

Abstract
The transient outward potassium current was studied in outside-out macropatches excised from the soma of CA1 pyramidal neurons and stratum (st.) oriens–alveus inhibitory interneurons in rat hippocampal slices. Arachidonic acid dose dependently decreased the charge transfer associated with the transient current, concomitant with an increase in the rate of current inactivation. Arachidonic acid (AA) did not affect the voltage dependence of steady state inactivation but did prolong the period required for complete recovery from inactivation. The effects of AA were mimicked by the nonmetabolizable analog of AA, 5,8,11,14-eicosatetraynoic acid, suggesting that metabolic products of AA were not responsible for the observed blocking action. In addition, AA blocked st. oriens–alveus–lacunosum-moleculare interneuron transient currents but not currents recorded from basket cell interneurons. In current clamp experiments, AA was without effect on the action potential waveform of CA1 pyramidal neurons under control recording conditions. In voltage-clamp experiments, the use of a test pulse paradigm, designed to mimic the action potential voltage trajectory, revealed that the transient current normally associated with a single spike deactivates too rapidly for AA to have an effect. Transient currents activated by longer duration “action potential” waveforms, however, were attenuated by AA. Consistent with this finding was the observation that AA broadened interictal spikes recorded in the elevated [K+]o model of epilepsy. These data suggest that AA liberated from hippocampal neurons may act to block the transient current selectively in both CA1 pyramidal neurons and inhibitory interneurons and to broaden action potentials selectively under pathological conditions.
The freely diffusible, lipid soluble fatty acid arachidonic acid (AA) is an important second messenger in a wide variety of cell types. AA and its metabolic products have been shown to modulate a large number of ligand- and voltage-gated ion channels in a variety of systems (for review, see Bevan and Wood, 1987; Ordway, 1991). AA can be liberated from cell membranes either through a direct action of phospholipase A2 or through the combined action of phospholipase C and diacylglycerol lipase. Metabolism of AA by a cyclooxygenase enzyme occurs extremely rapidly and can produce 20 distinct, short-lived but extremely potent intermediates (e.g., the prostaglandins, prostacyclins, and thromboxanes).
Modulation of a variety of voltage-dependent potassium channels by AA is well documented (Meves, 1994). AA has been shown to activate a large conductance (160 pS) K+ channel directly in cardiac atrial muscle (Kim and Clapham, 1989) and a small conductance (23 pS) K+ channel in smooth muscle (for review, see Ordway et al., 1991). The lipoxygenase metabolites of AA also activate the cardiac, muscarinic-activated K+ channel, stimulate BKCa channels in rat pituitary tumor cells (Duerson et al., 1996), and modulate S-type K+ channels in Aplysia (Piomelli et al., 1987). In central neurons, AA either depresses or enhances K+ currents through lipoxygenase or cyclooxygenase metabolites (Keyser and Alger, 1990; Schweitzer et al., 1990; Zona et al., 1993).
The molecular identities of the channels modulated by AA are largely unknown. Recently, however, AA has been shown to inhibit currents directly through channels formed by members of the Kv4 (Shal) subfamily of voltage-dependent potassium channels, whereas other members of Shaker subfamilies were relatively insensitive to AA (Villaroel and Schwarz, 1996; however, see Honore et al., 1994). Kv4.2 protein is strongly expressed across the somatodendritic axis of hippocampal CA1 pyramidal neurons (Sheng et al., 1992; Maletic-Savatic et al., 1995), and recent evidence suggests that the predominant A-type transient current throughout principal neurons of the CNS arises from channels formed by Kv4.2 (Serodio et al., 1994). Taken together this would suggest that a large fraction of the transient current in CA1 pyramidal neurons may be through Kv4.2-containing channels and therefore may be subject to AA modulation. To test this hypothesis, we made recordings from outside-out macropatches excised from the somata of a variety of neuronal types in the hippocampal slice preparation and studied the modulation of the transient current contained in these patches by AA.
MATERIALS AND METHODS
Hippocampal slices were prepared as described previously (Maccaferri and McBain, 1995). Briefly, Sprague Dawley rats (postnatal days 8–22) were killed by decapitation after deep anesthesia using isoflurane following National Institutes of Health animal welfare guidelines. The brain was removed and placed in ice-cold artificial CSF, composed of (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 1 CaCl2, 3 MgSO4, and 10 glucose, saturated with 95% O2and 5% CO2, pH 7.4. Transverse hippocampal slices ∼300μm thick were cut using a Vibratome (Oxford series 1000; Polysciences, Warrington, PA) and incubated at room temperature for a recovery period of at least 1 hr before use.
All voltage-clamp recordings were performed at room temperature using outside-out macropatches excised from the somata of the appropriate cell type. Patch electrodes were fabricated from thin-walled borosilicate glass (TW150; World Precision Instruments) and had resistances of 2–6 MΩ when filled with (in mm): 130 K-gluconate, 10 NaCl, 2 Na2ATP, 0.3 NaGTP, 10 HEPES, and 0.6 EGTA, buffered to pH 7.4 and ∼275 mOsm. In some experiments 10 mmbis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) was added to the internal solution (see text for details). Glutathione (5 mm) was included and Mg2+ was excluded from the intracellular solution to prevent a loss of N-type inactivation caused by cysteine oxidation (Ruppersberg et al., 1991). Biocytin (0.2–0.4%) was routinely added to the recording electrode for post hoc identification of the recorded cell. Individual neurons were visually identified using a near infrared charge-coupled device camera. First, tight seals (>1 GΩ) were obtained from visually identified cells in the CA1 stratum (st.) pyramidale or st. oriens–alveus as described previously (Maccaferri and McBain, 1995). After establishment of the whole-cell configuration, the electrode was slowly withdrawn from the slice over the course of several minutes to ensure the formation of a large macropatch of membrane. Slices and outside-out patches were perfused with media of the following composition (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 1 CaCl2, 3 MgSO4, 0.2 CdCl2, and 10 glucose and tetrodotoxin (TTX, 0.5–1 μm) saturated with 95% O2 and 5% CO2, pH 7.4. Series resistances in whole-cell voltage-clamp experiments were calculated from the capacitive current peak (unfiltered) in a 10 mV voltage step and were in the range of 10–30 MΩ; membrane potentials were not corrected for these errors. For current-clamp recordings, the whole-cell recording configuration was used. Solutions were identical to those described above, with the exception that TTX and Cd2+ were omitted. Data were collected using either an Axopatch 1D or an Axopatch 200A amplifier (Axon instruments, Burlingame, CA) modified to permit bridge balance compensation in the current clamp mode. Signals were filtered at 2 kHz and digitized at 5–10 kHz on a Pentium-based computer using the pClamp suite of programs (Axon Instruments). Data are presented as the mean ± SEM.
