

Abstract
cAMP is thought to be involved in learning process and known to enhance transmitter release in various systems. Previously we reported that cAMP enhances spontaneous transmitter release in the absence of extracellular Ca2+ and that the synaptic vesicle protein neuronal-synaptobrevin (n-syb), is required in this enhancement (n-syb-dependent; Yoshihara et al., 1999). In the present study, we examined the cAMP-induced enhancement of transmitter release in the presence of external Ca2+. We raised the intracellular concentration of cAMP by application of either forskolin, an activator of adenylyl cyclase, or by 4-chlorophenylthio-(CPT)-cAMP, a membrane-permeable analog of cAMP, in the presence of external Ca2+, while recording miniature synaptic currents (mSCs) at the neuromuscular junction in n-syb null mutant embryos. The frequency of mSCs increased in response to elevation of cAMP, and this effect of cAMP was completely blocked by Co2+ (n-syb-independent pathway). In contrast, in wild-type embryos the cAMP-induced mSC frequency increase was partially blocked by Co2+. In a mutant, DC0, defective in protein kinase A (PKA), nerve-evoked synaptic currents were indistinguishable from the control, but mSCs were less frequent. In this mutant the enhancement by cAMP of both nerve-evoked and spontaneous transmitter release was completely absent, even in the presence of external Ca2+. Taken together, these results suggest that cAMP enhances spontaneous transmitter release by increasing Ca2+ influx (n-syb-independent) as well as by modulating the release mechanism without Ca2+influx (n-syb-dependent) in wild-type embryos, and these two effects are mediated by PKA encoded by the DC0 gene.
- cAMP
- spontaneous synaptic currents
- neuronal-synaptobrevin
- DC0
- PKA
- forskolin
- neuromuscular junction
- Drosophila
- myosin heavy chain mutant
Synaptic plasticity is widely considered to be a basis for memory. The efficacy of synaptic transmission changes according to the history of activities in presynaptic and postsynaptic elements. The synapse maintains the imposed changes for a prolonged period, thus forming memory. InDrosophila memory mutants, dunce andrutabaga, the cAMP cascade is defective (Byers et al., 1981;Livingstone et al., 1984), and no post-tetanic facilitation was observed at the neuromuscular junction. Thus, changes in synaptic efficacy were suggested for the molecular mechanism of memory (Zhong and Wu, 1991). In Aplysia, cAMP mediates changes in synaptic transmission during dishabituation, sensitization, and classical conditioning (Byrne and Kandel, 1996). cAMP blocks various types of K+ channels, which in turn leads to membrane depolarization and/or a prolongation of presynaptic action potentials, and finally results in an activation of voltage-gated Ca2+ channels (Hawkins et al., 1983). The long-term potentiation (LTP) at the bullfrog sympathetic ganglion and at the rat hippocampal CA3 is also mediated by cAMP (Kuba and Kumamoto, 1986; Weisskopf et al., 1994) and requires Ca2+ influx at presynaptic terminals (Minota et al., 1991; Castillo et al., 1994). In other cases, cAMP directly enhances Ca2+ influx through modulation of Ca2+ channels (Artalejo et al., 1990; Gross et al., 1990). The Ca2+-independent effect of cAMP has also been demonstrated in the crayfish neuromuscular junction (Dixon and Atwood, 1989a,b) and in cultured mammalian CNS neurons (Chen and Regehr, 1997). Thus, the effects of cAMP on synaptic transmission are diverse. Multiple mechanisms might be operating in parallel in one synapse.
Using Drosophila genetics it is possible to separate the multiple mechanisms involved in the effects of cAMP on synaptic transmission. Previously we have examined synaptic transmission inDrosophila embryos lacking a synaptic vesicle protein, neuronal-synaptobrevin (n-syb; Deitcher et al., 1998), which is a v-soluble NSF attachment protein receptor (SNARE) protein (Söllner et al., 1993), and required for nerve-evoked transmitter release (Sweeney et al., 1995; Deitcher et al., 1998). Even though evoked release was absent, miniature synaptic currents (mSCs) were readily observed in n-syb null mutants. Their frequency increased in response to an increase of Ca2+ concentrations in high K+ saline. A Ca2+ ionophore, A23187, also increased the mSC frequency in the presence of external Ca2+. These findings indicate that then-syb null mutants are still capable of responding to elevations of internal Ca2+. Furthermore, in wild-type embryos cAMP increased the frequency of mSCs in the absence of external Ca2+, but did not in the n-syb null mutants (Yoshihara et al., 1999). Thus, requirements for spontaneous transmitter release and nerve-evoked release seem to be different.
