


Abstract
Chronic administration of nicotine increases the density of neuronal cholinergic nicotinic receptors in cells and in rodent brain, and similar increases have been reported in brains from human smokers. To further examine this phenomenon, we measured nicotinic receptor binding sites in brain regions from matched populations of smokers and nonsmokers. We first measured binding of [3H](±)epibatidine ([3H]EB) and [3H]cytisine in homogenate preparations from samples of prefrontal and temporal cerebral cortex. Binding of each radioligand was significantly higher (250–300%) in both cortical regions from brains of smokers. Frozen sections from each of the cerebral cortical regions and the hippocampus were used for autoradiographic analysis of [3H]EB binding. In cerebral cortex, binding was most dense in layer VI in the prefrontal cortex and layers IV and VI in the temporal cortex. Densitometric analysis of [3H]EB binding sites revealed marked increases of 300 to 400% of control in all cortical regions examined from smokers’ brains. Binding in the hippocampal formation was heterogeneously distributed, with dense areas of binding sites seen in the parasubiculum, subiculum, and molecular layer of the dentate gyrus, and the lacunosum-moleculare layer of the CA1/2. Binding of [3H]EB was significantly higher in all six regions of the hippocampus examined from brains of smokers compared with nonsmokers. These increases ranged from 160% of control in parasubiculum to 290% in the molecular layer of the dentate gyrus. The increase in nicotinic receptors in the cerebral cortex and hippocampus of smokers may modify the central nervous system effects of nicotine and contribute to an altered response of smokers to nicotine.
Nicotinic cholinergic receptors are widely distributed in the vertebrate central nervous system (CNS), where they appear to be associated with the axons and cell bodies of neurons that comprise several major neurotransmitter systems in the brain, including neurons that use dopamine, norepinephrine, acetylcholine, γ-aminobutyric acid, and glutamate. Because of the strategic locations of these receptors, nicotine may exert widespread influences on the function of several important neural pathways in the CNS.
Chronic administration of nicotine increases the density of nicotinic receptor binding sites in rat and mouse brain (Schwartz and Kellar, 1983; Marks et al., 1983). This increased receptor density is widespread in rodent brain, with more than two-thirds of the brain areas examined by autoradiography showing this effect after chronic nicotine (Kellar et al., 1989; Marks et al., 1992). But the extent of nicotine-induced increases in these receptors varies among brain regions; thus, binding to these receptors is increased by more than 60% in some layers of the cerebral cortex and by more than 100% in some regions of the hippocampus, whereas in other brain areas, notably some nuclei of the thalamus, there appears to be no significant nicotine-induced increase in receptors (Kellar et al., 1989; Marks et al., 1992; Flores et al., 1997). The possible relevance of these nicotine-induced receptor changes in rodent brain to long-term use of nicotine by humans was underscored by the finding that nicotinic receptors labeled by [3H]nicotine are increased in homogenates of autopsied brain samples from people who smoked cigarettes (Benwell et al., 1988; Breese et al., 1997).
Nicotinic receptors in the cerebral cortex and hippocampus are of particular interest because of their possible roles in cognition, memory, arousal, attention, and anxiety, all of which are reported to be affected by nicotine and/or withdrawal from nicotine (Levin, 1992). In the present study, we compared neuronal nicotinic receptor binding sites in the prefrontal cerebral cortex (area 10 and 11), temporal cerebral cortex (area 38), and the hippocampus from autopsied brains from smokers and age-matched nonsmokers. To do this, nicotinic receptors labeled by [3H](±)epibatidine ([3H]EB) and [3H]cytisine were measured in membrane homogenates of the two cerebral cortex areas and receptor autoradiography of [3H]EB binding sites was carried out in sections from the two cerebral cortex areas and the hippocampus. The autoradiographic studies allowed visualization and assessment of the effects of smoking on nicotinic receptor numbers in these brain areas in some anatomical detail. The homogenate binding measurements were made with both radioligands because [3H]cytisine, like [3H]nicotine, appears to label predominantly the α4/β2-nicotinic receptor subtype in brain (Whiting et al., 1991; Flores et al., 1992), whereas in contrast, [3H]EB appears to label several different neuronal nicotinic receptor subtypes (Houghtling et al., 1995; Perry and Kellar, 1995; Perry et al., 1997; Marks et al., 1998; Parker et al., 1998; Zoli et al., 1998). Not all nicotinic receptor subtypes are increased by chronic administration of nicotine in vivo (Flores et al., 1997) or by concentrations that can be achieved in vivo (Peng et al., 1997); thus, if smoking differentially altered nicotinic receptor subtypes in human brain, it could be revealed by comparing the binding of these two radioligands.