Drugs and solutions. Arachidonic acid was obtained as a sodium salt (Sigma, St. Louis, MO). Arachidonic acid was made fresh daily before use by dissolving in water; the solution was sonicated for 3–5 min and then vortexed for a further 2 min. The solution was aliquoted, frozen, and used as required. If used from frozen, arachidonic acid was thawed and sonicated for 5 min and vortexed for 1 min before use. Fresh arachidonic acid was made every 3 hr. 5,8,11,14-Eicosatetraynoic acid (ETYA; Calbiochem, La Jolla, CA) was dissolved in ethanol to make a stock solution of 10 mmimmediately before use. The final concentration of ethanol in solutions containing ETYA was 0.1%. For experiments involving ETYA, 0.1% ethanol was routinely added to all control solutions. Ethanol (0.1%) was without effect on the transient currents recorded from hippocampal neurons.
Histological methods. After electrophysiological recordings, slices were fixed in 4% paraformaldehyde (>24 hr), immersed in 30% sucrose/PBS and resectioned into ∼75 μm sections on a freezing microtome. Biocytin staining was revealed using an avidin–horseradish peroxidase reaction (Vectastain avidin–biotin complex standard kit) and enhanced using NiNH4SO4 (1%) and CoCl2 (1%). Slices were mounted and dehydrated on gelatin-coated glass slides for camera lucida reconstruction.
RESULTS
Transient outward potassium currents in outside-out macropatches
Because of the large physical profile and multiple dendritic branch points of hippocampal neurons, most voltage-clamp data obtained from whole-cell voltage-clamp experiments are subject to considerable space-clamp error (Spruston et al., 1994; Thurbon et al., 1994). In the present experiments, outside-out macropatches excised from the cell bodies of pyramidal neurons and inhibitory interneurons were used for all voltage-clamp experiments to minimize such errors. Typically, patches excised from hippocampal neurons possessed a sufficiently large number of channels to permit the study of macroscopic transient currents. In general, macropatches excised from cells deep within the slice yielded patches containing the largest transient currents compared with those excised from more superficial cells. Although transient currents usually dominated the total outward current in pyramidal neuron macropatches (Fig. 1), the transient current was typically studied in isolation by use of the voltage protocol shown in Figure 1. By alternating a prepulse to −110 or −30 mV (or −40 mV, 100 msec), the total current (sustained plus transient) or the isolated sustained current component, respectively, could be activated in the patch by a test pulse to +40 mV. The transient current was subsequently isolated by digital subtraction of the sustained current from the total current. In a typical experiment, 18 test pulses, which alternated between a prepulse to either −30 or −110 mV, were delivered to a patch in both control or drug-treated conditions. After isolation of each transient current trace, the nine traces were then summed and averaged to yield the isolated and averaged transient current (Fig. 1, right panel). Transient currents in outside-out macropatches possessed amplitudes ranging from 100 pA to 2 nA (mean amplitude, 465 ± 70 pA; Vtest = +40 mV; n = 47). Inactivation of the transient current could be fit by a single exponential with a time constant of 23.3 ± 1.2 msec (Fig. 1B, dotted line; n = 47), suggesting that only a single transient current component was present in the outside-out macropatches. Transient current amplitude was a steep function of the test potential (Fig.2A). Currents were activated at potentials positive to −60 mV, and in most patches the calculated conductances were nonsaturating at test potentials up to +70 mV (data not shown). Because of the nonsaturating nature of the transient current conductance, we were unable to determine the half-activation voltage from conductance–voltage plots of the voltage dependence of activation in the majority of patches. In four patches, however, maximal conductance was reached at the most positive potentials, and the mean half-activation potential of the transient current in these patches was −7.5 ± 2.2 mV (k, 20.7 ± 1.0;n = 4) (data not shown). Steady state inactivation was studied using depolarizing test pulses to a fixed voltage (+40 mV) preceded by a series of prepulse conditioning potentials ranging from −120 to −10 mV. The transient current was completely inactive at prepotentials positive to −40 mV. As the prepotential was made progressively more negative, the transient current amplitude increased (Fig. 2B). The relative amplitudes of the currents were plotted and fitted with a Boltzmann equation to generate the steady state inactivation curve (Fig. 2B) Half-inactivation of the transient current occurred at −72.1 ± 1.8 mV (k, 8.8; n = 8), a value close to that reported previously for transient currents in cultured hippocampal neurons (Ficker and Heinemann, 1992).

Isolation of the transient current in outside-out macropatches excised from hippocampal pyramidal neurons.A, Transient currents were isolated from the total outward current by alternating prepulses to −30 and −110 mV before a test pulse to +40 mV. Inclusion of a prepulse to −30 mV resulted in the complete inactivation of the transient current and left only the isolated sustained current phenotype. In contrast, inclusion of a prepulse to −110 mV activated both transient and sustained current phenotypes. Digital subtraction of the family of currents obtained with a prepulse at −30 mV (nine trials) from those obtained with a prepulse to −110 mV (nine trials) yielded the isolated transient current.B, The isolated transient currents were then averaged to generate the mean current for analysis. Under control conditions the current inactivation could be fit by a single exponential (dotted line) with a time constant of 21.5 msec.
