequence are able to cross cell membrane and are classified as cell-penetrating peptides (Magzoub and Graslund, 2004). Our results that both dynorphin A and dynorphin B did not enhance the expression of the new intracellular 51-kDa species after BFA treatment indicate that the two peptides do not penetrate plasma membranes. This finding is different from the recent report by Marinova et al. (2005) that dynorphin A and big dynorphin, but not dynorphin B, were able to translocate across plasma membranes in HeLa, COS-1, and PC12 cells. However, all peptide ligands tested for many other GPCRs did not display the chaperone-like ability (see above), demonstrating that they are membrane impermeable in CHO, HEK293, and COS-7 cells. Therefore, it is possible that the membrane permeability of dynorphin A and big dynorphin may be cell-specific.
In conclusion, treatment with different KOR ligands resulted in distinct regulatory effects on the mature FLAG-hKOR. Membrane-impermeable peptide full agonists lead to receptor down-regulation to greater extents as they do not act as pharmacological chaperones. Membrane-permeable nonpeptide antagonists act as pharmacological chaperones to induce receptor up-regulation without causing down-regulation. Membrane-permeable nonpeptide agonists have two opposing effects on cell surface receptors: to decrease the receptor by causing activation-dependent endocytosis and degradation and to increase the receptor by acting as pharmacological chaperones. These findings have great implications for the field of neuropeptides in general. Many nonpeptide agonists or antagonists of neuropeptide receptors are being developed as pharmacological tools and therapeutic agents, primarily because of better pharmacokinetic properties and penetration through the blood-brain barrier. The present study indicates that nonpeptide ligands have an added advantage over peptides in that they can act as pharmacological chaperones. Thus, nonpeptide agonists will cause less down-regulation of the receptors than peptide agonists, leading to less tachyphylaxis after long-term treatment.
Acknowledgments
We thank Adolor Corporation for generous gifts of several KOR ligands. We are grateful to Dr. Ronald J. Tallarida, Department of Pharmacology, Temple University School of Medicine, for kind help with kinetic analyses.
Footnotes
-
This work was supported by National Institutes of Health Grants DA17302 and DA04745.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.107987.
-
ABBREVIATIONS: KOR, κ opioid receptor; U50, 488H, 2-(3,4-dichlorophenyl)-N-methyl-N-[(2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide; U69, 593, (5α, 7α, 8β)-(+)-N-methyl-N-(7-[1-pyrrolidinyl]-1-oxaspiro[4,5]dec-8-yl)benzeneacetamide; ICI 204, 448, (RS)-[3-[1-[[(3,4-dichlorophenyl)acetyl]-methylamino]-2-(1-pyrrolidinyl)ethyl]phenoxy] acetic acid hydrochloride; TRK-820,17-cyclopropylmethyl-3,14-dihydroxy-4,5-epoxy-6-[N-methyl-trans-3-(3-furyl) acrylamido] morphinan hydrochloride; Nor-BNI, norbinaltorphimine; EBP50/NERF-1, Ezrin-radixin-moesin-binding phosphoprotein-50/Na+,H+-exchanger regulatory factor-1; ER, endoplasmic reticulum; CHO, Chinese hamster ovary; FLAG, DYKDDDDK; FLAG-hKOR, FLAG-tagged human κ opioid receptor; R(+)-SCH 23,0390, (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; PVDF, polyvinylidene difluoride; PAGE, polyacrylamide gel electrophoresis; CHO-FLAG-hKOR, CHO cell lines stably expressing FLAG-hKOR; BFA, brefeldin A; PBS, phosphate-buffered saline; Dyn, dynorphin; GPCR, G protein-coupled receptor; ICI 118,551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol.
- Received May 17, 2006.
- Accepted July 31, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}