Materials and Methods
Brain tissue was obtained at autopsy at the Cuyahoga County (Ohio) Coroner’s Office. The study was performed in compliance with policies of an institutional review board and written consent was obtained from the closest relative. The cause of death for each subject is listed in Table 1. Toxicology screens and retrospective psychiatric assessments revealed nothing remarkable. Information on smoking was collected in an interview with a knowledgeable informant within 6 months after death. Smokers were defined as people who smoked 20 or more cigarettes daily up to the time of death; nonsmokers were people who never smoked (except one subject who quit 24 years before death; see Table 1).
Subject data
Brains were collected from eight smokers and eight nonsmokers and were matched with respect to age (54 ± 4 years versus 54 ± 6 years) and postmortem interval (PMI; 21 ± 2 h versus 16 ± 3 h; see Table 1). Tissue samples were coded so that laboratory personnel were not aware of the smoking history until results were collected. There were seven males and one female in each group. Tissue blocks were dissected from a consistent region of the right hemisphere: the lateral surface of the anterior pole of the frontal lobe (prefrontal cortex, areas 10 and 11), the rostral 2 cm of the temporal pole (area 38), and the rostral 1.5 cm of the body of the hippocampus (immediately caudal to the uncus). In one case for each cerebral cortical region in nonsmokers we were unable to obtain tissue. The cerebral cortical samples were further divided into two pieces: one for homogenate binding studies and one for autoradiography. The tissue blocks for autoradiography were immersed briefly in isopentane, cooled in dry ice, completely frozen in powdered dry ice, and stored at −80°C until sectioning. The blocks were mounted on pedestals with Tissue-Tek (Miles Labs., Dallas, TX) and frozen sections (20 μm) were cut at −15°C with a microtome (IEC). Sections were thaw-mounted onto cold microscope slides coated with gelatin/chrom-alum, dried under a stream of room-temperature air, and refrozen at −80°C.
Homogenate binding was done as described by Houghtling et al. (1995)for [3H]EB and by Pabreza et al. (1991) for [3H]cytisine. Briefly, tissues were suspended in 50 mM Tris HCl buffer (pH 7.4), homogenized with a Brinkmann Polytron (Brinkman Instruments, Westbury, CT), and centrifuged at 35,000g for 10 min. The pellets were resuspended in fresh buffer, and aliquots equivalent to 10 mg tissue (−600 μg protein) were incubated in buffer for 4 h at 24°C with 1.3 nM [3H]EB (56.5 Ci/mmol; NEN, Boston, MA) or for 90 min at 0°C with 7.0 nM [3H]cytisine (39.6 Ci/mmol; NEN). In preliminary studies we found theKd of [3H]EB in human cerebral cortex to be 51 ± 5 pM (n = 3, data not shown); therefore, the single high concentration of [3H]EB used here should occupy >95% of the predominant subtype of nicotinic receptor in these tissues. Based on its binding affinities to nicotinic receptors expressed in frog oocytes (Parker et al., 1998) and human embryonic kidney cells (Y. Xiao and K. J. Kellar, unpublished observations), this concentration of [3H]EB should occupy a similar fraction of several neuronal nicotinic receptor subtypes, the known exceptions being α3/β4 receptors, of which ∼80% should be occupied (Parker et al., 1998; Xiao et al., 1998) and α7 receptors, of which there would be negligible occupancy. [3H]Cytisine binds in human cerebral cortex with aKd of ≈200 pM (Hall et al., 1993); therefore, the concentration of [3H]cytisine used should occupy >95% of the nicotinic receptor subtype(s) labeled by this ligand in brain. Nonspecific binding was determined in the presence of 300 μM (−)nicotine hydrogen tartrate, and specific binding was calculated as the difference between total binding (no nicotine added) and nonspecific binding. Incubations were terminated by vacuum filtration through Whatman GF/C filters that were prewet with 0.5% polyethylenimine and mounted on a Brandel cell harvester. Filters were then washed with three 4-ml aliquots of cold buffer and bound radioactivity was measured in a liquid scintillation counter. Protein was determined using the BCA reagent (Pierce, Rockford, IL) and measured at 570 nm.