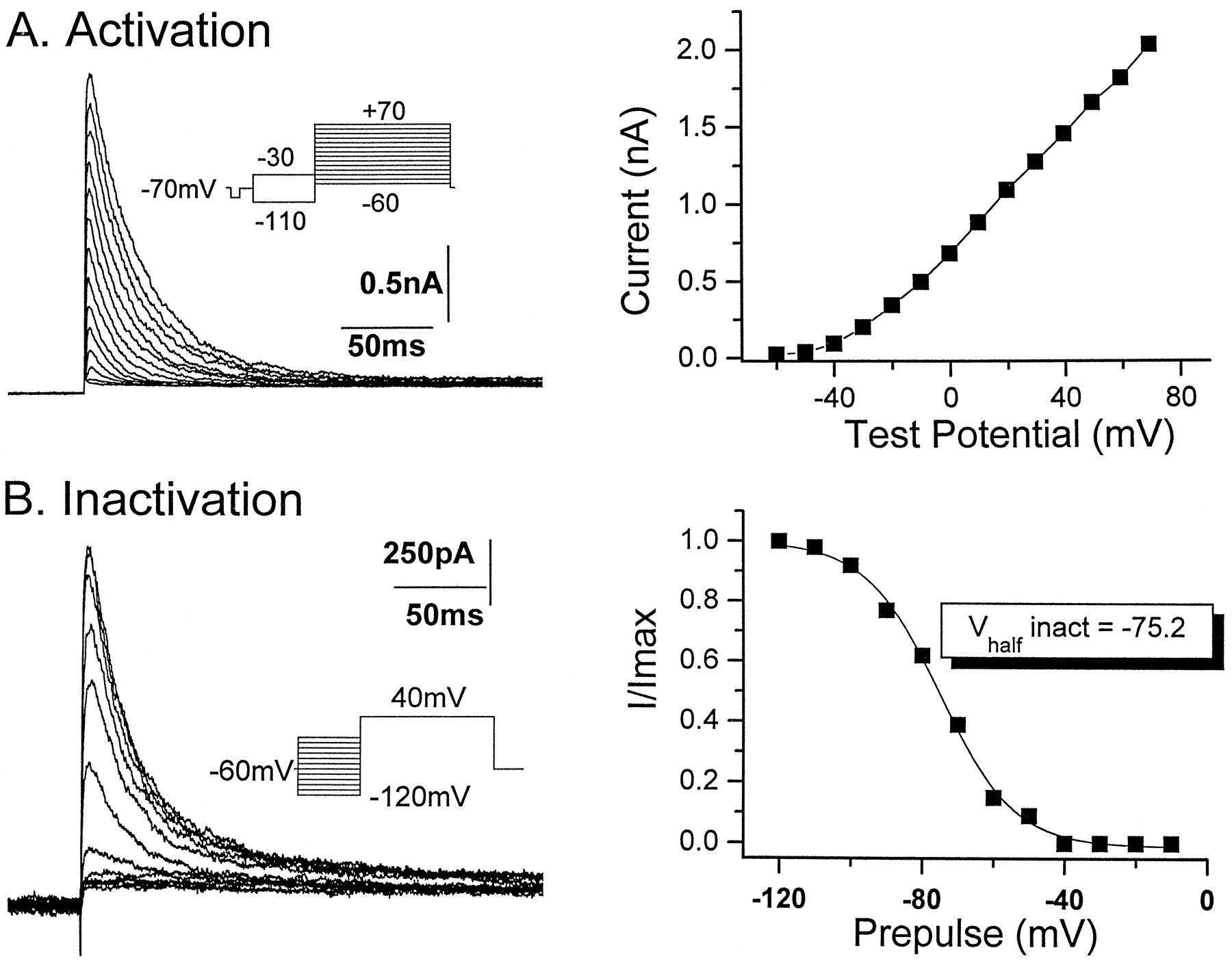
Activation and inactivation properties of CA1 pyramidal neuron transient currents in outside-out patches.A, Transient currents were activated at test potentials positive to −60 mV and up to +70 mV (10 mV increments). Interleaved with each cycle to activate the transient current (prepulse to −110 mV) was a test pulse cycle including a prepulse to −30 mV (seeinset). Inclusion of alternating prepulses to −110 and −30 mV allowed the isolation of the transient current from the total current for each test potential as described in Figure 1. A plot of the current–voltage relationship of the isolated transient current reveals that transient currents were a steep function of the test potential.B, Steady state inactivation of the transient current was determined by the use of a test pulse to +40 mV preceded by a series of prepulse conditioning potentials ranging from −120 to −10 mV. The transient current was completely inactivated at potentials positive to −40 mV. The relative amplitudes of the transient current were plotted as a function of the prepotential and fit with a Boltzmann equation to generate the steady-state inactivation curve (right panel). Half-inactivation of the transient current occurred at a voltage of −75.2 mV.
The transient current is blocked by arachidonic acid
The observation that recombinant channels formed by theShaker subfamily member Kv4.2 were blocked by low concentrations of AA (Villaroel and Schwarz, 1996) and that Kv4.2 was strongly expressed in the hippocampal CA1 subfield (Maletic-Savatic et al., 1995) prompted us to determine whether transient currents expressed in CA1 pyramidal neurons were sensitive to modulation by AA. Application of AA dose dependently attenuated the transient current in outside-out patches obtained from CA1 pyramidal neurons (Fig. 3). In the presence of AA both the peak amplitude and the time constant of inactivation were affected. The effects of AA on the current amplitude varied from patch to patch (range, 7–60% block by 1 μmAA). To obtain a more accurate estimate of the effects of arachidonic acid, we measured changes in the charge transfer associated with the transient current. The transient current charge transfer was obtained by the cumulative integration of the transient current with respect to time (Fig. 3B). Figure 3D shows the dose–response relationship for AA effects. The maximal reduction in the charge transfer was 87.0 ± 0.4% (n = 7) at a concentration of 10 μm arachidonic acid. The IC50 for the AA effect was 0.97 ± 0.03 μm (Fig. 3D). The block of the transient current occurred slowly over 2–3 min and reached steady state by ∼5 min. On removal of AA the current rapidly returned to near control values and did not require bovine serum albumin to facilitate the washout, as was reported for AA block of Kv4.2 expressed in oocytes (Villaroel and Schwarz, 1996). The reasons for the highly variable effect of AA on the current amplitude are at present unclear; it should be pointed out, however, that there was no correlation between the magnitude of current amplitude block and the reduction in the charge transfer.

Arachidonic acid dose dependently blocks the transient current in CA1 pyramidal neurons. A, Both the amplitude and time course of the transient current were attenuated by AA (3 μm). The effects of AA were readily reversible on washing. B, Plot of the cumulative charge transfer associated with the transient current. Integration of the transient current with respect to time allowed the calculation of the charge transfer. The transient current charge transfer was markedly attenuated in the presence of AA. The effect of AA was partially reversed on washing. C, AA dose dependently increased the rate of transient current inactivation. Normalization of the transient currents obtained in the control and in the presence of AA demonstrates an increase in the rate of steady-state inactivation in the presence of AA. The rate of current inactivation in both the control and in the presence of AA were fit by a single exponential function. In the presence of AA, the rate of current inactivation was 2.6 msec compared with 35 msec in the control (dotted line). D,Dose–response relationship of the effect of AA on the transient current charge transfer reveals an IC50 of ∼1 μm. The numbers indicated above each data point reflect the numbers of patches used to construct the points.
