

Abstract
The high-molecular-mass neurofilament subunit (NFH) is normally hypophosphorylated in the neuronal perikaryon and undergoes extensive phosphorylation after entering the initial axon segment. Aberrant hyperphosphorylation of perikaryal NFH is a common feature of many neurological diseases. In a previous study (Giasson and Mushynski, 1996), we demonstrated a correlation between phosphorylation of perikaryal NFH and induction of stress-activated protein kinase (SAPK)-γ. In this report, we present direct evidence showing that thein vivo activation of SAPKs by an upstream activator (MEKK-1) caused extensive NFH phosphorylation. We also show that stress-activated p38 kinases were not involved in the phosphorylation of perikaryal NFH in cultured dorsal root ganglion neurons and that this process was reversible. SAPKγ was shown to be located in both the cell body and the neurites of the cultured neurons, suggesting that it is likely to be involved in the phosphorylation of cytoplasmic substrates. These could include neuritic NFH, which is highly phosphorylated despite the demonstrated lack of cyclin-dependent kinase-5 activity in these neurons. Neuritic NFH was also highly phosphorylated in neuronal cultures devoid of Schwann cells, indicating that this form of post-translational modification does not require cues stemming from Schwann cell–axon contacts. Collectively, these findings provide significant new insights into mechanisms involved in NFH phosphorylation in normal neurons and in disease states characterized by aberrant phosphorylation of neurofilaments.
- cyclin-dependent kinase-5
- heavy neurofilament-subunit
- p38-kinases
- phosphorylation
- stress-activated kinases
Neurofilaments (NFs) are the principal intermediate filaments (IFs) found in many types of mature neurons. They are the most abundant structure in large myelinated axons (Hoffman et al., 1984) and are an important determinant of axonal caliber (Yamasaki et al., 1991; Ohara et al., 1993; Eyer and Peterson, 1994). NFs are composed of three proteins, the low (NFL)-, mid-sized (NFM)-, and heavy (NFH)-molecular-mass subunits (Hoffman and Lasek, 1975). In common with other IF proteins, each NF subunit contains a highly conserved α-helical rod domain, involved in dimer formation, flanked by an N-terminal head domain and a C-terminal tail domain (Fuchs and Weber, 1994).
NFH from myelinated axons is highly phosphorylated in vivo(Julien and Mushynski, 1982), predominantly at Lys-Ser-Pro (KSP) repeats in the tail domain (Julien and Mushynski, 1983; Lee et al., 1988; Elhanany et al., 1994). The role of NFH tail domain phosphorylation is not fully understood, although it has been shown to inhibit interaction between NFH and microtubules (Hisanaga et al., 1991, 1993a,b; Miyasaka et al., 1993) and to protect NFH from proteolysis (Goldstein et al., 1987; Pant, 1988). It may also regulate the distribution of NFs between stationary and mobile phases in the axon (Lewis and Nixon, 1988).
The use of monoclonal antibodies that could distinguish between phosphorylated and unphosphorylated epitopes in the tail domain of NFH has shown that axonal NFH is normally more highly phosphorylated than that located in the cell body and dendrites (Sternberger and Sternberger, 1983; Lee et al., 1987). Perikaryal NFH is maintained in a hypophosphorylated state with Mr 160 kDa on SDS-PAGE compared with a value of 200 kDa for axonal NFH (Glicksman et al., 1987; Oblinger, 1987; Nixon et al., 1989). The gel electrophoretic mobility of axonal NFH increases to that of perikaryal NFH after dephosphorylation of the tail domain (Julien and Mushynski, 1982;Carden et al., 1985), and this shift is reversed by phosphorylation at KSP repeats (Hisanaga et al., 1991, 1993b; Miyasaka et al., 1993). Of the neuronal proline-directed protein kinases that can phosphorylate NFH, only tau protein kinase II/cyclin-dependent kinase-5 (cdk-5) has been shown unequivocally to cause a reduction in its mobility on SDS-PAGE to levels seen for axonal NFH (Hisanaga et al., 1993b;Kobayashi et al., 1993; Miyasaka et al., 1993; Guidato, 1996a; Sun et al., 1996).