The preceding results, showing that in the absence of external Ca2+ cAMP enhances spontaneous transmitter release, suggest two basic features regarding the effects of cAMP on spontaneous transmitter release. First, this pathway involves n-syb, a protein that is essential for evoked release (n-syb-dependent pathway). Second, cAMP enhancement of release is not dependent on external Ca2+. In this report, we asked using then-syb null mutant whether cAMP also enhances spontaneous transmitter release through an increase of intracellular Ca2+ when Ca2+ is available. We further asked whether PKA encoded by DC0 is involved in this enhancing effect of cAMP on transmitter release.
MATERIALS AND METHODS
Fly stocks. The n-syb null allele isolated by Deitcher et al. (1998) is designated asn-sybΔF33B. A chromosome withn-sybΔF33Bwas balanced with TM6, [y+], a balancer chromosome for the third chromosome with wild-type yellow gene. The chromosome with the null allele of myosin heavy-chain gene,Mhc1, was a gift from Dr. Kaname Mogami (University of Tokyo, Tokyo, Japan). It was balanced withCyO, [y+], a balancer chromosome for the second chromosome with wild yellow gene, making a stock y w; Mhc1/CyO, [y+]; n-sybΔF33B/TM6, [y+]. Double mutant homozygotes were distinguished from embryos of other genotypes by their yellow mouth hooks. We confirmed the double mutant genotype by the absence of synchronized transmitter release induced by nerve stimulation for every preparation preceding the experiment. This procedure was based on our previous finding that n-sybΔF33Bhomozygotes lack nerve-evoked synaptic currents (Deitcher et al., 1998). The genotype of Mhc1homozygotes was confirmed by the complete lack of movement.DC0B3 was a gift from Dr. Daniel Kalderon (Columbia University, New York, NY) and was also balanced withCyO, [y+], inyellow background. A double mutant,Mhc1DC0B3, was made by recombination and rebalanced with CyO, [y+].
Physiology. Procedures for recording synaptic currents were the same as described previously (Kidokoro and Nishikawa, 1994;Nishikawa and Kidokoro, 1996). The external saline contained (in mm): NaCl, 140; KCl, 2; MgCl2, 5; CaCl2, 1; and HEPES-NaOH, 5, pH 7.1. For recording mSCs, 3 μm tetrodotoxin (Wako Chemicals, Osaka, Japan) was included in external saline to prevent generation of action potentials. When the Ca2+ concentration was reduced, Mg2+ was increased by the same amount. The internal solution in patch pipettes contained (in mm): CsCl, 158; ATP, 2; EGTA, 5; and HEPES-NaOH, 10, pH 7.1. With this internal solution the recording electrode had a liquid junction potential of −5.3 mV in the external solution. Because the electrode was held at −60 mV during the voltage-clamp recording, the actual membrane potential was −65.3 mV. Forskolin (Wako Chemicals) was dissolved in 100% ethanol at a concentration of 10 mm and diluted in external saline shortly before use. The amount of dilution varied as indicated in each experiment. We added 0.5 ml of diluted forskolin solution to the bath containing 0.5 ml external solution. Thus, the final concentration of forskolin was half of that in the original solution. CPT-cAMP (Boehringer Mannheim, Mannheim, Germany) was dissolved in the external solution at 2 mm and was sonicated. We added 0.5 ml of this solution to the bath containing 0.5 ml of the external solution. Thus, the final concentration of 4-chlorophenylthio-(CPT)-cAMP was 1 mm. External saline containing 2 mmCo2+ was prepared by reducing Mg2+ by the same amount and perfused into the bath after removing the bath solution to a minimal amount (∼0.2 ml). For nerve stimulation, the tip of a microelectrode filled with 4m K acetate and having resistance of ∼10 MΩ was placed in the ventral ganglion.
During repetitive stimulation with a microelectrode placed in the ventral ganglion, motoneurons sometimes became agitated and generated frequent spontaneous synaptic currents, probably because of neuronal activities within the ganglion. To avoid this situation, for experiments to evoke synaptic currents by nerve stimulation, pentobarbital (1 mm) was included in the bath solution to suppress CNS activities, and MCCG-I (100 μm; Wako Chemicals) was also added to the bath to block metabotropic glutamate receptors (Zhang et al., 1999). The falling phase of synaptic currents was measured by fitting with one exponential by the least squares method. The falling decay time constant depends on the amplitude of synaptic currents (Kidokoro and Nishikawa, 1994). Therefore, synaptic currents having an amplitude between 100 and 200 pA were selected, 5–20 synaptic currents were averaged, and one exponential was fitted to the averaged synaptic current. All electrophysiological experiments were performed at room temperature (21–26°C).