The effect of arachidonic acid on the pyramidal neuron transient current was associated with a marked increase in the rate of current inactivation occurring during a prolonged test pulse. Figure3C shows that when the transient current recorded in 3 μm arachidonic acid was scaled to the peak control current, currents in the presence of arachidonic acid inactivated more rapidly than those seen in the control. At concentrations of 1 and 10 μm arachidonic acid, the time constants of current inactivation were 12.0 ± 1.2 (n = 16) and 10.0 ± 1.8 (n = 8) msec, respectively, compared with 23.3 ± 1.2 msec (n = 47) in the control (Vtest = +40 mV). These data contrast with the effects of arachidonic acid on channels formed by recombinant Kv4.2 subunits, in which the effect of arachidonic acid on current amplitude was not associated with an alteration of the inactivation kinetics. Arachidonic acid has been shown previously to accelerate the kinetics of inactivation of currents through channels formed by theShaker subunit; however, these effects of AA were also associated with an enhancement of the peak amplitude (Villaroel and Schwarz, 1996). Arachidonic acid was without effect on the voltage dependence of inactivation. Figure 4 demonstrates that despite a large reduction in the charge transfer associated with the transient current, arachidonic acid did not shift the voltage dependence of inactivation. The mean half-inactivation voltage of transient currents in 10 μm arachidonic acid was −76.6 ± 2.8 (k, 10.7; n = 7) compared with −72.1 ± 1.8 mV (k, 8.8; n = 8) in the control. The effects of arachidonic acid on the voltage dependence of activation could not be studied in detail, because at test potentials of up to +70 mV the transient current did not reach maximal conductance (data not shown) in the majority of cells. In only two patches in which the maximal conductance was observed to saturate were we able to study the effects of AA on the voltage dependence of activation. In these patches the voltage dependence of the transient current was not altered in the presence of AA (1 μm; half-activation voltage, −2.1 mV; k, 19.3; compared with −5.5 mV; k, 19.3 in control) (data not shown).

AA does not alter the voltage dependence of inactivation. Despite increasing the rate of transient current inactivation, AA does not alter the voltage dependence of inactivation. AII, BII, The relative amplitudes of the transient current were plotted as a function of the prepotential and fit with a Boltzmann equation to generate the steady-state inactivation curve (see Fig. 2). In the presence of 10 μm AA, the half-inactivation voltage (−75.8 mV) is similar to that observed in the control (−77.7 mV). All currents were obtained from the same patch.
AA decreases the rate of recovery from inactivation
In striatal neurons, the block of voltage-dependent Na+ currents by AA is associated with a slowing of the rate of recovery from inactivation (Fraser et al., 1993). In the present experiments AA also delayed the rate of recovery of the transient current from inactivation (n = 5). Figure5 demonstrates that in the control, the rate of recovery from inactivation at −110 mV was best fit by a single exponential with a time constant of 13 msec. In the presence of 1 μmAA, the time constant of recovery was 24 msec. In all cells tested, 1 μm AA increased the time constant of the rate of recovery from inactivation at −110 mV by 170 ± 13% (n = 5) over control.

AA decreases the rate of recovery from inactivation. Outside-out patches were voltage clamped at −30 mV to fully inactivate the transient current. A prepulse to −110 mV of increasing duration preceded the test pulse (10 msec increments up to the 100 msec prepulse duration, then 50 msec increments up to 550 msec; the inset shows only 50 msec increments for clarity). A test pulse to +40 mV (200 msec duration) was then made to activate the transient current. A plot of the transient current peak amplitude against the prepulse duration allowed the determination of the rate of recovery from current inactivation. Under control conditions the rate of recovery from steady-state inactivation proceeded with a single exponential of 12.8 msec. In the presence of AA (1 μm) the time constant of the rate of recovery from inactivation increased by 88% to 24 msec.
ETYA mimics the effects of AA
The arachidonic acid-mediated inhibition of transient currents through recombinant Kv4.2 channels (Villaroel and Schwarz, 1996) and transient currents in sympathetic neurons (Villaroel, 1993, 1994) was mimicked by the nonhydrolyzable arachidonic acid analog ETYA. Because ETYA is not a substrate for metabolism by the cyclooxygenase, lipoxygenase, or cytochrome P450 (epoxygenase) pathways associated with arachidonic acid, these data suggested that arachidonic acid interacted directly with Kv4.2 channels without requiring the formation of a metabolic end product. In agreement with the previous data, ETYA had an effect on pyramidal neuron transient currents identical to that of arachidonic acid. ETYA maximally attenuated the transient current charge transfer by 63 ± 9% (n = 7) at a concentration of 10 μm (Fig. 6). ETYA was also without effect on the voltage dependence of inactivation but did increase the rate of steady state inactivation (Fig. 6). In the presence of 10 μm ETYA, the transient current inactivation time constant was 12.1 ± 1.5 msec compared with 21.4 ± 2.7 in the control (n = 7). The effects of ETYA did not require an elevation in [Ca2+]i, because recordings made with pipettes containing the calcium chelator BAPTA (10 mm) did not alter the magnitude of the block observed in the presence of ETYA (data not shown). The similarity of the effects of arachidonic acid and ETYA suggests that the two agents act via an identical mechanism. Furthermore, these data suggest that arachidonic acid has a direct effect on transient channels and does not act by being converted to prostaglandins or by the formation of the metabolites hydroxyeicosatetraenoic acids, epoxyeicosatrienoic acids, or hydroperoxyeicosatetraenoic acids.

ETYA mimics the effects of AA on the transient current. The nonmetabolizable analog of AA, ETYA, mimicked the effects of AA on transient current charge transfer. In the presence of 10 μm ETYA both the amplitude (A) and the charge transfer (B) of the transient current were inhibited. The effects of ETYA were partially reversed on return to control. C, Normalization of the transient current obtained in both the control and 10 μmETYA reveal that the rate of steady-state inactivation is increased in the presence of ETYA, similar to the effects of AA (Fig. 3). Single exponentials adequately described the current inactivation in both the control and 10 μm ETYA (dotted line). The time constants of inactivation in control and ETYA were 16.0 and 7.0 msec, respectively.
