

Abstract
This study examines the role of K channel segregation and fiber geometry at transition zones of mammalian nerve terminals in the peripheral nervous system. Mutant mice that are deficient in Kv1.1, a fast Shaker K channel normally localized beneath the myelin sheath, display three types of cooling-induced abnormal hyperexcitability localized to regions before the transition zones of myelinated nerves. The first type is stimulus-evoked nerve backfiring that is absent at birth, peaks at postnatal day 17 (P17), and subsides in adults. The second type is spontaneous activity that has a more delayed onset, peaks at P30, and also disappears in older mice (>P60). TEA greatly amplifies this spontaneous activity with an effective dosage of ∼0.7 mm, and can induce its reappearance in older mutant mice (>P100). These first two types of hyperexcitability occur only in homozygous mutants that are completely devoid of Kv1.1. The third type occurs in heterozygotes and represents a synergism between a TEA-sensitive channel and Kv1.1. Heterozygotes exposed to TEA display no overt phenotype until a single stimulation is given, which is then followed by an indefinite phase of repetitive discharge. Computer modeling suggests that the excitability of the transition zone near the nerve terminal has at least two major determinants: the preterminal internodal shortening and axonal slow K channels. We suggest that variations in fiber geometry create sites of inherent instability that is normally stabilized by a synergism between myelin-concealed Kv1.1 and a slow, TEA-sensitive K channel.
- potassium channel gene
- homologous recombination
- myelinated nerves
- transition zones
- nerve conduction
- mouse
In adult mammalian myelinated nerves, fast potassium channels (Kv1.1 and Kv1.2) are sequestered under the myelin sheath in the juxtaparanodal region, but the role of these K channels in regulating excitability has been a matter of speculation (Chiu and Ritchie, 1980; Wang et al., 1993). The mature pattern of localization is not achieved instantly at the beginning of myelination, but is the result of a gradual redistribution process whereby Kv1.1 first appears at the node, then gradually shifts from the paranodal to the juxtaparanodal positions as the nerve matures (Vabnick et al., 1999). This channel redistribution is also accompanied by morphological changes in myelin thickness, internodal length, and fiber diameter (Pfeiffer and Friede, 1985; Yamamoto et al., 1996). How do channel redistribution and morphological changes affect the excitability of the myelinated nerves? The gradual redistribution of Kv1.1 under the myelin sheath is accompanied by a gradual diminution of 4-AP sensitivity as the nerve matures (Kocsis and Waxman, 1983; Vabnick et al., 1999).Vabnick et al. (1999) suggested that the redistribution of Kv1.1 (node→paranode→juxtaparanode) prevents aberrant excitations during development.
An important issue not addressed by the above studies is whether channel segregation plays a more important role at the transition zones near the nerve terminal (where the myelinated segment ends and the nonmyelinated segment begins) than elsewhere in the nerve. This issue is interesting for various reasons. There is an impedance mismatch at this transition zone, and the shortening of internodes before the nerve terminal has been suggested to facilitate successful invasion of the nerve terminal by action potentials (Revenko et al., 1973). From a developmental standpoint, the transition zone may be the last site to mature (Yamamoto et al., 1996). Even in the adult, ongoing myelin remodeling of the internodal segments preceding the terminal may persist (Baker and Ip, 1965; Quick et al., 1979; Pfeiffer and Friede, 1985). Regulation of transition zone excitability also has implications for diseases such as multiple sclerosis, where pathological transition zones are created during demyelination and remyelination. The transition zone, because of its local geometrical heterogeneity, may be inherently unstable and particularly sensitive to excitability perturbations.
The functional role of K channel clustering at transition zones was first addressed using mutant mice that lack Kv1.1 (Zhou et al., 1998). We reported that in immature nerves postnatal day 14 (P14)–P21, the absence of Kv1.1, which is normally segregated to the paranode and concealed by the myelin sheath, produces a disproportionately large change in hyperexcitability at the transition zone compared with the rest of the nerve (Zhou et al., 1998). Here, we extend these studies to encompass a broader developmental range (P5–P101) to gain further insight into the determinants of excitability zone excitability. In the first part, we examine developmental changes in transition zone excitability, and ask whether Kv1.1 deletion unmasks the importance of other K channels. In the second part, we use a computer model to examine various determinants of the excitability of the transition zone. Our studies reveal two types of abnormal excitability changes with different developmental schedules, and, in addition, unmask a synergism between Kv1.1 and a TEA-sensitive channel or channels that critically determines the excitability of the transition zone.
MATERIALS AND METHODS
Kv1.1 null mice
Kv1.1 mutant mice in a mixed B6x129 genetic background (Smart et al., 1998) were generated by heterozygote–heterozygote mating and used for electrophysiological studies (Zhou et al., 1998). For electrophysiological experiments performed on mice P11 or older, the genotypes were predetermined by PCR analysis of tail DNA samples performed at P6–P8 as described previously (Zhou et al., 1998). For electrophysiological experiments performed on mice P10 or younger, the genotype was determined after the experiments by PCR analysis of postmortem tail DNA samples. Controls consisted of age-matched littermates that were Kv1.1+/+.
Electrophysiology
Phrenic nerve–diaphragm preparation. Whole diaphragm with phrenic nerve was excised from the wild-type and mutant mice and mounted in a recording chamber as described previously (Zhou et al., 1998). Briefly, the nerve was stimulated with a bipolar electrode while the nerve compound action potential was recorded from the cut end with a tight suction electrode. The nerve-evoked muscle compound action potential was recorded simultaneously with a surface electrode pressed gently against the diaphragm surface. We consistently recorded from the same location on the diaphragm surface in all experiments.
Intracellular recordings. Intracellular recordings from single muscle fibers were performed with sharp microelectrodes (10–20 MΩ, 3 m KCl) inserted near the endplate regions. The muscle was cut on both sides of endplate region near the tendon regions to prevent contraction and was incubated for ∼1 hr before experiments started.
Presynaptic current measurement. Extracellular potential measurement of the presynaptic current waveform was performed in thetriangularis sterni muscle–nerve preparation after the technique of Brigant and Mallart (1982), as described in our previous work (Zhou et al., 1998). Briefly, a sharp microelectrode (3–5 MΩ, 2m NaCl) was inserted into the perineural space near the endplate under visual guidance with Nomarski optics (400×). This thin muscle preparation allows precise placement of the electrode near the heminodal region. Our recorded extracellular field potential waveform, which consisted of two negativities, is consistent with the position of the recording pipette in the heminodal region as described previously [Brigant and Mallart (1982), their Fig. 9].
Solutions
The normal Ringer’s solution bathing the nerve–muscle preparation contained (in mm): NaCl 129, KCl 3.0, CaCl2 2.4, MgSO4 1.3, NaHCO3 20, glucose 20, and HEPES 3. The solution was vigorously bubbled with 95% O2 and 5% CO2 to a pH of 7.4, and the preparation was continuously perfused by this solution at 2–3 ml/min. Temperature of the bath was changed by a DC-feedback temperature controller. A thermistor probe was placed near the stimulating and recording site to monitor temperature changes. The nerve was stimulated with brief stimuli (0.01 msec) with a bipolar electrode connected to the voltage output of a Grass Stimulator S48.