Immunohistochemistry. Staining with anti-HRP, anti-synaptotagmin, or anti-β-galactosidase was performed as described previously (Yoshihara et al., 1997). We used FITC-conjugated goat IgG against HRP (Organon Teknika, West Chester, PA) at a dilution of 1:100, rabbit serum antibody against synaptotagmin (Littleton et al., 1993; kindly provided by Dr. Hugo Bellen, Baylor College of Medicine, Houston, TX) at a dilution of 1:500, and rabbit IgG against β-galactosidase (Organon Teknika) at a dilution of 1:1000.
RESULTS
Previously we examined the effect of cAMP on spontaneous transmitter release in the absence of external Ca2+ (Yoshihara et al., 1999; Zhang et al., 1999). In this study we wished to extend our work to the effect of cAMP on spontaneous transmitter release in the presence of external Ca2+ and on nerve-evoked synaptic currents. In the presence of external Ca2+, however, muscles in newly hatched wild-type larvae tended to contract vigorously after repetitive activation of presynaptic terminals. Muscle contraction stretched the nerve, resulting in frequent spontaneous synaptic events, which rendered analysis of synaptic transmission difficult. To avoid this complication we used a noncontracting muscle mutant,Mhc1, which is a null mutant of the myosin heavy chain (Mogami et al., 1986).
To ascertain that the phenotype ofMhc1 mutation is restricted to muscles we examined gene expression of the myosin heavy chain in embryos 21–24 hr after fertilization, using an Mhc-β-galactosidase transformant (Hess et al., 1989) and anti-β-gal antibody. The expression was restricted to muscles and was not found in neurons (data not shown). We also examined the morphology of neuromuscular junctions in those embryos using anti-HRP and anti-synaptotagmin antibodies. Presynaptic terminals were generally normal, although muscles were smaller and thinner than those of wild-type larvae (data not shown).
Neuromuscular synaptic transmission in a noncontracting myosin mutant, Mhc1
Synaptic currents were recorded from longitudinal body-wall muscles, mainly numbers 6 or 7 or occasionally 13, using the whole-cell patch-clamp technique. In the presence of tetrodotoxin (3 μm) mSCs were examined. In saline with 0.5 mmCa2+ the mSC frequency was 3.7 ± 2.2/min (mean ± SD; the number of cells examined,n = 28; data are expressed in this format throughout the text unless otherwise stated), which is similar to that in wild-type (2.0 ± 1.6/min; n = 5; Deitcher et al., 1998). The mean amplitude of mSCs was 179 ± 36 pA (n = 14), which is also similar to that in wild-type (168 ± 24 pA; n = 6; Deitcher et al., 1998). Nerve-evoked synaptic currents were examined in the presence of 0.5 mm Ca2+. The failure rate was 0.64 ± 0.14 (n = 6), and the mean amplitude was 289 ± 124 pA (n = 6, excluding failures) in the presence of a blocker of metabotropic glutamate receptors, 100 μm MCCG-I, and a general anesthetic, 1 mm pentobarbital (see Materials and Methods). Thus, we concluded that the properties of synaptic currents in this noncontracting mutant were similar to those in wild-type.
Forskolin, an activator of adenylyl cyclase, and CPT-cAMP, a cAMP analog, increased the mSC frequency inMhc1
Spontaneous synaptic currents were infrequent in the presence of TTX (3 μm) and 0.5 mmCa2+ (Fig.1A), and the frequency increased when forskolin was added in the bath (final concentration 100 μm) (Fig. 1B). The frequency of mSCs started to increase at ∼10 min after the addition of forskolin in the bath and continued to increase toward the end of observation period of 30 min (Fig.2Aa). The time course of the frequency change was similar in all five preparations examined, but the extent of increase was somewhat variable; the averaged frequency at 30 min after addition of forskolin was 403 ± 404/min (n = 5; Fig. 2Aa). A control solution, containing the same amount of ethanol that was used to dissolve forskolin, did not affect the mSC frequency (Fig.2Ab), nor did a solution containing a nonactive analog of forskolin, dideoxyforskolin (ddFSK; Seamon and Daly, 1986; Fig. 2Ac).