Autoradiography was carried out according to published methods (Perry and Kellar, 1995). Frozen tissue sections were incubated in Tris HCl buffer (pH 7.4) containing 120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, and 1 nM [3H]EB. Nonspecific binding was determined in adjacent sections in the presence of 300 μM (−)nicotine hydrogen tartrate. Sections were incubated at room temperature for 40 min, which is sufficient time for this concentration of [3H]EB to achieve equilibrium (Perry and Kellar, 1995), rinsed twice for 5 min in ice-cold buffer, and then dipped briefly in distilled water. After air drying, the sections were apposed to tritium-sensitive film ([3H]-Hyperfilm; Amersham, Arlington Heights, IL) along with [3H]standards (Amersham) for 133 days. Quantitative densitometric analysis of binding was done using the Loats INQUIRY system. To determine specific binding, nonspecific binding in adjacent sections was subtracted from the total binding in each anatomical region. A minimum of three measurements in two adjacent sections were averaged to determine the binding value for a given tissue. It should be noted, however, that the values for binding are determined by comparison of a two-dimensional image of a tissue section, which has variable quenching and consistency, to a plastic standard of known radioactivity. Therefore, the binding values from the autoradiographic samples presented here are best regarded as semiquantitative. They are, however, well suited for comparative studies across treatment groups assayed in parallel, such as presented here. Autoradiographic images were compared with Nissl stained adjacent tissue sections for identification of brain structures.
Paired Student’s t tests (with pooled variance) were used to compare the means of nicotinic receptor binding in brain homogenates from smokers and nonsmokers. Samples from smokers and nonsmokers were paired according to age (±6 years), and Bonferroni corrections were applied to correct for multiple comparisons. Grouped t tests and nonparametric statistical tests such as the Wilcoxon Rank Sum/Mann-Whitney U test were also employed and yielded similar statistical conclusions. Means of binding to multiple brain regions from the autoradiography studies were compared with an ANOVA using Bonferroni corrections for multiple comparisons. The relationship between age or PMI and receptor binding was examined with Pearson correlations, corrected for multiple comparisons. Although there were no significant relationships between age or PMI and receptors, an analysis of covariance was performed with age and PMI as covariates to compare binding measures in smokers and nonsmokers. The results of the analysis of covariance were essentially identical with paired comparisons.
Results
Binding of [3H]EB and [3H]Cytisine in Homogenates of Cerebral Cortex from Smokers and Nonsmokers.
Nicotinic receptor binding sites in homogenates from prefrontal and temporal cerebral cortex were measured using nearly saturating concentrations of [3H]EB and [3H]cytisine; thus, the number of binding sites labeled should closely approximate their density. As shown in Fig.1, in the prefrontal cerebral cortex, binding of both [3H]EB and [3H]cytisine was approximately three times higher in brain samples from smokers than from nonsmokers (p < .001). Similarly, in the temporal cerebral cortex (Fig. 1) binding of both ligands was markedly higher (approximately 2.5 times) in brain samples from smokers compared with nonsmokers (p < .001). Overall, the relative increase in nicotinic receptors in smokers’ brains appeared to be somewhat greater in the prefrontal cortex than in the temporal cortex.

Binding of [3H]EB and [3H]cytisine to homogenates of human cerebral cortex from prefrontal region (A) and temporal region (B). Bars represent means ± S.E.M. of the binding of saturating concentrations of [3H]EB (1.3 nM) or [3H]cytisine (7 nM) in smokers (n = 8) or nonsmokers (n = 7). In all four comparisons between tissues from smokers and nonsmokers, the binding was significantly higher in brains from smokers (***p < .001). In addition, in the temporal cortex from smokers [3H]EB binding was higher than [3H]cytisine binding (+p< .02).