Perikaryal NFH is highly phosphorylated in many neurodegenerative diseases, such as Alzheimer’s disease (Cork et al., 1986; Zhang et al., 1989), Parkinson’s disease (Forno et al., 1986; Pollanen et al., 1994), and amyotrophic lateral sclerosis (ALS) (Manetto et al., 1988;Munoz et al., 1988; Sobue et al., 1990). We previously presented correlative evidence indicating that stress-activated protein kinase-γ (SAPKγ) could be responsible for the aberrant phosphorylation of perikaryal NFH (Giasson and Mushynski, 1996). SAPKs are proline-directed kinases belonging to the mitogen-activated protein (MAP) kinase family, which also includes extracellular signal-regulated kinases (ERKs), p38 kinases (Cano and Mahadevan, 1995; Kyriakis and Avruch, 1996), and a novel member, SAPK-3 (Mertens et al., 1996). The MAP kinases are related structurally and are activated by similar cascades in response to diverse stimuli (Cano and Mahadevan, 1995;Kyriakis and Avruch, 1996).
In this report, we present direct evidence that the in vivoactivation of SAPKs by constitutively active MAP kinase/ERK kinase kinase-1 (MEKK-1) induces phosphorylation of the NFH tail domain. We also show that p38 kinases are not involved in the hyperphosphorylation of perikaryal NFH and that this process is completely reversible. These findings provide basic information that enhances our understanding of mechanisms causing aberrant NF phosphorylation in neurological diseases.
MATERIALS AND METHODS
Materials. Nerve growth factor (NGF) (2.5S) was purchased from Prince Laboratories (Toronto, Ontario, Canada). Anti-NF antibodies SMI 31 and SMI 34 were obtained from Sternberger Monoclonals (Baltimore, MD). Anti-SAPKγ (C-17), anti-ERK-1 (C-16), anti-ERK-2 (C-14), anti-p38α (C-20), anti-cdk-5 (C-8) polyclonal antibodies, anti-cdk-5 (DC17) monoclonal antibody, and glutathioneS-transferase (GST)-cJun (amino acids 1–79) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Histone H1 was obtained from Life Technologies (Gaithersburg, MD). The pRC/CMV eukaryotic expression vector was purchased from Invitrogen (San Diego, CA). Anti-NFH (N52) and anti-NFL (NR4) monoclonal antibodies were from Sigma (St. Louis, MO). Polyclonal anti-vimentin antibody andN-acetyl-Leu-Leu-norleucinal (CI) were from ICN (Mississauga, Ontario, Canada). SB 203580 was generously provided by SmithKline Beecham.
Cell culture. Embryonic day 15–16 dorsal root ganglia (DRGs) were dissected, dissociated, and maintained in culture as described previously (Giasson and Mushynski, 1996). To allow for the manual separation of cell bodies from neurites, the dissociated DRGs were plated in a small area at the center of a 35 mm culture dish. Neurites extended radially to form a halo surrounding the cell body mass. For cultures treated with antimitotic agents, the cells were cycled among 10−5m5-fluoro-2′-deoxyuridine, 10−6mcytosine β-d-arabino-furanoside and 5 × 10−6m 5-fluoro-2′-deoxyuridine, 5 × 10−7m cytosine β-d-arabino-furanoside every 4 d for 16 d, starting 24 hr after plating.
NIH 3T3 cells were obtained from the American Type Culture Collection (Rockville, MD) and cultured in 85% DMEM (high glucose), 10% heat-inactivated horse serum, 5% fetal bovine serum (Life Technologies), and antibiotics. The cells were transfected using lipofectamine reagent (Life Technologies) according to the manufacturer’s instructions.
Immunoprecipitation kinase assays. SAPKγ activity was assayed as described previously (Giasson and Mushynski, 1996). Briefly, after cell lysis in the presence of Triton X-100, cell debris was removed by centrifugation at 13,000 × g and the protein concentration of each supernatant was determined to equalize the amount of protein used in each immunoprecipitation. SAPKγ was immunoprecipitated, the immunoprecipitates were washed extensively, and activity was assayed using [γ-32P]ATP and GST-cJun as a substrate. Phosphorylation of GST-cJun was visualized after SDS-PAGE (Laemmli, 1970) by autoradiography of dried gels and quantified using a Fujix BAS2000 Bio-Imaging Analyzer (Fuji Bio-Imaging).