Computer modeling
The simulation model combines Halter and Clark’s (1991) model for myelinated axons and Miralles and Solsona’s (1996) model for the nerve terminal (see Fig. 7). For the nerve trunk portion, the axon diameter/fiber diameter (d/D) ratio is 0.70 and the internodal length/axon diameter (L/d) ratio is 255, which is near the optimal dimensions for peripheral myelinated nerves (Waxman, 1975). The intraaxonal and periaxonal voltages are computed as in Halter and Clark (1991), with the voltage potential difference across the axonal membrane determining the activation of ion channels distributed on the axonal membrane. Our model consists of a myelinated fiber with 15 internodes, which ends at a nerve terminal with four branches (see Fig.7). There are four types of ion channels in this model (Na channels, fast K channels, slow K channels, and leakage channels).
Na channels. We used the nodal Na channel permeability value (33.9 × 10−5 m/sec) from Halter’s model(1991) and distributed it at the nodes and heminode. At the internode we assume that the Na channel density is 4% of the nodal value (Shrager, 1989). Note that the Halter model (1991) uses a higher Na channel density than other models because of the need to reproduce an appropriate conduction velocity. At the presynaptic terminal, the Na channel density is gradually reduced to 0.4% of the nodal value at the tip of the terminal branch.
Fast K channels. We distributed fast, noninactivating K channels in the internode with peak density at the juxtaparanodal region [which in our model is over the fluted paranodal axon segment (FLUT) region]. The kinetics of the fast K channel is taken fromHalter (1991), and is similar to the fast delayed rectifier in most computer models of the myelinated fiber. The permeability of fast K channels at the juxtaparanodal region is calculated from published patch-clamp data (3.85 × 10−6 m/sec) [Vogel and Schwarz (1995), their Table 1]. The density in the other internodal regions [stereotype internodal region (STIN) and paranodal myelin sheath attachment axon segment (MYSA)] declines to 8% of the peak value at FLUT. We assume there are no fast K channels at the node. At the nerve terminal, the fast K channel density is highest at the region right before the branches, then declines to 0.8% of the FLUT value at the tip of the branch, with an intermediate value in between. The kinetics of the presynaptic fast K channel is identical to that in the axon.
Slow K channels. Slow K channel permeability and kinetics were taken from the Halter model (9.37 × 10−7m/sec). The kinetics of the slow K channels were obtained by Halter and Clark (1991) by curve fitting to published slow potassium current data from amphibian myelinated nerves. Slow K channels have a different distribution than the fast K channels in having a major representation at the nodal membrane. The channel density is highest at the node and heminode and declines to 33% elsewhere along the axon (STIN, FLUT, MYSA). Along the presynaptic terminal, the slow K channel density is highest at the heminode, then gradually declines to 3.3% of the heminode value at the terminal tip. Unlike the fast K channels, the slow K channels are already activated at the resting potential. This K channel profile is similar to that used in Miralles and Solsona (1996).
Leakage conductance. For the axon, the value for the leakage conductance is 303 S/m2 for the node and 5 S/m2 for the internode (Hines and Shrager, 1991). For the presynaptic terminal membrane, the leakage conductance is the same as that for the internodal axon. The specific membrane capacitance for the axonal membrane and each single myelin lamellae is 0.01 F/m2.
Ca channels. Although Ca influx is critical for neurotransmitter release from the presynaptic terminal, the current is very small and spatially limited to the terminal membrane region (Peres and Andrietti, 1986). We ignored calcium current in our simulations.
Simulations
The Crank–Nicholson integration method was used, and the time steps for calculation are 0.5 μsec at 20°C and 0.2 μsec at 37°C. The simulation program was written in C and compiled by Borland C++5.02.
RESULTS
In this report, we document three types of abnormal hyperexcitability in the PNS of Kv1.1 mutant mice, localized to the transition zones of the nerve terminal. The three types of hyperexcitability are stimulus-evoked nerve backfiring, spontaneous activity, and synergism between Kv1.1 and TEA-sensitive channels. The first type has already been described in our previous report, in relation to a narrow developmental window (P14–P21; Zhou et al., 1998). In the present study, we addressed a broader developmental range (P5–P101) during which myelination progresses from immature to mature status. We used many of the same techniques for localization of excitability changes described in the previous report (Zhou et al., 1998).
First type of hyperexcitability: evoked nerve backfiring peaks at P17
The key phenotypes of Kv1.1-null mice at different ages are illustrated in Figure 1. Nerve and muscle compound action potentials were recorded from the phrenic nerve–diaphragm preparation. Figure 1A shows typical responses, recorded at room temperature, of Kv1.1 preparations taken from mice of three age groups (P10, P18, and P40). In the young mice (P10), there is a one-to-one nerve-muscle transmission in Kv1.1 nulls, which is indistinguishable from the age-matched, wild-type response (wild-type data not shown). As the mice develop (P18), the first type of hyperexcitability appears, demonstrated in our previous work to be evoked nerve backfiring from the transition zone near the nerve terminal (Zhou et al., 1998). This type of hyperexcitability peaks at P17, and subsides with age (Fig. 1B,left).

Developmental changes in excitability in the Kv1.1 null mutants. A shows typical nerve stimulation-evoked muscle (top) and nerve (bottom) compound action potentials from Kv1.1-null mutants of three age groups. The muscle action potential was measured by a pipette pressed against the diaphragm surface, whereas the nerve action potential was measured by a tight suction electrode from the cut end of the phrenic nerve. To highlight the delayed repetitive discharge in the nerve response, the nerve signal has been amplified 10 times. The bath temperature was 20°C. With aging, the mutant phenotype changed from normal (P10), to evoked repetitive activity (P18) and then to spontaneous activity in the baseline before the stimulation (P40). B shows plots of the evoked repetitive activity and spontaneous activity as a function of animal age. Evoked nerve backfiring was quantified by counting the number of spikes in the nerve compound action potential trace over a fixed time interval (10–40 msec) after the stimulation. Spontaneous activity was quantified by counting the number of nerve spikes in a fixed baseline segment (30 msec) before stimulation. In most experiments, the freshly dissected preparation was first warmed to 35°C for 5–10 min, and then the bath temperature was cooled to 20°C for the main parts of the experiments. The evoked and spontaneous activity in most experiments were measured within 5–30 min after this cooling. The smooth solid curves are fits (by eye) to the data, showing peaks at P17 and P30 for the evoked nerve backfiring and the spontaneous activity, respectively.
Second type of hyperexcitability: spontaneous activity peaks at P30
The disappearance of the evoked nerve backfiring was followed by a new form of hyperexcitability consisting of spontaneous activity. This spontaneous activity can be easily detected in the baseline of both the muscle and nerve action potential traces before stimulation (Fig.1A, P40). This spontaneous activity peaked at P30 but also subsides with further maturation (Fig.1B, right). The inset in Figure1B (right) compares the developmental schedule of the evoked and spontaneous activity, showing the emergence of the spontaneous activity coincides with the disappearance of the evoked activity. The spontaneous activity does not require previous nerve stimulation and can be induced by monitoring both the resting nerve and muscle activity while the bath temperature is lowered from 35 to 18°C (Fig. 2, left two panels, P32, −/−). The warm–cool cycle was repeated twice to illustrate the reproducibility of the phenomenon (Fig. 2, left two panels). In age-matched wild-type mice, no spontaneous activity can be observed at either bath temperature (Fig.2, right two panels). An interesting feature about this spontaneous activity is that it tended to be quite labile, particularly in older mice. For example, in several mice within the age range P40–P53, the spontaneous activity, although present during the early part of an experiment, disappeared after several cycles of temperature change; in several older mice (P60–P101), spontaneous activity was virtually absent to begin with. In contrast, in the younger null mutants (P23–P30), the spontaneous activity persisted through multiple cycles of temperature change during experiments that lasted 1–2 hr.