In the prefrontal cortex, [3H]EB and [3H]cytisine labeled approximately the same number of receptor binding sites in both smokers and nonsmokers (Fig.1). In the temporal cortex, however, there was a trend for [3H]EB to label more sites than [3H]cytisine in samples from both groups, and this difference was statistically significant in smokers (p < .02). In nonsmokers, the density of nicotinic receptors measured with either ligand appeared to be higher in the prefrontal cortex than in the temporal cortex; this difference was statistically significant for measurements with [3H]cytisine (p < .001), but not with [3H]EB (0.05<p < .10).
Pearson’s correlational analysis was performed to test for the influence of age or PMI on binding of either ligand to cortical homogenates. No significant correlation with age was seen with either ligand in the prefrontal cortex (Fig. 2). In the temporal cortex (Fig. 2), there was a trend for decreased [3H]EB binding with age in smokers and nonsmokers, but this trend did not reach statistical significance. In these studies, PMI was negatively correlated with binding of [3H]EB in temporal cortex from nonsmokers (data not shown), but no other correlations with PMI were detected. To further examine the possibility of an influence of PMI on the binding values, an analysis of covariance was performed to determine whether there was still a statistically significant difference in the means of receptor binding values between smokers and nonsmokers when age or PMI are covaried. In both tissues and with both ligands, the differences in nicotinic receptor binding between smokers and nonsmokers remained highly significant (p < .001).

Binding of [3H]EB and [3H]cytisine as a function of age of subjects in homogenates of human cerebral cortex from prefrontal region (A) and temporal region (B). Influence of age on [3H]EB and [3H]cytisine binding sites in these tissues was examined using Pearson’s correlational analysis. In prefrontal cortex samples from nonsmokers and smokers, respectively, correlation coefficients were: for [3H]EB binding 0.008 and 0.091, and for [3H]cytisine binding 0.048 and 0.195. In temporal cortex samples, from nonsmokers and smokers, respectively, correlation coefficients were: for [3H]EB binding 0.852 and 0.779, and for [3H]cytisine binding 0.05 and 0.20. After Bonferroni corrections for multiple comparisons, no statistically significant effect of age on binding of either ligand in either region was detected.
Autoradiographic Analyses of [3H]EB Binding in Cerebral Cortex and Hippocampus from Smokers and Nonsmokers.
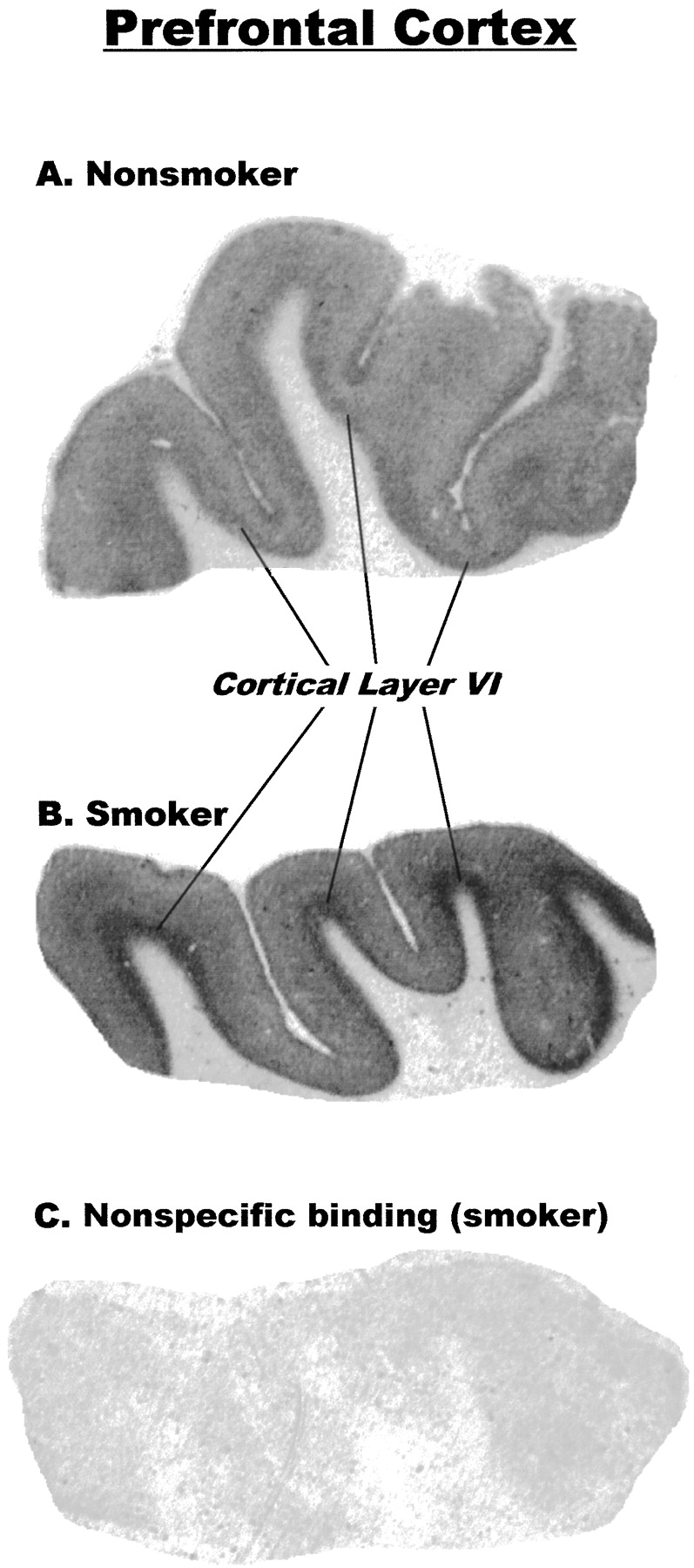
Representative autoradiographic images of nicotinic receptor binding sites labeled by [3H]EB in prefrontal cortex and in temporal cortex are shown in Figs.3 and 4, respectively. Autoradiography with [3H]EB in human brain yielded excellent images, with low to nearly undetectable nonspecific binding (Fig. 3C). Nicotinic receptor binding sites were confined almost entirely to gray matter in both regions of cerebral cortex. In the prefrontal cortex (Fig. 3), the highest nicotinic receptor density was seen in the deepest cortical layer, which appeared to correspond to layer VI. In the temporal cortex (Fig. 4), there were two densely labeled cortical layers, corresponding approximately to layers IV and VI.