Sample traces of mSCs in anMhc1 mutant embryo in the presence of 0.5 mm Ca2+ and 3 μm TTX.A, Before application of forskolin. Downward deflections indicate inward currents. B, 28 min after application of forskolin (100 μm). Calibration bars shown inB apply also to A.

Effects of forskolin (A) and CPT-cAMP (B) on the frequency of mSCs at the neuromuscular junction of Mhc1 mutant embryos in the presence of external Ca2+ (0.5 mm) and 3 μm TTX. Aa, At theleft end of the horizontal bar, forskolin was infused in the bath. The final concentration was 100 μm. The sample number is 5. Error bars attached to each symbol indicate the SEM. Ab, Control. The solution containing 2% ethanol, which was used to dissolve forskolin, was added to the bath. The final concentration was 1%. The sample number is 6. Error bars attached to each symbol indicate the SEM. Some bars are smaller than the symbol. Ac, Control. The solution containing 200 μm ddFSK was added to the bath. The final concentration was 100 μm. All error bars are smaller than the symbol. The sample number is 5. Ba,Data obtained from one cell in anMhc1 embryo before and during application of CPT-cAMP (1 mm). Four of six cases showed a similar effect, although the time course was somewhat variable among individual cases, whereas in two samples the effect was not observed. Because of this variability among cells, the data from a cell are depicted. Bb, Control. The sample number is 5. All error bars are smaller than the symbol.
To confirm that the increase of mSCs induced by forskolin was mediated by an elevation of cAMP, we examined the effect of a membrane-permeable analog of cAMP, CPT-cAMP, and found that in a majority of cases (four of six cases tested) this compound at 1 mm also transiently increased the mSC frequency (Fig. 2Ba, data from one cell are depicted because of the variability of time course among cells). No effects were observed when saline was used instead of the CPT-cAMP-containing solution (Fig. 2Bb). The time course was variable, but in all cases in which the response was observed, the change was transient. The transient increase in the mSC frequency was also observed previously in wild-type larvae with another analog of cAMP, dibutyryl cAMP (Yoshihara et al., 1999). The transient changes in the mSC frequency induced by cAMP analogs are in contrast to the steady and robust increase induced by forskolin. These cAMP analogs might be degraded promptly in the terminal by enzymes, such as phosphodiesterases, whereas cAMP may be generated continuously by activation of adenylyl cyclase by forskolin. The mean peak frequency was 436 ± 516/min (n = 4). Thus, the cAMP analog also increased the mSC frequency.
To determine whether an influx of Ca2+ was contributing to the mSC frequency increase induced by forskolin, we used a general voltage-gated Ca2+ channel blocker, Co2+. We first confirmed that the mSC frequency increase induced by high K+was completely blocked by Co2+ at 2 mm (20 mm KCl, six cells tested, data not shown). This result indicated that Co2+does block voltage-gated Ca2+ channels inMhc1 mutant presynaptic nerve terminals. After the increase of mSC frequency induced by forskolin became evident, 2 mmCo2+ was perfused into the bath (Fig.3A; data from one cell are depicted because the timing of Co2+application was not the same among cells examined). The mSC frequency decreased immediately to a lower level (indicated by an arrow). The mean mSC frequency after application of Co2+ was 222 ± 225/min (n = 3). The remaining level of mSCs in the presence of Co2+ suggested that forskolin increased the mSC frequency without influx of Ca2+in Mhc1, as was previously found in wild-type (Yoshihara et al., 1999). However, it was equally plausible that Ca2+ that had entered before application of Co2+ may have contributed to an elevated level of internal Ca2+concentration or that there was Co2+-insensitive Ca2+ influx. To evaluate the contribution of these possible mechanisms, we added Co2+ (2 mm) in the external medium before application of forskolin. Still the mSC frequency increased, but to a lower level (94 ± 125/min;n = 4; Fig. 3B), as was observed in wild-type embryos after application of forskolin in the absence of external Ca2+ (Yoshihara et al., 1999). It appeared that there are two components in the effect of forskolin on the mSC frequency. One is external Ca2+-dependent, which is a Co2+-blockable major component, and the other is a minor component that is either dependent on Co2+-insensitive Ca2+ influx or external Ca2+-independent. The latter two possibilities were tested in the next section by using a double mutant,Mhc1; n-sybΔF33B.