Spontaneous nerve activity in old null mutants originated from the nerve terminal region. Top shows the schematic drawing of the experimental configuration. Compound muscle and nerve activities were measured simultaneously in the phrenic nerve–muscle preparation from a P32 null mutant. A lidocaine pipette, consisting of a pair of closely spaced pipettes, one for ejecting lidocaine (150 mm) and the other for sucking it up, was used to selectively block conduction on the phrenic nerve halfway between the nerve cut end and the nerve terminal. At 20°C, spontaneous muscle and nerve activity could be observed in the null mutant (left two panels) but not in the age-matched wild type (right two panels). The spontaneous activity in the mutant was induced by cooling, as shown by changing the bath temperatures. Curare (15 μm) blocked the muscle activity without affecting the nerve activity. Local conduction block with lidocaine abolished the spontaneous activity, demonstrating its origin from the nerve terminal region.
Localization of spontaneous activity to the nerve terminal region
To delineate nerve versus muscle contributions to the spontaneous activity, curare was added to immobilize the muscle fibers (Fig. 2,left two panels). This eliminated the muscle spontaneous activity, but left intact the nerve spontaneous activity, suggesting that the hyperexcitability originated from the nerve. Where in the nerve does the spontaneous activity originate? The activity detected by the recording pipette at the cut end could represent spontaneous activity of the nerve trunk, injury activity from the cut end, or activity originating from the nerve terminal region that backpropagates along the nerve. To distinguish between these possibilities, a lidocaine pipette was used to selectively block nerve conduction at a site located roughly halfway between the recording pipette and the nerve terminal (Fig. 2, top scheme). Our lidocaine pipette was fabricated and tested in numerous control experiments to allow a local nerve blockade without leakage of the drug into the bath to block other parts of the nerve. If the spontaneous nerve activity was uniformly distributed over the nerve trunk, this interruption of conduction at the midpoint should block half of the activity reaching the recording pipette. If the spontaneous activity was caused by injury excitation occurring at the cut end of the nerve, this local block should produce no effect on the recorded spontaneous activity. Neither result was observed. Instead, the spontaneous activity was completely abolished by the lidocaine block (Fig. 2,left two panels, bottom). The best explanation of the complete silencing of the spontaneous activity is that the activity originated from the nerve terminal region. Further corroborative evidence on this point will be presented below when we explore whether myelinated or nonmyelinated fibers generate the spontaneous activity.
The spontaneous activity originates from myelinated fibers
Phrenic nerves contain both myelinated and nonmyelinated fibers, and an important issue is which fiber type gives rise to the spontaneous activity. To resolve this issue, a brief, supramaximal stimulation (0.01 msec) was applied to the nerve trunk to activate only the fastest conducting myelinated fibers, which produced a compound action potential derived only from these fibers. Interestingly, the spontaneous activity, which was present in the baseline before the action potential, was completely occluded for ∼100 msec after the action potential (Fig. 1A, P40 nerve trace). Furthermore, the spontaneous activity was occluded in a graded manner, as more myelinated fibers are recruited by gradually increasing the stimulation strength (data not shown). We are confident that only myelinated fibers were being stimulated because a slow conducting compound action potential corresponding to nonmyelinated fiber activation was not observed with our brief stimulations. Unless there was an interaction between myelinated and nonmyelinated fibers (some kind of ephaptic interactions), which we think highly unlikely, we believe that this interaction between the spontaneous activity and myelinated nerve activation is best explained by the spontaneous activity arising from myelinated nerves. As will be discussed later (see Figs. 8-11), the computer simulations showing that myelinated nerves indeed have the capability of generating spontaneous discharge, at least for the evokable type further strengthen this argument. The fact that ∼100 msec had to elapse before the spontaneous activity resumed suggests a refractoriness of ∼100 msec for the generation of the spontaneous activity. This could reflect refractoriness either in the nerve trunk or in the generation of spontaneous activity from the nerve terminal. Twin-pulse experiments showed that the nerve trunk is fully capable of generating a second impulse 10 msec after the first pulse (data not shown). Hence, the refractoriness in the spontaneous activity reflects the refractoriness of the site of origin of the spontaneous activity, presumably the nerve terminal region. This argument for the terminal origin of the spontaneous activity is indirect, but is consistent with our earlier, more direct evidence based on local conduction block. We conclude that the spontaneous activity originates near nerve terminal regions of myelinated fibers.
TEA amplifies the spontaneous activity
Besides fast K channels, myelinated fibers also express slow K channels that have been suggested to modulate repetitive discharge of the nerves. Unlike fast K channels that are absent from the node, slow K channels have a major representation at the nodal membrane, although they are also expressed in the internode (Kocsis et al., 1986; Baker et al., 1987). An interesting issue is whether spontaneous activity in the Kv1.1-null mice is linked to slow K channels. Because slow K channels are TEA sensitive, we examined the effect of TEA on the spontaneous activity. We found the developmental onset of spontaneous activity in the null mutants was also accompanied by an increasing ability of TEA to induce or amplify spontaneous activity. Figure3A (left) shows a P29 mutant mouse where the baseline spontaneous activity was not very robust. Application of TEA gradually increased the baseline spontaneous activity (Fig. 3A, left, nerve records), and ultimately transformed the response to one that resembled the severe spontaneous activity phenotype observed in some null mutants. This effect of TEA in amplifying the endogenous nerve spontaneous activity is highly age-dependent, occurring only in old (P23–P100) but not in young (P12–P14, Fig. 3B) mutant mice. In the young mice, the only observed effect of TEA was a potentiation of the evoked nerve backfiring that is normally insignificant in these young mutants (Fig.3B); spontaneous activity was never induced in the baseline. In all age groups tested, TEA (0.5–5 mm) had no effect on the wild-type mice (Fig. 3A,B).