Cdk-5 activity was assayed by immunoprecipitation kinase assay as described previously (Tsai et al., 1993) using an anti-cdk-5 polyclonal antibody (C-8) and histone H1 as the substrate. Visualization of the phosphorylated substrate was achieved as described for SAPKγ.
Gel electrophoresis and Western blot analysis. Cells were harvested in PBS (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, and 1.8 mm KH2PO4) and lysed in 2% SDS and 62.5 mm Tris, pH 6.8, and protein concentration was determined using the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). Glycerol and β-mercaptoethanol were added to concentrations of 10 and 5%, respectively. The cell extracts were diluted to the appropriate concentrations with SDS-sample buffer (2% SDS, 62.5 mm Tris, pH 6.8, 10% glycerol, and 5% β-mercaptoethanol), and the proteins were resolved on slab gels by SDS-PAGE (Laemmli, 1970). Proteins were transferred electrophoretically to Immobilon-P membrane (Millipore, Bedford, MA) in buffer containing 48 mm Tris, 39 mm glycine, and 5% methanol. The membranes were blocked with 1% skimmed milk powder in Tris-buffered saline/Tween (20 mm Tris, pH 7.7, 137 mm NaCl, and 0.1% Tween 20), incubated with primary antibodies, and developed using the ECL Western Blotting Detection Kit (Amersham).
RESULTS
Transfection of cells with MEKK-1Δ induces NFH tail domain phosphorylation
MEKK-1Δ, a constitutively active form of MEKK-1 that serves as an activator of the SAPK cascade (Minden et al., 1994; Yan et al., 1994; Xu et al., 1995), was tested for its ability to induce NFH phosphorylation in vivo. NIH 3T3 cells transfected with the expression vector pRC/CMV alone did not express NFH (Fig.1A, lane 1). In extracts from cells transfected with the expression vector containing the mouse NFH gene (Julien et al., 1988) beginning 15 nucleotides upstream from the translational start site, NFH was detected with N52 antibody as a predominantly hypophosphorylated isoform(s), judging from its mobility on SDS-PAGE and from its failure to bind monoclonal antibodies SMI 31 or SMI 34 (Fig.1A, lane 2). Monoclonal antibody N52 can detect both hypo- and hyperphosphorylated forms of NFH (Shaw et al., 1986), although the relevant epitope can be blocked by cdk-5 phosphorylation (Guidato et al., 1996b). SMI 31 and SMI 34 are both phosphorylation-dependent monoclonal antibodies that react with different epitopes in the tail domain of NFH (Sternberger and Sternberger, 1983; Lee et al., 1988; Shea and Beermann, 1993). Cotransfection of NIH 3T3 cells with pRC/CMV vectors expressing NFH and MEKK-1Δ yielded hyperphosphorylated NFH, as determined by its reduced mobility on SDS-PAGE and by its reactivity with both SMI 31 and SMI 34 (Fig. 1A, lane 3). The expression of MEKK-1Δ also resulted in the activation of SAPKγ (Fig.1B).