Representative autoradiographic images of [3H]EB binding to comparable sections of human prefrontal cerebral cortex from a nonsmoker (A) and a smoker (B). Dark areas represent binding of [3H]EB. Cortical layer VI, which contains highest level of binding, is indicated. Image in C, which is a section adjacent to section shown in B, was incubated with [3H]EB in the presence of 300 μM nicotine to determine nonspecific binding.

Representative autoradiographic images of [3H]EB binding to comparable sections of human temporal cerebral cortex from a nonsmoker (A) and a smoker (B). Dark areas represent binding of [3H]EB. Cortical layers I-III, IV, V, and VI are indicated in section B.
The marked increase in nicotinic receptor binding sites measured in homogenized samples from smokers’ brains was readily apparent in the autoradiographic images of [3H]EB binding sites in the sections from both cerebral cortical regions (Figs. 3 and 4). Analysis of these images by quantitative densitometry indicated that in the prefrontal cortex, the density of these receptors was more than 400% higher in brains from smokers than from nonsmokers (Fig.5); and in the temporal cortex, the density was more than 300% higher in brains from smokers than from nonsmokers (Fig. 5). Within the cortical regions, the magnitude of the increases was similar in all layers examined: 440% and 490% in frontal cortex layer and from 310% to 350% in temporal cortex layer. All increases were highly significant (p < .001).

Quantitative comparison of [3H]EB binding analyzed densitometrically in human cerebral cortical sections from nonsmokers and smokers. Binding in sections from prefrontal cortex (F) was analyzed separately in layers I-V (1–5) and layer VI (6). In sections from temporal cortex (T), binding was analyzed separately in layer I-III (1–3), layer IV (4), layer V (5), and layer VI (6). Bars represent means ± S.E.M. of [3H]EB-specific binding in sections from nonsmokers (n = 7) and smokers (n = 8). In all cases, binding in brains from smokers was higher than that in the same region of brains from nonsmokers, ***p < .001.
Representative autoradiographic images of nicotinic receptor binding sites labeled by [3H]EB in human hippocampus are shown in Fig. 6. Specific binding was detected throughout the hippocampal formation, with distinct and dense areas of nicotinic receptor binding apparent in the parasubiculum, subiculum, lacunosum moleculare layer of the CA1/CA2, and the molecular layer of the dentate gyrus (Fig. 6). The receptors in each of these areas, as well as in the CA4 region (polymorphic hilar region) and the oriens, pyramidale, and radiatum layers of the CA1 appeared to be markedly increased in brains from smokers.