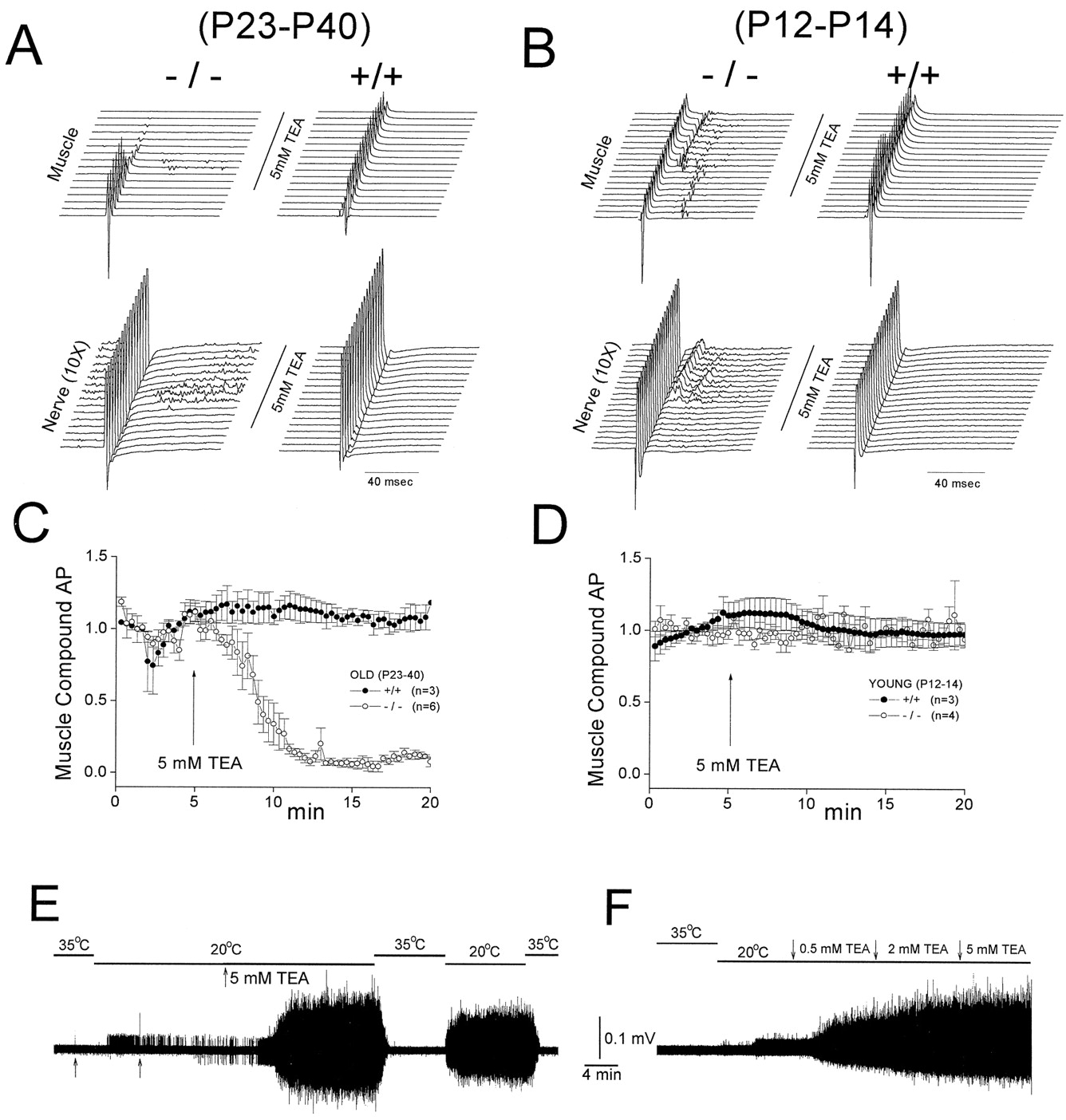
Differential TEA sensitivity in old and young null mutants. Nerve stimulation-evoked nerve and muscle compound action potentials were measured from phrenic nerve–diaphragm preparations of two age groups: old (P23–P40; A, C) and young (P12–P14; B, D). Wild-type littermates from the same age groups were used as controls.A and B show typical muscle and nerve responses to 5 mm TEA. The responses were generated by supramaximal nerve stimulation with the time between two traces being 1 min. The nerve traces have been amplified 10 times to illustrate the spontaneous nerve activity. TEA was without effect on the muscle response in the wild type but showed an age-dependent effect in the null mutants. In the young mutants, the evoked backfiring was potentiated while in the older mutants spontaneous activity was amplified. TEA did not affect the initial nerve compound action but caused a gradual decline in the muscle compound action in the old mutant. C and D show the averaged results of the TEA-induced decline in the muscle action potential in the old (C) but not young (D, open symbols) mutants. Wild-type data are represented byclosed symbols. E and Fshow the temperature sensitivity of the TEA-induced spontaneous activity. Old mutants were used that had little or no endogenous spontaneous activity. Resting compound nerve activity was recorded from the nerve cut ends (as in Fig. 2) as the temperature of the bath was switched between 35 and 20°C. In E (P61), adding 5 mm TEA to the bath dramatically induced spontaneous activity. In F (P101), spontaneous activity was induced with TEA concentrations as low as 0.5 mm. Curare was present to immobilize the muscle (E,F).
An important difference between the endogenous spontaneous activity and the TEA sensitivity is that whereas the endogenous spontaneous activity declines with age (Fig. 1B, right), the effect of TEA persists through all ages tested. Figure 3, Eand F, shows two old null mutants that displayed little or no spontaneous activity. TEA (5 mm) dramatically induced a spontaneous activity that is highly temperature-sensitive (Fig.3E, P101). In the other null mutant (Fig. 3F, P68), a stepwise increase in TEA concentrations showed that as low as 0.5 mm TEA was enough to induce the spontaneous activity.
One striking effect of TEA in the older mutants was a gradual abolishment of neurotransmission, as seen in the elimination of the evoked muscle compound action potential (Fig. 3A,left, muscle records). The mechanism for this TEA action is unclear. Because TEA has been suggested to block choline uptake (Roed, 1989), one possibility is that the neurotransmission block is caused by a dual action of TEA in inducing spontaneous discharge as well as slowly depleting the ACh release pool. Although we have not resolved the mechanism for the neurotransmission block induced by TEA, the gradual reduction of the evoked muscle compound action potential becomes a very convenient assay for the age-dependent severity of the action of TEA, as shown in the summarized plot in Figure 3 for old (C) and young (D) null mutants.
Single muscle recordings
To gain further quantitative information into the action of TEA on the null mutant nerves, we recorded the postsynaptic membrane potential (PSP) from single muscle fibers in cut muscle preparations. Figure4A shows recordings of a Kv1.1-null PSP with no baseline spontaneous activity. Nerve stimulation was given every 30 sec. Adding TEA (5 mm) produced a prolonged phase of PSP discharge at ∼10 Hz (Fig.4A, bottom, inset at expanded time scale). In other experiments, this spontaneous activity was induced by TEA without using nerve stimulation. The amplitude of PSP gradually reduced to zero, with a time course similar to that observed in the reduction of the muscle action potential in uncut muscle preparations (Fig. 3C). The gradual disappearance of the PSP was not caused by failure of nerve action potentials because both the compound nerve action potential and the refractory period of nerve transmission were totally unaffected by TEA treatment (data not shown). TEA (5 mm) was without effect on the wild-type PSP (Fig.4A).

Measurement of TEA-induced repetitive discharge in single muscle fiber. A shows evoked PSPs recorded with sharp electrodes in cut-muscle preparations. The PSP was evoked by stimulating the phrenic nerve with a single stimulation every 30 sec. TEA (5 mm) had no effect on the evoked PSP of wild type (top trace). In the null mutant, TEA led to spontaneous PSPs with a regular frequency of ∼10 Hz (bottom,inset, expanded time scale). The stimulation was turned off when the spontaneous discharge occurred. The amplitude of the spontaneous PSP slowly declined to zero. B, Dose–response relationship of TEA. Presynaptic current waveforms were measured extracellularly from the triangular sterni nerve–muscle preparation in which resolvable single spikes from four to eight single fibers can be measured as TEA concentration was increased. A sample extracellular presynaptic current waveform was shown (B,top right), illustrating that TEA depressed a negativity corresponding to activation of a presynaptic K conductance. The increase in the spike frequency (probably caused by recruitment of more fibers) with increasing TEA concentration was measured. TEA was applied for 10 min at each concentration. The sigmoidal dose– response curve was fitted to the data with a half-maximal effective concentration of 0.77 ± 0.05 mm (n = 3).
Dose–response relation
Using the triangularis sterni preparation, we recorded presynaptic current waveforms from a bundle of four to eight fibers by inserting a microelectrode into the preterminal region of Kv1.1-null endplates. This preparation produced well resolved single spikes whose frequency increased with TEA concentrations (Fig. 4B,left), probably resulting from recruitment of more fibers into spontaneous activity. The dose–response relationship between TEA concentration and the spontaneous activity revealed a half-maximal dosage of 0.77 mm (Fig. 4B,right).