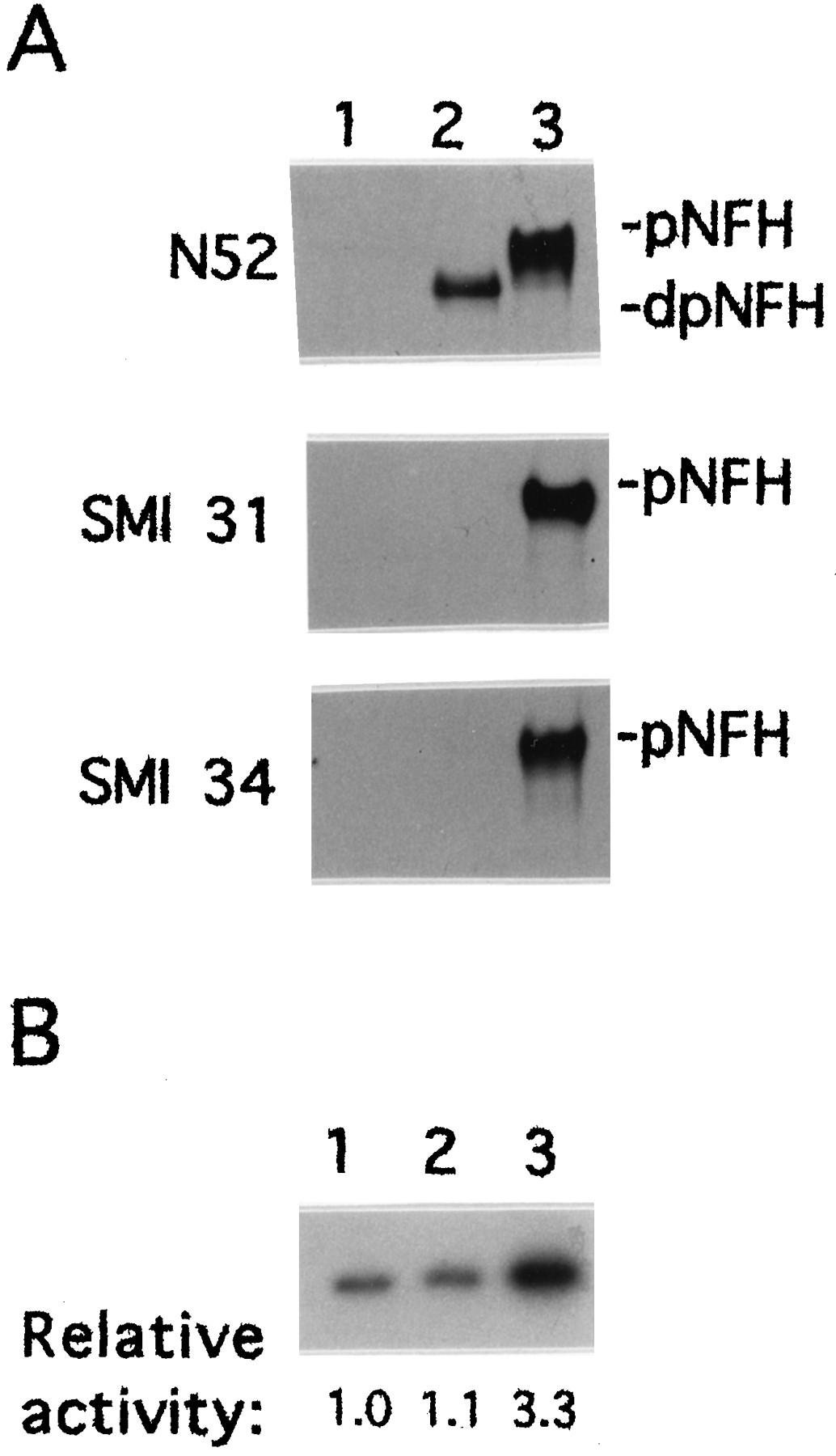
Transient transfection with constitutively active MEKK-1Δ induces NFH tail domain phosphorylation. NIH 3T3 cells were transfected with the pRC/CMV eukaryotic expression vector (lane 1), with the mouse NFH gene cloned into the pRC/CMV vector (lane 2) or both the mouse NFH gene and the MEKK-1Δ cDNA, each cloned into pRC/CMV (lane 3).A, NFH was detected by Western blot analysis using monoclonal antibodies N52, SMI 31, or SMI 34. pNFH anddpNFH refer to hyper- and hypophosphorylated NFH, respectively. Equal amounts of protein were loaded in each lane.B, The activity of SAPKγ was determined by immunoprecipitation kinase assays as described in Materials and Methods. 32P-phosphorylation of GST-cJun was visualized by autoradiography and quantified by image analysis. The relative activity of the immunoprecipitated kinase is indicate below each lane.
P38 kinases are not involved in stress-induced NFH phosphorylation
Proline-directed p38 kinases are often activated simultaneously with SAPKs (Cano and Mahadevan, 1995; Raingeaud et al., 1995). To test whether p38 kinases are also involved in the hyperphosphorylation of perikaryal NFH, we used a specific inhibitor, SB 203580 (IC50 0.6 μm), which does not inhibit SAPKs (Cuenda et al., 1995). Cultured DRG neurons were treated with 30 μm CI, a calpain (Saito and Nixon, 1993) and proteasome inhibitor (Tsubuki et al., 1993; Rock et al., 1994), which has been shown to activate SAPK and induce hyperphosphorylation of perikaryal NFH (Giasson and Mushynski, 1996) (Fig.2, lane 2). The addition of 20 μm SB 203580 had no effect on the CI-induced reduction in mobility, and therefore phosphorylation, of NFH (Fig. 2, lane 3).