Representative autoradiographic images of [3H]EB binding to comparable sections of human hippocampus from a nonsmoker (A) and a smoker (B). Dark areas represent binding of [3H]EB. Abbreviations: dg, molecular layer of the dentate gyrus; Ent Ctx, entorhinal cortex; hil, polymorphic hilar region (CA4); l-m, lacunosum-moleculare layer of CA1/CA2; o-p-r, oriens, pyramidale, and radiatum layers of CA1/CA2; PaS, parasubiculum; Sub, subiculum.
[3H]EB autoradiographic images were compared by densitometry in six areas of the hippocampus from smokers and nonsmokers (Fig. 7). There was an approximate 4-fold range in the levels of nicotinic binding sites among the different areas analyzed in nonsmokers, with the highest levels detected in the parasubiculum and the lowest in the oriens/pyramidale/radiatum layers of the CA1/CA2 region. Densitometric analysis confirmed that the density of sites was markedly higher in all six of these areas in the hippocampus from smokers (Fig. 7). The largest increases were found in the molecular layer of the dentate gyrus (290% of control binding), the oriens/pyramidale/radiatum layers of the CA1/CA2 region (270%), and the subiculum (220%), whereas the smallest percent increase was found in the parasubiculum (160%). All of the increases were statistically significant.

Quantitative comparison of [3H]EB binding analyzed densitometrically in human hippocampal sections from smokers and nonsmokers. Binding to separate hippocampal regions (see Fig. 6) was analyzed. Abbreviations: DG, molecular layer of the dentate gyrus; CA4, polymorphic hilar region; l-m, lacunosum-moleculare layer of CA1/CA2; o-p-r, oriens, pyramidale, and radiatum layers of CA1/CA2; Sub, subiculum; PaSub, parasubiculum. Bars represent means ± S.E.M. of [3H]EB specific binding in sections from nonsmokers (n = 8) and smokers (n = 8). Binding in smokers statistically different from that of nonsmokers in same region: *p < .05; **p < .01; ***p < .001.
Discussion
Previous studies have found a higher level of nicotinic receptors labeled by [3H](−)nicotine in brain samples from smokers compared with nonsmokers in the gyrus rectus of the frontal lobe, the hippocampus, and the thalamus (Benwell et al., 1988;Breese et al., 1997). The current study extends these findings to nicotinic receptors measured with [3H]cytisine as well as with [3H]EB. Both ligands have high affinity for the α4/β2 receptor subtype, which is likely to predominate in the cerebral cortex (Whiting et al., 1991; Flores et al., 1992), but [3H]EB also labels a broad spectrum of other nicotinic receptor subtypes not readily labeled by other nicotinic ligands (Houghtling et al., 1995; Gerzanich et al., 1995; Parker et al., 1998; Xiao et al., 1998). In addition, autoradiography of [3H]EB binding sites has allowed visualization and quantitative comparisons of the nicotinic receptor density across layers of the prefrontal and temporal cerebral cortex and within specific areas of the hippocampus.
Markedly higher densities of nicotinic receptor binding sites were found in both areas of cerebral cortex and in the hippocampus in brains from smokers. The relative increases in smokers’ brains were somewhat greater in the prefrontal cortex than in the temporal cortex in both the homogenate binding studies and the quantitative autoradiographic studies; however, within each of the two cerebral cortical regions the relative increases appeared to be similar across the cortical layers.
There was a relatively wide range of receptor densities among the specific regions of the hippocampus; certain regions, particularly the parasubiculum, subiculum and the CA1:lacunosum-moleculare layer, have a high number of receptor binding sites, whereas other regions, such as the molecular layer of the dentate gyrus, CA4 region (hilus), and the oriens/pyramidale/radiatum layers of the CA1/2 have a more modest density of sites. This distribution of nicotinic receptor binding sites labeled by [3H]EB in the human hippocampus is similar to the distributions of sites previously reported using [3H](−)nicotine (Perry et al., 1992) and [3H]cytisine (Aubert et al., 1995). The autoradiographic density of [3H]EB binding sites was higher in all regions of the hippocampus from smokers compared to nonsmokers. These increases ranged from approximately 160% of control binding in the parasubiculum, which has the highest levels of receptors in nonsmokers, to nearly 300% in the dentate gyrus and the oriens/pyramidale/radiatum layers of the CA1/2. The basis for these regional differences in response is not known. One possibility, however, is that the regions contain different populations or proportions of receptor subtypes that differ in their susceptibility to be increased by chronic nicotine exposure (see below).