Temperature sensitivity and origination from the nerve terminal region
Using the technique of local conduction block with lidocaine (Fig.2), we demonstrated that the TEA-induced spontaneous activity, like the endogenous spontaneous activity, originated from nerve terminals of myelinated fibers (data not shown). Further, the TEA-induced spontaneous activity, like the endogenous one, was temperature-sensitive (Fig. 3E). From these similarities, we suggest that the TEA-induced activity and the spontaneous activity have a common origin. It appears that in the absence of Kv1.1, the excitability of the mutant nerve terminals is dominated by a TEA-sensitive K channel or channels.
The repetitive discharge is not related to Ca-activated K channels at the nerve terminal
It is possible that blocking calcium currents or blocking calcium-activated potassium channels at the nerve terminal may lead to repetitive discharge in the mutant. We therefore applied blockers of calcium-activated potassium channels (apamin, which blocks a different class of calcium-activated K channels than that blocked by TEA, 250 nm, for 1 hr), or removed bath calcium, to test these possibilities. We found that neither manipulation mimicked the effect of TEA (data not shown), suggesting that the action of TEA in the null mutants was unrelated to blocking calcium-activated potassium channels.
Effect of elevating bath potassium
Another possible mechanism for the action of TEA is that it causes a membrane depolarization, leading to repetitive discharge in Kv1.1 nulls whose transition zones are already destabilized by the absence of Kv1.1. However, we found that raising the bath potassium concentration from the normal value of 3 to 9 mm did not reproduce the effect of TEA.
4-AP reproduces the spontaneous activity phenotype in the wild type
An interesting issue is whether the spontaneous activity phenotype in the null mutants is a direct result of Kv1.1 gene deletion or a secondary effect caused by alteration in expression of other genes or changes in myelin morphology. In preliminary studies we have found no changes in the myelin morphology by ultrastructural examination of longitudinal sections of Kv1.1 mutant sciatic nerves (data not shown). Furthermore, at least in brain, the expression of a closely related member, Kv1.2, was not altered by Kv1.1 deletion (Smart et al., 1998). Nevertheless, we felt our case for a primary effect of Kv1.1 deletion would be strengthened if we could reproduce the mutant phenotype by acutely blocking Kv1.1 in a wild-type mouse. DTX would have been a preferred drug because of its specific action on Shaker K channels such as Kv1.1. However, its access to paranodal K channels under the myelin sheath is restricted (Zhou et al., 1998; Vabnick et al., 1999). Hence, we used 4-AP, a broad-spectrum blocker of fast K channels that can penetrate the myelin sheath. Figure 5shows the effect of 4-AP (250 μm) on an old (P35) and a young (P13) wild-type phrenic nerve–diaphragm preparation. In the older wild-type mouse (top), cooling-induced spontaneous activity was absent before 4-AP, but appeared after 4-AP application. Using the technique for local blockage of nerve conduction with lidocaine (Fig.2), we demonstrated that the 4-AP induced spontaneous activity originated from the nerve terminal regions. In the younger wild-type mouse (bottom), no effect of 4-AP was seen. Hence, the key features of the Kv1.1-null phenotype with respect to spontaneous activity (delayed developmental onset, temperature-sensitivity, origination from the nerve terminal region) were all reproduced by 4-AP in the wild type, supporting the case that the mutant phenotype reflects a primary effect of losing Kv1.1.

Developmental appearance of spontaneous nerve activity in null mutants could be mimicked by applying 4-AP to the wild type. Left panels, Absence of temperature dependence of resting nerve activity in an old (P31, top) and young (P13, bottom) wild-type mouse. Right panels, 4-AP (250 μm) induced temperature-sensitive spontaneous activity in the old, but not young, wild-type mouse. Resting nerve activity was measured from the cut end of a phrenic nerve as in Figure 2. Using selective lidocaine blockade (as in Fig. 2), we demonstrated that the 4-AP-induced spontaneous activity originated from the nerve terminal region (data not shown).
We hasten to point out that the 4-AP studies, unlike our gene deletion studies, lack the specificity to pinpoint the site of hyperexcitability. For example, 4-AP blocks both myelin-concealed K channels and fast K channels on the presynaptic membrane. In contrast, our gene deletion removes the former while leaving the latter intact. The 4-AP experiment alone does not allow the relative role of terminal versus myelin-segregated K channels in controlling the transition zone excitability to be sorted out. In contrast, this conclusion was possible with our Kv1.1 null mutants.
Synergism between Kv1.1 and TEA-sensitive channels
Besides inducing spontaneous activity, TEA also produces an evokable response in the Kv1.1 mutants that is best characterized as a synergism between TEA-sensitive and Kv1.1 channels. This is best studied in the heterozygotes whose phenotype is indistinguishable from the wild type before TEA application, as described below.
Heterozygotes as a model system to study synergism between Kv1.1 and TEA-sensitive channels
The heterozygotes are ideal for examining synergistic interaction between TEA and Kv1.1 deficiency, for several reasons. First, the half-normal gene dosage present in Kv1.1 heterozygotes is associated with ∼50% reduction in mRNA level (Smart et al., 1998), suggesting that the expression of Kv1.1 protein, though not eliminated, is reduced. Second, this partial Kv1.1 deficiency alone causes no overt phenotypes. Thus, the PNS excitability is virtually indistinguishable from the wild type (Smart et al., 1998). In the present study, the transition zone excitability of the heterozygotes is normal (both evoked nerve backfiring and spontaneous activity were absent). Figure6, A and B, shows a phrenic nerve–diaphragm preparation from a P35 heterozygote (+/−) before TEA application. In these experiments, a fixed paradigm was applied to both wild type and heterozygotes. After dissection, the preparation was allowed to stabilize at 20°C for 60 min before the experiments began. The experiment started by raising the temperature to 35°C for 6–10 min during which a single stimulation was given. The temperature was then lowered to 20°C, and another single stimulation given (Fig. 6A). Both stimulations triggered a single nerve response in the heterozygote (Fig. 6A). TEA (5 mm) was then added (Fig. 6B). The response to the single stimulation at 35°C remained unaffected. When the system was cooled to 20°C, no spontaneous activity appeared (Fig.6B). However, a single stimulation triggered a long-lasting phase of repetitive discharge (∼15 min) that could be terminated by warming (Fig. 6B). This phenomenon could then be reproduced by cooling, in which case a single stimulation again was needed to trigger the long-lasting repetitive discharge. In some heterozygotes (Fig. 6E), spontaneous activity appears before the stimulation (asterisk). However, in a total of four heterozygotes tested, stimulation was needed to initiate the discharge. We suspect that with TEA present, the system is at such a critical balance that a single “accident” or ectopic firing can initiate the long-lasting repetitive discharge. That the evoked repetitive discharge originates from the nerve terminal was demonstrated by using local lidocaine blockade (Fig. 2) to decouple the nerve terminal from the nerve trunk. In this “nerve-trunk-only” preparation, no long-lasting discharge was seen after a single nerve stimulation (Fig.6F). Finally, TEA had no effects on the wild-type preparation under similar experimental conditions (Fig.6C,D). In other experiments, we observed that higher TEA concentrations (10 mm) or prolonged TEA incubation did produce a similar, but less robust phenomenon in some wild types. This suggests that this synergism phenomenon involving TEA might also be present in the wild type, but the threshold for developing it is much lower in the heterozygotes. Collectively, the data in Figure 6 clearly suggests a synergistic phenomenon. Thus, though Kv1.1 deficiency alone (Fig. 6A) and TEA alone (Fig. 6D) produce no overt phenotypes, their combination (−/+ plus TEA) produces a dramatic effect on the evoked excitability (Fig. 6B).