P38 kinases are not involved in perikaryal NFH hyperphosphorylation. Localized DRG cultures were prepared as described in Materials and Methods. The cultures were maintained for 20 d (lane 1, control) and treated with 30 μmCI for 10 hr (lane 2). A culture was pretreated with 20 μm SB 203580 for 2 hr before the addition of 30 μm CI for 10 hr (lane 3). The neuronal cell bodies were manually separated from the neurites and subjected to Western blot analysis using the anti-NFH monoclonal antibody N52.pNFH and dpNFH refer to hyper- and hypophosphorylated NFH, respectively.
Distribution of MAP kinases in DRG neurons
The distribution of MAP kinases within DRG neurons was assessed by Western blot analysis as shown in Figure3. DRG cultures maintained in medium containing antimitotic agents were fractionated into neurite (lane 1)- and cell body (lane 2)-enriched fractions as described in Materials and Methods. The antimitotic agents eliminated all of the Schwann cells normally found in DRG cultures and prevented the proliferation of fibroblasts. However, the cultures still contained a population of quiescent fibroblasts resistant to antimitotic treatment. To compensate for contamination by these fibroblasts, we prepared DRG cultures treated with antimitotic agents and maintained without NGF to eliminate neurons (Giasson and Mushynski, 1997). Lane 3 in Figure 3 was loaded with an amount of protein from fibroblast cultures equal to that for neurite (lane 1) and cell body (lane 2)-enriched fractions. Lanes 4–6 were loaded, respectively, with two-, four-, and eightfold less fibroblast protein than lane 3. The inclusion of lanes 3–6 allowed us to determine whether the proteins detected in lanes 1 and 2 were neuronal in origin or from contaminating fibroblasts. Vimentin and NFL were used as specific markers for fibroblasts and DRG neurons, respectively. There were equivalent amounts of NFL in the neuronal cell body- and neurite fractions, and NFL was not detected in cultures maintained without NGF. Two other DRG neuronal markers, peripherin and α-internexin (Athlan et al., 1997), also were not detected in the fibroblast cultures (data not shown). There were approximately equal levels of fibroblast contamination in the neuronal cell body and neurite fractions as determined by their vimentin content, and these fractions contained <12% fibroblast protein. P38α was expressed at low levels in DRG neurons and only in the cell body fraction. ERK-1/-2 and SAPKγ were equally distributed between the cell body and neurite fractions.

SAPKγ and ERKs are located in both perikaryon and neurites of DRG neurons. DRG neurons were maintained in culture in the presence of antimitotic agents as described in Material and Methods. The cultures were separated into neurite (lane 1)- and cell body (lane 2)-enriched fractions. Protein extracts from DRG cultures maintained with antimitotic agents and without NGF were loaded in lanes 3–6. Lanes 1–3were loaded with 5 μg of protein, whereas lanes 4–6 were loaded with 2.5 μg, 1.25 μg, and 0.62 μg of protein, respectively. The proteins were detected by Western blot analysis. NFL, Vim,SAPKγ, ERK1, ERK2, andp38 refer to the low-molecular-mass neurofilament subunit, vimentin, SAPKγ, ERK-1, ERK-2, and p38α kinase, respectively.
The hyperphosphorylation of perikaryal NFH is reversible
Cultured DRG neurons were treated with 30 μm CI to induce the hyperphosphorylation of perikaryal NFH (Giasson and Mushynski, 1996), as reflected in its reduced mobility on SDS-PAGE (Fig. 4, lane 2). After removal of CI from the culture medium, perikaryal NFH was seen to undergo progressive dephosphorylation. Approximately half of the protein had returned to its normal mobility on SDS-PAGE within 2 d (Fig. 4, lane 3); by 4 d, almost all of the NFH had return to a normal hypophosphorylated state (Fig. 4, lane 4).