[3H]EB and [3H]cytisine appeared to label a similar number of receptors in the prefrontal cortex, but in the temporal cortex, there was a trend for [3H]EB to label more receptors than [3H]cytisine, and this was statistically significant in smokers. This suggests that in the temporal cortex, and probably in other brain regions as well, [3H]EB can label one or more additional populations of nicotinic receptors not labeled by [3H]cytisine (Perry and Kellar, 1995; Perry et al., 1997; Marks et al., 1998; Zoli et al., 1998). In fact, Albuquerque and colleagues (Alkondon and Albuquerque, 1993) and Changeux and colleagues (Zoli et al., 1998) have proposed four classes of neuronal nicotinic receptors. Thus it is possible that comparisons of [3H]EB and [3H]cytisine binding sites in cerebral cortex could reveal differences in receptor populations.
Chronic administration of nicotine increases the density of brain nicotinic receptor binding sites in both rats (Schwartz and Kellar, 1983) and mice (Marks et al., 1983). Thus, it is highly likely that the increased receptors in brains from smokers result from their chronic exposure to nicotine, rather than any other component of tobacco. Moreover, the controlled studies in rodents clearly imply that the increased nicotinic receptors in smokers’ brains are a consequence of smoking, rather than smoking behavior being a consequence of an intrinsically higher number of nicotinic receptors. Strong evidence for this comes from a recent study by Leonard and her colleagues (Breese et al., 1997) that found that although the density of [3H](−)nicotine binding sites was significantly higher in the hippocampus and thalamus from people who smoked up to the time of death, the density of these sites in people who had quit smoking at least 2 months before death was not significantly different from nonsmokers.
In rats and mice exposed to nicotine for 1 to 3 weeks, nicotinic receptors are usually found to be increased by 30 to 60% in most brain areas examined, including the cerebral cortex, and up to 100% in a few subcortical brain structures measured autoradiographically (Kellar et al., 1989; Marks et al., 1992). The much larger increases in brain nicotinic receptors from humans who smoked might reflect, at least in part, the much longer duration of exposure to nicotine in human smokers. For example, in the studies reported here the brain samples were from subjects who were on average 54 years old at the time of death; therefore, a conservative estimate of the average duration of chronic nicotine exposure in these smokers is >30 years. However, it is also possible that nicotinic receptors in human brain have a greater capacity to increase in response to nicotine.
The increase in receptor binding sites in rodent brain does not appear to involve an increase in the steady-state level of nicotinic receptor subunit mRNA (Marks et al. 1992; Peng et al., 1994; Bencherif et al. 1995), suggesting that new protein synthesis is not required. Instead, studies in a mouse fibroblast cell line expressing the avian α4/β2 receptor subtype indicate that the nicotine-induced increase in receptors is most likely related to a decrease in the rate of receptor degradation (Peng et al., 1994), or possibly to conversion of low affinity receptors to a conformation with high affinity for agonists (Bencherif et al., 1995). Furthermore, the increased receptor binding sites in this cell line appear to be mostly internalized, rather than on the cell surface (Whiteaker et al., 1998). Taken together, the data suggest that, in addition to the duration of exposure to nicotine, the magnitude of nicotine-induced receptor increases in a particular brain area may reflect the turnover rate of the receptor (and/or possibly other cellular events) which, in turn, may be heavily influenced by the level of activity of the cells on which the receptor is located, as well as differences in receptor subtypes.
Because nicotine is classified as an agonist, the increase in nicotinic receptor density induced by chronic exposure to nicotine may appear to be paradoxical. But, in fact, after its initial agonist action, nicotine can produce marked and protracted desensitization and even inactivation of some neuronal nicotinic receptors in vivo (Langley and Dickinson, 1889; Paton, 1954; Sharp and Beyer 1986; Hulihan-Giblin et al., 1990a,b). Therefore, although the initial action of nicotine is stimulatory, its time-averaged effect, at least at some nicotinic receptor subtypes, is predominantly inhibitory. Because of this dual effect, nicotine can be considered to be a time-averaged antagonist (Hulihan-Giblin et al., 1990a,b).