Demonstration of synergism between Kv1.1 deficiency and TEA in Kv1.1 heterozygotes (+/−). Thetraces represent continuous recordings of resting phrenic nerve activity recorded by suction electrodes (see Fig. 2). Curare (15 μm) was present in the bath to eliminate muscle activity. Arrows indicate nerve action potentials in response to single nerve stimulations applied to the middle of the nerve trunk. The synergism under study is a single-stimulation evoked, long-lasting repetitive discharge that requires a combination of partial Kv1.1 deficiency (+/−) and TEA. This synergism was demonstrated by several steps. First, Kv1.1 deficiency (+/−) alone was without effect (A). Second, TEA alone (i.e., in +/+ with normal Kv1.1 expression) also was without effect (C, D). However, when Kv1.1 deficiency (+/−) was combined with TEA (B), a single stimulation evoked a dramatic phase of repetitive discharge that could be terminated by warming. E shows an example of a step-wise increase in the baseline activity (asterisk) before the single stimulation in a TEA-treated Kv1.1-heterozygote.F, Demonstration that the evoked repetitive discharge in TEA (+/−) did not arise from the nerve trunk. The nerve terminal was decoupled from the main nerve trunk by a local lidocaine block halfway between the recording pipette and the nerve terminal (Fig. 2). This left a functional nerve-trunk-only preparation that responded to stimulation, but did not produce the long-lasting repetitive discharge. Same (+/−) preparation as in E.
Computer simulations
In this section, a computer model is developed to examine the determinants of transition zone excitability. We are particularly interested in geometrical factors, as well as the synergism between Kv1.1 and slow K channels suggested by our studies above. Our goal is to see whether some of these phenomena can be reproduced by our model. The model (see Materials and Methods) has 14 nodes and a nerve terminal with four branches (Fig. 7). The model has Na channels (for action potential generation), two types of K channels (Kfast and Kslow) and a leakage channel. Axonal Kfast channels (intended to simulate Kv1.1) have the highest density at the paranodal–juxtaparanodal region, low density at the rest of the internode, and are absent from the node. Axonal Kslow channels have major nodal representation and have reduced density in the internode. Kfast channels are closed at the resting potential but are activated during an action potential. In contrast, Kslow channels are already open at the resting potential. The four presynaptic terminal branches have both Kfast and Kslow channels with kinetics identical to the axonal channels.

Schematic drawing of the computer model. The model for myelinated nerve– presynaptic terminal is based on Halter and Clark (1991) for the myelinated axon and Miralles and Solsona (1996) for the nerve terminal. Dimensions are in micrometers.A, The nerve fiber is represented as a triaxial distributed-parameter model (Halter and Clark, 1991), and we follow their terminology for the various nerve regions. The node, paranode (MYSA), juxtaparanode (FLUT), and internode (STIN) are represented by 1, 4, 4, and 5 segments, with a total distance of 1.5 μm (node), 10 μm (MYSA), 8 μm (FLUT), and 2000 μm (STIN), respectively. Each segment has its own morphological and ion channel parameters to describe the two compartments, the intra-axonal and periaxonal space. The distance between the axon and the myelin sheath is 0.001 μm (MYSA), 0.003 μm (FLUT), and 0.004 μm (STIN). These values are taken fromHalter and Clark (1991). The axon diameter at the node is 5 μm. For the MYSA region, the axon diameter gradually increases from 4.8 to 5.6 μm. The axon diameter at STIN is 8 μm. B, For the preterminal region, we progressively shortened the internodal length to mimic the actual morphology, with a progressive ratio of 1:0.5:0.25:0.125:0.0675 (α = 0.5) starting at the fourth internode before the presynaptic terminal. This shortening ratio is taken from Miralles and Solsona (1996), whose ratio in turn was based on morphological data (Quick et al., 1979). Internodal shortening is achieved by only shortening STIN, leaving all other parameters (length of FLUT and MYSA, axonal diameter, myelin sheath thickness, etc.) unaffected. The nerve terminal is represented by four branches.
Myelin-segregated Kfast channels are more important than terminal K channels in maintaining transition zone stability
Figure 8B shows an action potential that propagates along the myelinated nerve (from top to bottom) and invades the nerve terminal. The gradual rightward shift of the action potential traces reflects the time delays in arriving at successive sites caused by conduction delay. Cooling from 37 to 20°C broadens the action potential, slows the conduction velocity, but does not alter the faithful, one-to-one invasion of the nerve terminal (Fig.8B). We now try to destabilize this transmission by one of two ways. First, we selectively delete all K channels on the nerve terminal branches. This broadens the action potential at the nerve terminal as expected, but the faithful one-to-one invasion ensures (Fig. 8C). Second, we selectively delete all axonal Kfast channels segregated under the myelin sheath (Fig.8A). This produces little or no change in the shape of the action potential, but dramatically destabilizes the system. The action potential first successfully invades the terminal branches. However, ∼15 msec later, a second action potential is spontaneously initiated at the third node upstream from the nerve terminal (asterisk), causing two action potentials, one back-propagating along the nerve, and one forward-propagating to the terminal branches, causing a second invasion of the nerve terminal. After this second wave of excitation, no subsequent re-excitation is observed, even after at long times (data not shown).

Myelin-concealed Kfast channels, not presynaptic K channels, are important stabilizers against backfiring. A single stimulation was given to the 14th node (data not shown) upstream from the terminal, eliciting an action potential that propagates from top to bottom toward the nerve terminal. Only the last portion of the nerve near the terminal is shown. Action potentials at successive sites along the nerve are shown. The rightward shift of the traces represents conduction velocities of 25 m/sec (37°C) and 14 m/sec (20°C), respectively. The standard model (i.e., wild-type) contains Kfast on both the axon and presynaptic terminal branches.A, Selective deletion of all axonal Kfastchannels concealed by the myelin (setting Kfast = 0 at MYSA, FLUT, and STIN). B, Standard model (wild-type) with normal Kfast on both axons and nerve terminal branches. C, Selective deletion of all K channels (Kfast and Kslow) on the presynaptic terminal branches. Only deletion of the myelin-concealed Kfast produces backfiring, which is initiated at the third node upstream from the terminal (marked with anasterisk). The second action potential propagates both backward along the nerve and forward toward the nerve terminal.