AA blocks the transient current in st. oriens–alveus interneurons but not st. pyramidale basket cells
The subpopulation of inhibitory interneurons which reside in the st. oriens–alveus have been well characterized with respect to their morphology (McBain et al., 1994), voltage-dependent K+current properties (Zhang and McBain, 1995a,b), and lack of synaptic plasticity (Maccaferri and McBain, 1995, 1996). Previously we have demonstrated that transient currents recorded from st. oriens–alveus interneurons possess biophysical and pharmacological properties similar to those of CA1 pyramidal neurons, suggesting that similar or related channel subunits may underlie both transient currents (Zhang and McBain, 1995a,b). In support of this hypothesis, AA had an identical effect on the transient current in macropatches excised from the somata of visually identified st. oriens–alveus inhibitory neurons (n = 7; Fig. 7). At a concentration of 1 μm, AA blocked the st. oriens–alveus inhibitory interneuron transient current charge transfer by 47 ± 9.0% (n = 4). Similar to pyramidal neurons, AA had a maximal effect at 10 μm, reducing the charge transfer by 57 ± 1% (n = 4). In all st. oriens–alveus interneurons, AA also increased the rate of current inactivation (Fig. 7). The time constant for current inactivation in the presence of AA (1 μm) was 22.7 ± 2.4 msec (n = 4) compared with to 34.8 + 6.2 msec seen in the control. Where possible, post hoc morphological identification of these cells revealed them to be horizontally oriented interneurons, the axons of which ramify in the strata lacunosum and moleculare (McBain et al., 1994; Maccaferri and McBain, 1995).

The transient current in CA1 st. oriens–alveus interneurons but not basket cell interneurons is blocked by AA.A, An outside-out macropatch excised from a morphologically confirmed interneuron of the stratum oriens–alveus possesses a transient current similarly modulated by AA. At a concentration of 1 μm AA, the charge transfer of the transient current is blocked by 41% (B). In contrast, an outside-out patch excised from the soma of a confirmed CA1 pyramidal cell layer inhibitory interneuron possessed a transient current insensitive to AA (1 μm).
In contrast, a population of morphologically distinct inhibitory interneurons located at the border between the CA1 pyramidal subfield and the st. oriens, the so-called basket cells, were relatively insensitive to AA (n = 9). Figure 7 shows data obtained from one basket cell in which 1 μm AA did not block the transient. At a concentration of 10 μm, the mean block by AA was 17 ± 7% (n = 8) of the transient current charge transfer. Where possible, post hoc morphological identification of these cells confirmed them as typical basket cells with axons that ramified throughout the pyramidal cell layer (Buhl et al., 1994; Du et al., 1996).
AA does not alter the time course of single action potentials
The transient potassium current has been suggested to possess a variety of roles in CNS neurons, including setting the frequency of neuronal discharge by influencing the interspike interval, influencing the threshold for action potential initiation, and contributing to the spike repolarization and afterhyperpolarization (AHP) (for review, see Rogawski, 1985; Halliwell, 1990). Because the transient current is thought to have a role in action potential repolarization in central neurons (Storm, 1987), we next performed whole-cell recordings from CA1 pyramidal neurons under current clamp conditions to determine whether AA block of the transient current influenced the properties of the evoked action potential waveform. Figure 8A demonstrates that although the threshold for action potential initiation was raised 1–2 mV in the presence of both 1 and 10 μm AA, neither concentration of AA altered the action potential waveform. In these experiments, because CA1 pyramidal neurons do not fire spontaneous action potentials, action potentials were initiated by holding the cell at −60 mV, and depolarizing current pulses of sufficient amplitude to trigger action potential activity were delivered through the electrode. AA (10 μm), at a concentration that blocked >80% of the transient current charge transfer in outside-out patches, was without effect on action potential activity recorded in six neurons (Fig. 8). The waveforms of action potentials occurring late in the train similarly were not affected by AA. In addition, AA did not alter the cell membrane potential or input resistance of hippocampal neurons (data not shown). Similar data were obtained using ETYA (10 μm; data not shown). Despite the lack of effect of AA on single spike activity, AA did reduce the frequency of action potential firing in response to a depolarizing current pulse (data not shown). This effect on spike frequency is unlikely to result from a blockade of the transient current, because any reduction of the transient current would be expected to increase the spike discharge frequency. These latter effects and the effects on spike threshold are more probably attributable to an effect of AA on voltage-dependent Na+ channels, similar to that described for striatal neurons (Fraser et al., 1993), or alternatively by activation of the M current (Schweitzer et al., 1990).

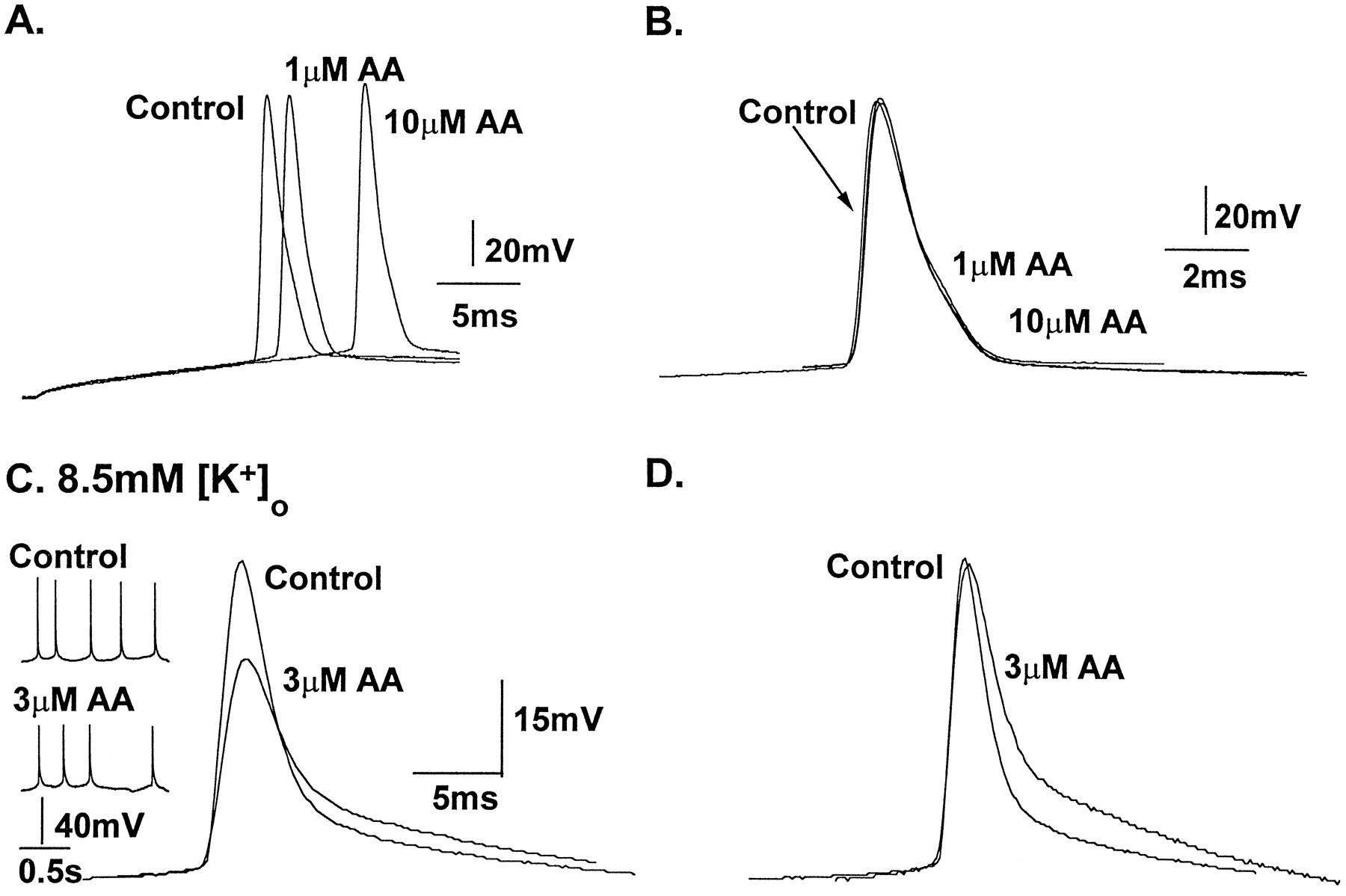