However, the characteristics of the acute response of neuronal nicotinic receptors to nicotine (e.g., their conductances and rate of desensitization) depend on their subunit composition (Luetje and Patrick, 1991; Fenster et al., 1997). Furthermore, nicotinic receptor subtypes are affected differentially by chronic exposure to nicotine, both in cell models (Hsu et al., 1996; Olale et al., 1997;Peng et al., 1997) and in vivo (Flores et al., 1997). For example, in rats chronic administration of nicotine increases the density of the α4/β2-nicotinic receptor subtype in the cerebral cortex, as determined by immunoprecipitation of [3H]cytisine-bound receptors with antibodies directed at specific nicotinic receptor subunits (Flores et al., 1992). Similarly, nicotinic receptors labeled by [3H]EB in rat cerebral cortex are increased by chronic administration of nicotine, whereas in contrast, the nicotinic receptors in the rat adrenal gland, which contains few if any α4/β2 receptors, are not increased by this treatment (Flores et al., 1997). Thus, the α4/β2 receptor binding site in rat brain appears to be particularly prone to increase during chronic administration of nicotine, possibly because it has a higher affinity for nicotine and other agonists compared with other receptor subtypes. Consistent with this, α3-containing receptor subtypes in the human neuroblastoma cell line SH-SY5Y are up-regulated only after exposure to much higher nicotine concentrations (≥10 μM) than would normally be attained in a smoker’s brain (Peng et al., 1997). The nearly identical pharmacology of the rat and human α4/β2 receptor (Gopalakrishnan et al., 1996) and the high-affinity labeling of nicotinic receptors in human brain by [3H]cytisine (Hall et al., 1993;Aubert et al., 1995; this study), as well as by [3H]acetylcholine (Whitehouse et al., 1986) and [3H](−)nicotine (Whitehouse et al., 1986;Benwell et al., 1988; Perry et al., 1992; Breese et al., 1997), suggest that the nicotinic receptor that is increased in the brains of human smokers is predominantly the α4/β2 receptor, although this has not been demonstrated directly.
In conclusion, nicotinic receptors are markedly increased in brains from smokers compared with nonsmokers. Nicotinic receptors are thought to mediate the CNS effects of nicotine on motor function, arousal, emotional state, neuroendocrine function, and cognition (for reviews, see Levin, 1992; Brioni et al., 1997). In addition, because these receptors mediate nicotine-induced effects within important pathways of the so-called “reward system” (Rowell et al., 1987; Di Chiara and Imperato, 1988), they are associated with, and, in fact, may directly mediate many of the reinforcing and addictive properties of nicotine. Although it is possible that more than one subtype of nicotinic receptor is involved in the addictive properties of nicotine and in the long-term consequences of smoking, it is clear from studies of the brains from smokers that chronic exposure to nicotine produces marked changes in at least one of these receptor subtypes.
Acknowledgments
The excellent assistance of the Cuyahoga County Coroner’s Office, Cleveland, Ohio, is greatly appreciated. We gratefully acknowledge the work of Ginny E. Dilley, James C. Overholser, Ph.D., and Herbert Y. Meltzer, M.D. in the tissue collection and retrospective psychiatric assessments. The assistance of Laura A. Shapiro in sectioning the tissues and Jacqueline Raizin in quantitative densitometry is gratefully appreciated.
Footnotes
-
Send reprint requests to: Kenneth J. Kellar, Ph.D., Department of Pharmacology, Georgetown University School of Medicine, 3900 Reservoir Rd. NW, Washington, DC 20007. E-mail:kellark{at}gunet.georgetown.edu
-
↵1 This work was supported by National Institutes of Health Grants DA06486, MH45488, and NS34706. Portions of this work were presented at meetings of the Society for Neuroscience (1996) and of the Society for Research in Nicotine and Tobacco (1996).
- Abbreviations:
- EB
- epibatidine
- [3H]EB
- [3H](±)epibatidine
- PMI
- postmortem interval
- Received October 30, 1998.
- Accepted February 4, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}