The effect of preterminal internodal length
The above simulation suggests that a major determinant of nerve backfiring is the gradual internodal shortening that precedes the nerve terminal. Thus, backfiring occurs after the shortening begins, but upstream from the transition zone site (the heminode). Teleologically, the shortening of the internodes is needed for impedance matching to facilitate invasion of the nerve terminal (Revenko et al., 1973; Waxman and Brill, 1978). However, this shortening might also elevate the inherent excitability of the last few myelinated segments (with more nodes per unit length). We suggest that this, coupled with an increase in electrotonic coupling between the nodal and the internodal membrane caused by internodal shortening (Barrett and Barrett, 1982), makes the preterminal myelinated segment prone to re-excitation. The myelin-segregated Kv1.1 might act to stabilize the transition zone by damping out excessive nodal and internodal electrotonic coupling in the preterminal regions. This teleological argument is corroborated by the simulation in Figure 9, A andB, showing that if the internodes are forbidden to undergo preterminal shortening (Fig. 9A), deletion of Kv1.1 no longer causes backfiring. However, the price is that action potentials now fail to invade the nerve terminal because of impedance mismatch. Another example in which local shortening of internodes is important is remyelinated nerves. Figure 9C shows a myelinated nerve with a remyelinated segment consisting of eight abnormally short internodes. With normal presence of fast K channels under the myelin sheath, propagation over the remyelinated region is highly stable except for a minor slowing of conduction velocity over the short internodes (Fig.9C). However, deletion of fast K channels under the myelin causes instability as the action potential propagates over the remyelinated segment (Fig. 9D), resulting in nerve backfiring initiated at one of the short internodes (asterisk). The results of Figure 9 suggest that the myelin-concealed Kv1.1 plays an important role in preserving electrical stability whenever a local variation of internodal length occurs, both physiologically and pathophysiologically.

Deletion of Kfast fails to induce backfiring if there is no preterminal internodal shortening. Effects of deleting Kfast with (B) or without (A) internodal shortening before the nerve terminal. In the absence of preterminal internodal shortening, deletion of myelin-concealed K channels fails to destabilize the system. However, because of impedance mismatch, the action potential fails to invade the nerve terminal. C, D, Remyelinated fibers. The remyelinated segment is represented by eight short internodes, each one-fifth of the normal internodal length. C, Normal fast K channels present under the myelin sheath. D, Deletion of all fast K channels under the myelin sheath. Nerve backfiring is initiated at the asterisk. Temperature of the simulation is 20°C.
Kv1.1 redistribution
Recently, Vabnick et al. (1999) demonstrated a paranodal-to-juxtaparanodal redistribution of Kv1.1 during development and suggested that this channel redistribution prevents aberrant excitation during development. An interesting issue is whether the disappearance of evoked backfiring during myelin maturation may reflect K channel redistribution. However, we found that in our standard model (Fig. 8), redistribution of Kfast channels from the paranode to the juxtaparanode has no effect on the evoked nerve backfiring.
Slow K channels are crucial in transition zone excitability only when expression of Kv1.1 is compromised
Our empirical studies suggest that TEA-sensitive channels become a critical modulator of transition zone excitability once Kv1.1 is deleted or reduced. Because one probable candidate for the TEA-sensitive channel is the Kslow channel, we use our model to examine the effects of different combinations of Kfast and Kslow on transition zone excitability. When Kfast is normal (Fig.10A,left), reduction of Kslow (80% remaining to 20%) has no effect on transition zone excitability. However, deletion of Kfast (Fig. 10A, right) markedly sensitizes the system to reduction of Kslow, resulting in an increasing phase of evoked repetitive discharge as Kslow is progressively reduced. This is consistent with our observation that the null mutant is very sensitive to TEA (∼0.5 mm; Fig. 3F), whereas the wild type is insensitive up to 5 mm. Our computer simulations raise three important points about this stabilizing role of Kslow (in the absence of Kfast). First, only reduction in axonal Kslow, but not the presynaptic terminal Kslow, produces the evoked repetitive discharge (data not shown). Second, the repetitive discharge is localized to a site just preceding the transition zone. The rest of the fiber is not destabilized by the Kslow reduction. Third, a local reduction of Kslow (restricted to the three short internodes just preceding the terminal) is enough to dramatically destabilize the system to the same level as a global reduction (Fig.10B).

Kslow channels are important only when Kfast expression is compromised. Only axonal K channels are manipulated in this simulation. A,Left, Reduction of Kslow in the presence of intact Kfast. The percentage refers to the percentage of Kslow remaining. No destabilizing effect was seen.Vertical solid bar next to the nerve diagram illustrates the region over which Kslow is reduced.Right, The same reduction of Kslow after a deletion of Kfast. The system becomes highly unstable, with repetitive backfiring (evoked by a single stimulation) initiated from the third node upstream from the transition zone. B, Local Kslow reduction. Only axonal Kslow over three shortened internodal segments (indicated by the vertical solid bar) just proximal to the nerve terminal is reduced. This causes the same destabilization as a global Kslow reduction in A. Temperature of the simulation is 20°C.
Our model also can reproduce the synergism between TEA and Kv1.1 deficiency seen in the heterozygotes (Fig. 6B). Figure 11A shows that a 85% reduction (15% remaining) in Kfast and 99% reduction (1% remaining) in Kslow, each performed alone, has no effect on the excitability. However, when the two reductions are performed in combination, a single action potential, after approaching the nerve terminal, evokes repetitive nerve backfiring (Fig. 11B). The site of backfiring is the third node upstream from the terminal (Fig. 11B,asterisk) and, once evoked (by a single action potential), lasts indefinitely (Fig. 11C). Thus, major features of the synergism between TEA and Kv1.1 deficiency in the heterozygotes are reproduced by our model.

Synergism between Kslow and Kfast at the transition zone. Only axonal K channels are manipulated in this simulation. A, Left,Composition of axonal K channels: Kfast = 15% of normal; Kslow = 100% of normal. The conduction is normal. Right, Composition of axonal K channels: Kfast = 100% of normal; Kslow = 1% of normal. The conduction is also normal. B, The manipulations in A performed in combination: Kfast = 15% of normal; Kslow = 1% of normal. A dramatic synergistic interaction is revealed. After a single stimulation, action potentials are spontaneously initiated at the third node (asterisk) upstream from the terminal, traveling both backward along the nerve and forwards toward the terminal. C represents a plot of the response from the initiation site (third node) at long time scale, showing that this single-stimulation evoked repetitive discharge, once initiated, shows no sign of abating. Note the similarity to the synergism demonstrated empirically for TEA in the heterozygotes (Fig.6B). Temperature of the simulation is 20°C.
DISCUSSION
Myelinated fibers exhibit local variation in geometry as well as regional channel segregation. The transition zone between the myelinated and the nonmyelinated segment near the nerve terminal is a site of impedance mismatch that is particularly vulnerable to excitability perturbation, both physiologically and pathologically. This paper shows, through the use of a genetic mutant, that deleting a fast K channel (Kv1.1) that is normally segregated under the myelin sheath produces profound changes in the transition zone excitability not seen elsewhere in the nerve. Our empirical and theoretical study gives insight into how Kv1.1 channels function to stabilize a zone of inherent instability just proximal to the terminal and how synergistic interaction with a TEA-sensitive K channel or channels is indispensable to this function.
Kv1.1 is an axonal channel critical for stabilizing transition zones in axonal trees
Transition zones in general refer to the regions in an axonal tree where there is a local change in geometry caused by certain functional requirements. Important examples are the branch points or the nerve terminal region where the myelinated segment ends and the nonmyelinated segment begins. The safety factor for nerve conduction is altered at these sites because of impedance mismatch (Swadlow et al., 1980). Impedance mismatching is normally minimized by local variation in fiber geometry. For example, the internodes shorten as the nerve terminal is approached (Quick et al., 1979), and this has been shown theoretically to facilitate invasion of the nerve terminal (Khodorov and Timin, 1975). Even in normal adult, the disproportionately short internodes preceding the terminal may reflect active Schwann cell remodeling (Quick et al., 1979). In pathological situations, remyelination proceeds by forming short internodes preceding lesion sites, providing impedance matching that contributes to successful propagation (Waxman and Brill, 1978).