AA does not alter the time course of single action potentials but broadens spikes during electrographic interictal events. A, Under control physiological recording conditions, whole-cell current-clamp recordings were made from CA1 pyramidal neurons. AA (1 and 10 μm) raised the threshold for action potential firing (A) without altering the time course of single action potentials (B). Action potentials were activated by depolarizing pulses delivered from a holding potential of −60 mV. Typically, the action potential threshold occurred close to −45 mV in the control.B, Alignment of the action potential waveforms shown inA clearly shows that AA at concentrations that block 50 and 80% of the transient current in outside-out patches fail to modify the action potential waveform. C, When extracellular K+ was elevated to 8.5 mm, electrographic interictal bursts of action potentials were observed (insets). These action potentials were of a longer duration than seen under control physiological conditions (>10 msec) because of the reduced driving force for K+ current repolarization. Spike duration was prolonged and the amplitude was reduced in the presence of 3 μm AA.D, Normalization of the two action potentials shown inC shows that despite a similar time to peak, the entire repolarization phase of the action potential was prolonged. In the presence of AA, interictal events of similar durations were observed. The number of spikes usually contained in these episodes was reduced (inset). These data suggest that action potentials occurring during electrographic interictal events possess an increased fraction of transient current, which is a target for AA modulation.
The lack of effect of AA on the action potential waveform suggested that either the transient current does not play a role in action potential repolarization or, alternatively, that the duration of a single action potential is insufficient to activate a large enough fraction of transient current to be a target for AA modulation. To determine the transient current component elicited by a single action potential, we used a test pulse waveform designed to mimic the average parameters of single action potentials recorded during current-clamp experiments to activate transient currents in outside-out macropatches (see Fig. 9, inset). The “action potential” test pulse comprised a 1.2 msec depolarizing ramp with a “spike” amplitude of 85 mV (Vhold = Vthreshold = −45 mV; test pulse, +40 mV); the repolarization phase was a 3.8 msec ramp back to −45 mV. The “spike waveform” had an overall duration of 5 msec. Because the inactivation of the transient current is almost complete at resting membrane potentials (Figs. 2 and 4), the test pulse was preceded by a waveform designed to mimic the spike afterhyperpolarization. This waveform comprised a test pulse to −90 mV (Vhold, −60 mV) followed by a ramp to −45 mV (spike initiation threshold) of a duration of 150 msec (see Fig. 9,inset). The inclusion of this waveform permitted the partial removal of transient current inactivation, by an amount equivalent to that removed during an AHP immediately preceding the action potential of interest. Figure 9 demonstrates that in a macropatch containing a transient current of greater than 1 nA in amplitude (Vtest = +40 mV) when the test pulse duration was 200 msec (associated charge transfer of 40 pC; Fig.9AI), the action potential pulse paradigm activated only ∼1% of the total transient current charge transfer available during a single spike of 5 msec duration (Fig.9AII,AIII). Increasing the spike duration by 1 msec increments increased the transient current component (Fig.9AII). A “spike duration” of 10 msec activated 2.5 pC of transient current charge transfer, which corresponded to ∼6.5% of the maximum charge transfer available in this patch. These data suggest that only a modest transient current component is activated during a single action potential waveform (5 msec duration) in CA1 pyramidal neurons.

AA does not block the fraction of transient current underlying short duration “single action potentials” but does inhibit transient currents activated by longer duration spikes. The lack of an effect of AA on action potentials evoked in physiological conditions suggests that the transient current associated with a single spike may deactivate too rapidly for AA modulation. To determine the contribution of the transient current during a single spike, a test pulse paradigm designed to simulate the time course of pyramidal neuron action potentials (inset; see text for details) was used to activate transient currents in an outside-out patch containing a transient current >1 nA in amplitude during a 200 msec duration test pulse (Vtest = +40 mV). A, Current inactivation in the control was fit by a single exponential with a time constant of 23 msec (dotted line). AII,Increasing the duration of the spike test potential from 5 to 10 msec (1 msec increments) recruited an increasing fraction of the transient current (AII). The data shown inAII represent only the transient current component activated during the test pulse to +40 mV (Vhold = −45 mV).AIII, Plots of the charge transfer associated with each transient current activated by increasing action potentials reveals that doubling the duration of the action potential yields a fivefold increase in the transient current component obtained in the patch. Note that even when the duration of the action potential was 10 msec, the transient current activated was <5% of the total current activated by a 200 msec duration test pulse. B, In the same patch, AA (1 μm) blocked 50% of the transient current activated by a 200 msec test pulse to +40 mV and increased the rate of current inactivation (τ = 11 msec;dotted line). BII, BIII, In the presence of AA, short duration action potentials (5–7 msec) activated a similar fraction of transient current charge transfer to that seen in control. In contrast, transient currents activated by longer duration action potentials were subject to modulation by AA. The transient current charge transfer associated with a 10 msec action potential was reduced by 24% compared with control. These data are consistent with the data illustrated in Figure 8, which showed that AA was without effect during short duration action potentials but increased the duration of the broader action potentials recorded during electrographic interictal events.