Aberrant phosphorylation of perikaryal NFH is reversible. Localized DRG cultures were prepared as described in Materials and Methods. The cultures were maintained for 20 d (lane 1) and treated with 30 μm CI for 10 hr (lanes 2–6). After treatment with CI, the cultures were maintained in CI-free medium for 2 d (lane 3), 4 d (lane 4), 6 d (lane 5), and 8 d (lane 6). The neuronal cell bodies were manually separated from the neurites and subjected to Western blot analysis using the anti-NFH monoclonal antibody N52. pNFH and dpNFH refer to hyper- and hypophosphorylated NFH, respectively.
Axonal NFH in DRG neurons is hyperphosphorylated despite the inactivity of cdk-5
The Western blots in Figure5A show that most of the NFH in the neuronal cell body-enriched fraction was hypophosphorylated, whereas that in the neurite-enriched fraction was mostly hyperphosphorylated. The small amount of hypophosphorylated NFH in the neurite-enriched fraction originates from neuronal cell bodies localized outside of the circumference of the circular punch used to separate the two neuronal compartments. The hyperphosphorylated NFH in cell body-enriched extracts derives from the initial segment of neurites and from neurites criss-crossing the area occupied by the cell body mass. The slowly migrating, highly phosphorylated isoforms of NFH reacted with both phosphorylation-dependent antibodies, SMI 31 and SMI 34. Therefore, NFH in cultured DRG neurons demonstrated the normal phosphorylation pattern (Sternberger and Sternberger, 1983; Glicksman et al., 1987; Lee et al., 1987; Oblinger, 1987; Nixon et al., 1989), which was also observed in DRG cultures treated with antimitotic agents and devoid of Schwann cells (Fig. 5B).

Distribution of phosphorylated NFH isoforms in DRG neurons. Localized DRG cultures were prepared as described in Materials and Methods and separated into cell body (C)- and neurite (N)-enriched fractions. The two subcellular fractions were subjected to Western blot analysis using the anti-NFH monoclonal antibodies N52, SMI 31, and SMI 34.pNFH and dpNFH refer to hyper- and hypophosphorylated NFH, respectively. A, DRG cultures were maintained for 20 d. Cell body- and neurite-enriched fractions were harvested in equal volumes of SDS-sample buffer, and the same volume was loaded in each lane. B, DRG cultures were maintained in the presence of antimitotic agents as described in Materials and Methods. Cell body- and neurite-enriched fractions were lysed in 2% SDS and 62.5 mm Tris, pH 6.8, protein concentrations were determined, and equal amounts of protein were loaded in each lane.
The activity of cdk-5 in cultured DRG neurons was determined by immunoprecipitation kinase assays using rat brain extract as a positive control (Fig. 6A,lanes 1–6) (Tsai et al., 1993). Because the relative amounts of immunoprecipitable cdk-5 in brain extract compared with extract from DRG cultures were unknown, different amounts of rat brain extract were used in the immunoprecipitation kinase assays, and levels of immunoprecipitated cdk-5 were determined by Western blot analysis (Fig. 6B). Despite the fact that comparable amounts of cdk-5 were immunoprecipitated from 200 μg of DRG extract and 100 μg of brain extract (Fig. 6B, lanes 4, 8), no histone H1-phosphorylating activity was detected in DRG samples (Fig. 6A, lane 8). Western blot analysis of total protein extracts from rat brain and DRG cultures revealed that, on an equal protein basis, rat brain contained approximately twice as much cdk-5 as did the DRG cultures (data not shown).

Analysis of anti-cdk-5-immunoprecipitable histone H1 kinase activity from DRG cultures and adult rat brain.A, Fifty micrograms (lanes 1,2), 100 μg (lanes 3, 4), and 200 μg (lanes 5, 6) of protein from brain and 200 μg of protein from DRG cultures maintained for 20 d (lanes 7, 8) were immunoprecipitated with nonimmune serum (lanes 1, 3, 5, 7) and anti-cdk-5 (C-8) polyclonal antibody (lanes 2, 4, 6, 8). The activity of cdk-5 was assayed with [γ-32P]ATP and histone H1. B, The cdk-5 immunoprecipitated fromA was detected by Western blot analysis using anti-cdk-5 monoclonal antibody (DC17).