Other variations in local fiber geometry may also prove important. First, post-branching internodes are significantly smaller than the rest of the fiber (Pfeiffer and Friede, 1985). Second, in dorsal root ganglion (DRG) neurons, the first internode after the initial segment has an unusually thin myelin sheath (Spencer et al., 1973). At the branch point of DRG neurons, the caliber of the CNS-directed axon is different from the PNS-directed axon (Spencer et al., 1973). Third, action potentials could fail at branch points (Wall, 1995), and frequency-dependent failures at branch points (Grossman et al., 1973) could act as safety measures to prevent an injurious level of axonal activity from permeating other regions of the axonal tree. Collectively, transition zone excitability has profound implications for signal integration in axonal trees, both physiologically and pathophysiologically (Swadlow et al., 1980). Our paper suggests that Kv1.1 is important at these sites.
Is axonal segregation of K channels a critical factor in determining transition zone excitability? If so, what K channel subtypes are important? Recent physiological analysis of Kv1.1-deficient mice has produced compelling evidence that the axonal Kv1.1 channel is a major determinant of transition zone excitability, both in myelinated and nonmyelinated fibers. In the nonmyelinated axonal tree of a CNS neuron (the cerebellar basket cell), deletion of axonal Kv1.1 has been suggested to confer hyperexcitability on neurons by specifically reducing conduction failures at branch points (Zhang et al., 1999). In myelinated axons of the PNS, Kv1.1 is segregated under the myelin sheath and exerts a profound stabilizing effect on the action potential when it reaches the transition zone near the nerve terminal (Zhou et al., 1998; this paper). Computer simulations in this paper suggest that the preterminal shortening of the internodes, while functioning to facilitate invasion, also concomitantly predispose the system to re-excitation. Damping out re-excitation by the use of myelin-concealed K channels has the advantage of preserving a one-to-one transmission without altering the waveform of the action potential, the duration of which determines the amount of release and which can be independently controlled by K channels located right at the terminal itself.
Developmental changes in transition zone excitability
All the excitability changes observed in this study occur at or near the transition zone and not elsewhere in the myelinated fiber. There are three lines of argument to support this conclusion. First, none of the excitability changes originate from the nerve fiber, as demonstrated by the experiment in Figure 2 for the spontaneous activity and by Figure 6F for the evoked repetitive activity. Second, the nerve terminal membrane cannot be responsible for the abnormal excitability, because Kv1.1 is normally absent on the nerve terminal membrane in the wild type (before the gene deletion). This has been well demonstrated by immunohistochemistry in our previous paper (Zhou et al., 1998, their Fig. 4). Therefore, by elimination, the site of abnormal excitability is most likely at or near the myelinated segments before the transition zone. Third, this is further given strong support by the computer simulations in this paper, which demonstrate that the evoked repetitive discharge indeed originates from the myelinated nerve segment just before the transition zone (Figs.8-11).
During postnatal development of Kv1.1-null mice, two types of abnormal hyperexcitability (evoked backfiring and spontaneous activity) emerge from the transition zone region in succession. The stimulus-evoked evoked backfiring peaks at P17 and then declines, but is followed by a period of spontaneous activity that peaks at P30 and then also subsides. What causes the disappearance of evoked backfiring as myelination proceeds? One possibility is that it reflects developmental redistribution of Kv1.1 from the paranodal region (MYSA in our computer model) deeper to the juxtaparanodal region (FLUT in our model) (Vabnick et al., 1999). Indeed, Kv1.1 has a peak occupancy at the paranode at ∼P15–P20 and thereafter starts to shift to the juxtaparanode (Vabnick et al., 1999). However, our computer simulation does not support this explanation, at least for the particular geometry chosen in our model. We suspect a more important factor may be developmental changes in fiber geometry or morphology near the transition zone. For example, there might be a tightening of the paranodal seal during maturation. However, we are already using an adult paranodal seal (1 nm) in our simulations. Another important change in geometry might be an elongation of the terminal branches as the animal ages. Indeed, in our computer modeling, deletion of Kv1.1 no longer produces nerve backfiring when the three terminal branches are lengthened by a factor of 10 (data not shown).
It is unclear what mechanisms underlie the emergence of spontaneous activity in the older Kv1.1 mutant mice. The parallel appearance of TEA sensitivity suggests the involvement of a TEA-sensitive K channel. This TEA-sensitive channel normally is not important when Kv1.1 is present, but becomes critical when expression of Kv1.1 is compromised. In the null mutants, TEA induces spontaneous activity that does not require stimulation. It is interesting that the sensitivity to TEA remains unabated even in old mutants in which endogenous activity has all but subsided (Fig. 3E,F). This raises the possibility that the improvement of the excitability phenotype with age (>P60, Fig. 1B) may be related to a compensatory upregulation of a TEA-sensitive K channel. In the heterozygotes, in which Kv1.1 expression is reduced, TEA produces no phenotype until a single stimulation is given, in which case a virtually indefinite phase of repetitive discharge resulted. This synergistic interaction between TEA-sensitive and Kv1.1 channels might have profound implications in clinical syndromes involving Kv1.1 mutations such as episodic ataxia (Browne et al., 1994). For example, a patient may lack an overt phenotype because of the stabilizing action of residual a TEA-sensitive channel or channels. However, any interference with these TEA-sensitive channels, perhaps associated with metabolic stress, would allow a single ectopic action potential to incite a dramatic and protracted clinical symptom of hyperexcitability. The delicate balance is further illustrated by our computer modeling demonstrating that local perturbation of TEA-sensitive K channel in two or three internodal segments preceding the nerve terminal is sufficient to completely destabilize the system. The important point in this study is not only establishing the importance of Kv1.1, but also the unmasking of a delicate balance hinging on a residual K channel whose dysregulation may provoke the manifestation of clinical symptoms.
The nature of the TEA-sensitive channels
What is the channel subtype and location of the TEA-sensitive K channel that interacts with Kv1.1 to modulate hyperexcitability? One possibility is that TEA targets a presynaptic K channel, inhibition of which undermines the transition zone stability when Kv1.1 is deleted. An argument against this is that in our computer simulations, deleting only K channels in the presynaptic terminal branches makes no difference to the excitability of the transition zone, with or without Kv1.1. We believe a more likely possibility is that TEA targets slow K channels located on the axons. At present, the support for such an explanation is primarily theoretical. The synergism between Kv1.1 and TEA can be simulated in our model by manipulating only the fast and slow K channels on the axons. No combination of manipulations involving only presynaptic K channels, or with only fast axonal K channels, can reproduce this synergism. Slow K channels have been suggested to be blocked by TEA with an effective dose of ∼1 mm (Baker et al., 1987), which is consistent with our dose–response studies on TEA (Fig. 4B). We cannot rule out TEA exerting its effect by blocking residual fast K channels in our Kv1.1 mutants. However, as stated earlier, blocking all fast K channels alone in our computer model never reproduces long-lasting discharges. The idea that TEA-sensitive, slow K channels are important in modulating repetitive discharge in myelinated axons has already been suggested (Baker et al., 1987; Waxman, 1995). Our results suggest that this slow K channel is crucial at transition zones whenever Kv1.1 expression is compromised.
Footnotes
This work was supported by National Institutes of Health Grant RO1–23375 to S.Y.C. and A.M. We thank Tammy Robbins and Denise Springman for expert technical assistance.
Correspondence should be addressed to S. Y. Chiu, Department of Physiology, University of Wisconsin School of Medicine, 1300 University Avenue, 285 Medical Science Building, Madison, WI 53706.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}