Addition of AA (1 μm) to this macropatch reduced the transient current amplitude activated during a 200 msec duration test pulse (Vtest = +40 mV) and blocked the associated charge transfer by 50% (Fig. 9BI). In the presence of AA, however, a similar fraction of the transient current charge transfer was activated during a 5 msec action potential to that seen in the control, i.e., 0.4 pC (Fig. 9BII,BIII). Only when spike waveforms of greater durations (>8 msec) were delivered was an effect of AA observed. Figure 9BIIIshows that when the spike duration was increased to 10 msec, AA reduced the associated charge transfer by 24%. Similar results were observed in patches excised from five other CA1 pyramidal neurons. The apparent lack of an effect of AA on shorter duration action potentials results presumably because the transient current is forced to deactivate soon after the depolarizing test pulse. This rapid deactivation of the channel will prevent the occurrence of significant steady state inactivation, one of the processes that would seem to be a major target of the effects of AA. The lack of effect of AA on transient currents activated during shorter duration simulated action potentials is consistent with the lack of effect of AA on action potentials recorded during current-clamp experiments described above (Fig. 8).
The results shown in Figure 9 suggest that AA will only affect action potentials when the spike duration is long enough to activate an appreciable fraction of the transient current charge transfer. Action potentials of >10 msec duration are not normally observed under physiological conditions but readily occur during electrographic interictal events observed in the “high-[K+]o” model of epilepsy (for review, see McBain et al., 1993). In this model extracellular K+ is elevated to 8.5 mm (from 3.5 mm), a manipulation that reliably induces stereotypical electrographic seizure activity in CA1 pyramidal neurons, attributable in part to the reduction in the K+-driving force, impaired GABA-mediated inhibition, enhanced NMDA receptor-mediated activation, and glial swelling (see McBain et al., 1993, for a more detailed description of the elevated [K+]o model). Interictal activity in CA1 pyramidal neurons comprises a short paroxysmal depolarizing shift superimposed on which is a brief discharge of three to five action potentials (see Fig. 8B, inset). Action potential durations recorded during these events are significantly longer than those seen under control conditions of 3.5 mm [K+]o (Fig.8B) (see also McBain, 1994). The voltage-clamp data shown in Figure 9 predict that the longer duration action potentials (10–15 msec) routinely observed during interictal electrographic events (see Fig. 8B, inset) would possess an appreciable transient current component for AA modulation. Figure 8B demonstrates that in the presence of 8.5 mm K+ and 3 μm AA, spontaneous action potential duration was increased 148% over the control (mean duration, 23.5 ± 0.8 vs 15.9 ± 1.5 msec seen in 8.5 mm [K+]o only;n = 4). Likewise, the time to 50% repolarization was increased by 125% in all neurons tested (5.6 ± 0.2 vs 4.5 ± 0.3 msec in control; n = 4). In the presence of AA (3 μm) the spike amplitude was also attenuated (50.3 ± 4.9 vs 69.0 ± 1.6 mV in control); however, the action potential time to peak remained unchanged. In the continued presence of AA, action potential and interictal activity increased in frequency and then abruptly ceased after 10–15 min. This cessation of action potential activity was not associated with a change in the membrane potential or input resistance. Action potentials could be induced by direct depolarization through the electrode only if a large hyperpolarization of the membrane potential preceded the depolarizing stimuli (data not shown). These effects were only partially reversible on washing. These data confirm the prediction that action potentials of longer duration would be a target for AA modulation and suggest that AA released during pathological but not physiological conditions will strongly influence action potential activity.
DISCUSSION
The principal findings of this study are: (1) arachidonic acid inhibited the transient current in hippocampal CA1 pyramidal neurons and st. oriens–alveus inhibitory interneurons; (2) the block of the transient current by AA was associated with an increased rate of current inactivation and prolongation of the rate of recovery from inactivation. AA did not, however, affect the voltage dependence of inactivation. (3) The effects of AA were mimicked by the nonmetabolizable analog ETYA and suggest that the effect of AA on transient current channels is direct and does not involve arachidonate metabolism; and (4) AA was without an effect on action potentials recorded under physiological conditions but did prolong action potentials recorded during electrographic interictal episodes. Consistent with this observation was the finding that transient currents activated by test pulses designed to simulate single action potential waveforms were not a target for AA modulation until the action potential waveform was of a sufficient duration to activate a larger transient current component.
The block of the hippocampal transient current by AA extends the already considerable list of voltage- and ligand-gated ion channels that are targets for AA or AA metabolite modulation (for review, see Meves, 1994). In the present experiments, several lines of evidence suggest that the effects of AA on the hippocampal transient currents were direct: (1) The use of outside-out patches excised from the somata of pyramidal neurons will dialyze out any cytoplasmic constituents rapidly, minimizing the production of metabolic end products of AA. (2) The similar effect of the nonmetabolizable analog ETYA supports the hypothesis of a direct effect. ETYA is not a substrate for metabolic pathways involving cyclooxygenase, lipoxygenase, and epoxygenase and inhibits many of these enzymes. We cannot, however, unequivocally rule out the possibility that both ETYA and AA are metabolized into more potent metabolites. (3) The effects of AA were readily reversible on washout of AA, arguing against the participation of a phosphorylation (or dephosphorylation) process, because reversal of the AA effect would require the action of a phosphatase (or a kinase). (4) Finally, the lack of action of intracellularly applied BAPTA rules out a requirement for an elevation of intracellular Ca2+ for the effects of AA.
The similarity between the effects of AA described here and those of AA on channels formed from recombinant Kv4.2 (Villaroel and Schwarz, 1996) suggest that the major transient current component present on cell bodies of pyramidal neurons occurs through channels comprising Kv4.2 subunits. Consistent with this hypothesis is the strong expression of the Kv4.2 subunit protein in CA1 pyramidal neuron somata and dendrites (Sheng et al., 1992; Maletic-Savatic et al., 1995). In the present experiments, the reduction of the charge transfer by AA was associated with a increase in the rate of current inactivation and a slowing of the recovery from inactivation. In contrast, AA did not alter the rate of current inactivation in recombinant Kv4.2 channels. This suggests that channels responsible for transient currents in hippocampal neurons may comprise additional subunits that possess additional sites for AA modulation. In support of this hypothesis, a recent study by Serodio et al. (1994)suggested that native channels responsible for the transient current are probably assembled from Kv4.2 subunits and an as yet unidentified low molecular weight accessory protein (Serodio et al., 1994).