DISCUSSION
This study presents direct evidence that SAPKs can phosphorylate the tail domain of NFH, as reflected both in the reduced mobility of NFH on SDS-PAGE and in its immunoreactivity with the phosphorylation-dependent monoclonal antibodies SMI 31 and SMI 34 (Fig.1). SAPK activation was accomplished by transfection of a vector expressing constitutively active MEKK-1Δ, which activates JNK kinase (JNKK/MKK4/SEK4), the upstream regulator of SAPKs (Yan et al., 1994;Dérijard et al., 1995; Lin et al., 1995). Although MEKK1 can also activate the ERK pathway (Lange-Carter and Johnson, 1994; Xu et al., 1995), it is a more efficient activator of the SAPK cascade (Minden et al., 1994; Yan et al., 1994). Furthermore, we previously demonstrated that ERK activation did not result in a detectable increase in thein vivo phosphorylation of NFH (Giasson and Mushynski, 1996), and others have shown that the in vitrophosphorylation of NFH by ERKs did not cause a significant reduction in its mobility on SDS-PAGE (Roder and Ingram, 1991; Roder et al., 1995). Use of a specific inhibitor of p38 kinases, SB 203580, demonstrated that the latter enzymes are not involved in the hyperphosphorylation of perikaryal NFH (see Fig. 2). These results support our previously reported correlative study (Giasson and Mushynski, 1996) and strongly suggest that SAPKs are involved in the aberrant phosphorylation of perikaryal NFH.
The stress-activated phosphorylation of perikaryal NFH is completely reversible (see Fig. 4), indicating that a protein phosphatase(s) in the neuronal perikaryon can maintain the protein in a hypophosphorylated state. A protein phosphatase-2A-like activity has been reported to dephosphorylate KSP repeats in NFH (Veeranna et al., 1995). However, attempts to dephosphorylate NFH in vitro to an extent that would alter its electrophoretic mobility using any of the major neuronal protein phosphatases—1, 2A, 2B, and 2C—were unsuccessful (Hisanaga et al., 1993a). This discrepancy may be attributable to differences between the in vivo and in vitro conformations of NFH, to differences in substrate specificity conferred by regulatory subunits associated with the catalytic phosphatase subunit (Sola et al., 1991), or to the involvement of a different protein phosphatase such as PP-X (Brewis et al., 1993). In any case, our results are consistent with the presence of an NFH tail domain phosphatase in the neuronal perikaryon. This enzyme may be absent from or less active in axons, where NFH is highly phosphorylated.
We have also demonstrated that ERKs and SAPKγ are equally distributed between the cell body and neurite compartments of DRG neurons. The localization of ERKs in neurites is consistent with a recent study demonstrating the axonal transport of these enzymes (Johanson et al., 1995). Although SAPKs have been reported to phosphorylate primarily transcription factors (Cano and Mahadevan, 1995; Kyriakis and Avruch, 1996), the axonal localization of SAPKγ suggests that it may also be involved in the phosphorylation of cytoplasmic substrates, such as NFH.
Cdk-5 is the only neuronal kinase (Hisanaga, 1993b; Miyasaka et al., 1993), other than SAPKs, that has been shown to phosphorylate NFH to the point of reducing its mobility on SDS-PAGE to that of axonal NFH. Cdk-5 has been reported to phosphorylate NFH in vitro to the extent of 3–5 moles (Miyasaka et al., 1993) or 10 moles (Hisanaga et al., 1993b) of phosphate per mole of NFH, preferentially at KSPXK repeats (where X is not an acidic residue) (Beaudette et al., 1993). We have observed that neuritic NFH in cultured DRG neurons is highly phosphorylated despite the demonstrated lack of cdk-5 activity (see Fig. 6), which is likely to be because its activator ligand, p35/p25 (Lew et al., 1994; Tsai et al., 1994), is not expressed in these neurons (Tsai et al., 1994). This may explain the apparent sparing of DRG neurons in cdk-5-deficient mice, whereas many types of CNS neurons in these animals are adversely affected (Ohshima et al., 1996). Consequently, it is possible that KSPXK motifs are not phosphorylated in DRG neurons, as is suggested by the finding that NFH is more highly phosphorylated in ventral root motor neurons than in dorsal root neurons (Soussan et al., 1996). Cdk-5 is likely to be active in and required for motor neuron survival because the latter show a number of abnormalities in mice lacking the enzyme, including ballooned perikarya, dispersed Nissl substance, and cytoplasmic vacuoles (Ohshima et al., 1996).
NFH is reported to undergo high levels of phosphorylation in the initial axon segment, the site at which myelination begins (Hsieh et al., 1994; Nixon et al., 1994a). We have demonstrated that NFH follows the normal pattern of phosphorylated isoform distribution in cultured DRG neurons despite the lack of myelination (see Fig. 5A). Furthermore, cultures devoid of Schwann cells also exhibited the normal NFH phosphorylation profile (see Fig. 5B). These experiments clearly demonstrate that phosphorylation of neuritic NFH is not initiated solely by cues stemming from Schwann cell–axon interactions. As mention above, there may be differences in phosphatase activity levels between the cell body and neuritic compartments. However, it is also likely that NFH-kinase(s) is (are) activated in the initial axon segment, and these enzymes conceivably could include SAPKs.
Phosphorylation of the NFH tail domain does not occur exclusively during the entry of NFs into axons. Phosphate addition continues during NF transport (Lewis and Nixon, 1988; Archer et al., 1994; Nixon et al., 1994b), and regional differences in tail domain phosphorylation within myelinated axons have also been reported. There is a reduced level of NF phosphorylation at the nodes of Ranvier (Mata et al., 1992) and a decrease in axonal NFH phosphorylation in hypomyelinating transgenic or Trembler mice (de Waegh et al., 1992; Cole et al., 1994). The latter observation indicates that axonal properties, including NFH phosphorylation, are modulated by signals transmitted from myelinating Schwann cells to axons. NFH phosphorylation in myelinated regions thus may be augmented through the activation of proline-directed kinases such as ERKs and SAPKs.
There is evidence suggesting that aberrant NF metabolism may be involved in the etiology of ALS (Côté et al., 1993; Xu et al., 1993). Motor neurons containing abnormally hyperphosphorylated perikaryal NFH and proximal axonal enlargements filled with NFs are characteristic of the disease (Carpenter, 1968; Hirano et al., 1984;Manetto et al., 1988; Munoz et al., 1988; Sobue et al., 1990). Furthermore, ALS is a neurodegenerative disease that targets the large, NF-rich motor neurons predominantly and large sensory neurons to a lesser degree (Tsukagoshi et al., 1979; Kawamura et al., 1981). If NFs are involved in ALS pathogenesis, it is more likely because of an impairment of axonal transport rather than simple accumulation of NFs in the perikaryon (Collard et al., 1995; Marszalek et al., 1996).
The mechanism underlying the axonal transport of NFs remains unsettled, although experiments with the neurotoxin β,β′-iminodipropionitrile (IDPN) suggest that microtubules may be involved. IDPN causes NFs and microtubules to segregate (Griffin et al., 1978; Papasozomenos et al., 1981) and, at appropriate doses, blocks NF movement, but it has only a modest effect on the transport of microtubules (Griffin et al., 1978). This causes large masses of NFs to accumulate in the proximal axon (Chou and Hartman, 1965). It is interesting to note in this respect that NFH interacts with microtubules only when its tail domain is hypophosphorylated (Hisanaga et al., 1991, 1993a,b; Miyasaka, 1993), suggesting that tail domain phosphorylation may be important in regulating the transport of NFs from the cell body to the axon. Aberrant hyperphosphorylation of perikaryal NFH in neurons subjected to some form of stress thus may be responsible for the formation of neurofilamentous accumulations that characterize many neurological diseases.
Footnotes
This research was supported by a grant from the Medical Research Council of Canada. B.I.G. is the recipient of a studentship from the Fonds de la Recherche en Santé du Québec. We thank Dr. Michael Karin for his generous gift of the MEKK-1 cDNA.
Correspondence should be addressed to Dr. W. Mushynski, McGill University, Department of Biochemistry, 3655 Drummond Street, Montréal, Québec, Canada H3G 1Y6.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}