The precise mechanism for the effects of AA on the transient current is at present unknown. The faster rate of current inactivation observed in AA suggests, however, an action on the inactivation process. Mutagenesis experiments on Kv4.2 channels have shown that the intracellular S4–S5 linker, which has been proposed to act as the receptor site for the N-terminal inactivation ball peptide, participates in AA inhibition (Villaroel and Schwarz, 1996). Access to an intracellular binding site limited by diffusion across the lipid bilayer would explain the time required for AA effects to reach steady state.
The present study demonstrated that currents through channels excised from st. oriens–alveus interneurons were also targets for modulation by AA. Although the Kv4.2 protein is strongly expressed in principal neurons of the CA1 hippocampus, it is largely absent from inhibitory interneurons in the st. oriens–alveus (Maletic-Savatic et al., 1995). It would seem unlikely, therefore, that modulation of Kv4.2 channels underlies the effects seen in st. oriens–alveus interneurons. Consistent with this observation was the finding that transient currents in st. oriens–alveus interneurons had a time course of steady state inactivation slower than pyramidal neurons and were significantly less sensitive to AA than those of pyramidal neurons (57 vs 87% maximal block). It is possible that other members of the Kv4 subfamily (Kv4.1 and Kv4.3) are expressed in these cells, although Kv4.1 expression is low in the hippocampus. Recently, a new member of this subfamily was cloned, Kv4.3, which is strongly expressed in the hippocampus (Serodio et al., 1996). Expression of this subunit is highest in interneurons of the CA1 st. oriens–alveus (P. Serodio and B. Rudy, unpublished observation) and is largely absent from CA1 pyramidal neurons. Expression of recombinant Kv4.3 subunits, such as Kv4.1 and Kv4.2, also results in the formation of channels carrying a transient current phenotype (Serodio et al., 1996). Recovery from steady state inactivation occurs at a slower rate in Kv4.3 than in Kv4.2 channels (Serodio et al., 1996), consistent with the slower rate of recovery seen in st. oriens–alveus–lacunosum-moleculare interneurons (Zhang and McBain, 1995a) compared with pyramidal neurons (Fig. 5). Whether channels formed by these subunits underlie the transient current seen in st. oriens–alveus interneurons or whether currents through homomeric Kv4.3 channels are subject to modulation by AA remains to be tested.
The concentrations of AA used in the present experiments are within the physiological range for modulation by AA (Anderson and Welsh, 1990; Meves, 1994). Concentrations of exogenous AA in excess of 10 μm are generally required to mimic the actions of neurotransmitter-induced AA effects. In addition, theKm values of cyclooxygenase and 12-lipoxygenase for AA are both 5 μm (Needleman et al., 1986). In CNS neurons, various neurotransmitters act to release AA by a mechanism involving phospholipase A2 or through the combined action of phospholipase C and diacylglycerol lipase (Axelrod, 1990). How might AA be released in the hippocampal formation? In the hippocampus, a variety of principal neurotransmitters are candidates for activation of the AA induction cascade. Muscarinic receptor activation has been shown to induce release of AA (Kanterman, 1990) and muscarinic agonists block transient potassium currents (Nakajima et al., 1986). Activation of hippocampal somatostatin receptors has been shown to liberate AA (Bito et al., 1993), although at present it is unknown whether somatostatin modulates transient potassium currents. Both metabotropic glutamate receptors (mGluR) and NMDA-preferring glutamate receptor activation induce the AA cascade system (Dumuis et al., 1990a,b), and NMDA receptor currents have also been shown to be potentiated by AA (Miller et al., 1992). A role for AA in the induction phase of hippocampal long-term potentiation (LTP) has been demonstrated (Williams et al., 1989; Drapeau et al., 1990), suggesting that AA may be liberated during the high-frequency tetanic stimulation used to induce LTP. AA production has been associated with ischemic cell damage in CNS neurons (Madden et al., 1986) and seizure generation (for review see Bazan et al., 1986), conditions associated with increased neuronal activity. Both NMDA receptor and mGluR activation are intimately associated with the generation of electrographic seizures in a variety of epilepsy models (for review, see Schwartzkroin, 1993), including the High-K+ model used in the present experiments (Traynelis and Dingledine, 1988; McBain et al., 1993; McBain, 1994). It is entirely plausible, therefore, that AA may be liberated during electrographic seizure activity as a consequence of either mGluR or NMDA receptor activation. Because action potential durations are prolonged during electrographic interictal activity, AA may act on transient current channels to broaden the action potential waveform further, consequently enhancing Ca2+ influx into synaptic terminals, resulting in a greater release of neurotransmitter. Consistent with this hypothesis, glutamate release from hippocampal mossy fiber terminals has been demonstrated to be facilitated by AA (Freeman et al., 1990). Synaptically released glutamate may then activate an increased number of mGluR or NMDA receptors, leading to a self-sustaining cycle. In the present experiments, however, the continued presence of AA led to the cessation of all electrographic activity and the termination of action potential firing, suggesting that prolonged AA exposure ultimately prevents action potential activity presumably by altering the activity of a variety of ion channels in hippocampal neurons. These effects may arise in part from the action of AA on both voltage-dependent Na+channels (Fraser et al., 1993) and/or activation of the M current (Schweitzer et al., 1990). An understanding of the modulation of the currents responsible for these combined cellular mechanisms, together with the action of AA on the transient current, will ultimately provide insight into the total functional significance of AA modulation of hippocampal neurons.
In conclusion, these data suggest that arachidonic acid modulates the transient potassium current in both CA1 pyramidal neurons and st. oriens interneurons by a mechanism involving an increase in the inactivation rate and a slowing of the recovery from inactivation. Current-clamp experiments suggest that the transient current normally associated with single action potentials in normal physiological conditions would not be a target for AA modulation. In contrast, the longer duration action potentials occurring during electrographic seizure activity are strongly modulated by AA, suggesting that AA liberated during intense neuronal activity may act to exacerbate pathological conditions such as seizure and ischemic cell damage.
Footnotes
S.K. was a National Institute of Child Health and Human Development pre-Intramural Research Training Award (IRTA) fellow. We thank Drs. Mark Mayer, Gianmaria Maccaferri, and Vittorio Gallo for their constructive criticism of this manuscript.
Correspondence should be addressed to Chris J. McBain, Laboratory of Cellular and Molecular Neurophysiology, Room 5A72, Building 49, National Institute of Child Health and Human Development, National Institutes of Health, 49 Convent Drive, Bethesda, MD 20892-4495.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}