The effect of Co2+ on the mSC frequency increase induced by forskolin. A, Forskolin was applied 30 min before the first data point in the graph. An example of perfusion of 2 mm Co2+ during the forskolin (500 μm) application on anMhc1 mutant embryo in the presence of external Ca2+ (1 mm). All five samples in the same series of experiments showed a similar effect. Because the timing of Co2+ application was not the same in all five cases, only one sample is shown here. A horizontal arrow indicates the level of mSC frequency in the presence of 2 mm Co2+. B, The effects of forskolin (500 μm) in theMhc1 mutant in the presence of 2 mm Co2+ and 1 mmCa2+. The sample number is 4, and data from each sample were averaged. Error bars attached to each symbol indicate the SEM. Some error bars are smaller than the symbol.
During the series of experiments described below we unexpectedly observed a clear shortening of mSC time course after application of 500 μm forskolin in Mhc1. The time course of mSC falling phase was 4.9 ± 1.3 msec before and 2.0 ± 0.2 msec (n = 5) after application of forskolin (these two means are significantly different atp = 0.01, t test for paired data; see Materials and Methods for details of measurement). The mean amplitude was not significantly different (183 ± 39 pA, n = 8, and 163 ± 26 pA, n = 7, for before and after forskolin application; p > 0.05). Because this finding was not along the main theme of this study we did not pursue it any further.
Spontaneous transmitter release in a double mutant (Mhc1;n-sybΔF33B)
Above we showed that the major effect of forskolin on mSC frequency was mediated by Ca2+ influx through voltage-gated Ca2+ channels. To determine whether the remaining increase in the mSC frequency in the presence of Co2+ in the external solution is caused by an external Ca2+-independent effect or a Co2+-insensitive Ca2+ influx, we used n-sybmutant embryos. We have shown previously in a neuronal-synaptobrevin-null mutant, n-sybΔF33B, that an elevation of internal Ca2+ by various means increases the mSC frequency (Yoshihara et al., 1999). If forskolin increases the mSC frequency inMhc1 by increasing internal Ca2+ through a Co2+-insensitive pathway, it should do so in a double mutant, Mhc1; n-sybΔF33B. On the other hand, if the remaining increase of mSC frequency induced by forskolin is an external Ca2+-independent component in Mhc1, no increase is expected to occur in the double mutant in the presence of Co2+ (n-syb-dependent).
First, we confirmed the effect of forskolin on the mSC frequency in the double mutant. In 1 mm Ca2+saline forskolin gradually increased the mSC frequency to 73 ± 66/min (n = 4) (Fig.4Aa) in the double mutant. The time course of the mSC frequency change was somewhat different from that in Mhc1. After the initial onset, at ∼10 min after addition of forskolin, the mSC frequency did not continue to increase throughout the 30 min recording period, resulting in a lesser increase at the end. When the infusing solution contained only ethanol without forskolin, no change in the mSC frequency was observed (Fig. 4Ab). Furthermore, a nonactive analog of forskolin, ddFSK, had no effect (Fig.4Ac). Thus, we concluded that the effect on the mSC frequency in this double mutant was specific to forskolin acting.

Effects of forskolin and CPT-cAMP on the mSC frequency in the double mutant Mhc1;n-syb ΔF33B, in the presence of external Ca2+ (1 mm). Forskolin (500 μm, Aa), 5% ethanol used as a solvent for forskolin as a control (Ab), as a nonactive analog of forskolin, ddFSK (500 μm,Ac), an analog of cAMP, CPT-cAMP (1 mm,Ba), or saline for control of CPT-cAMP (Bb) was applied during the time indicated by thehorizontal bars below the abscissa. The error bars attached to each symbol indicate the SEM. Some bars are smaller than the symbol. The external Ca2+ concentration was 1 mm for Aa, Ab, andAc, and 0.5 mm for Ba andBb. Three micromolar tetrodotoxin was included in the external saline to prevent generation of action potentials in the nerve. Sample numbers are 4 in Aa, 3 inAb, 3 in Ac, 5 in Bb, and data from each sample were averaged. In Ba, because the time course varied from one sample to another, a representative example is shown. Three examples of five cases showed a similar effect, whereas two samples showed no effects. Bb, Control. The error bars attached to each symbol indicate the SEM. Some bars are smaller than the symbol.
CPT-cAMP had an enhancing effect (Fig. 4Ba), but saline did not (Fig. 4Bb), suggesting that an elevation of cAMP level in the presynaptic nerve terminal causes an increase in Ca2+ influx which, in turn, results in the higher mSC frequency. However, the magnitude of the frequency change in the double mutant was smaller than that inMhc1 (Fig. 2Aa). The smaller increase in the double mutant was probably attributable to a lower sensitivity to Ca2+ and to a lack of the n-syb-dependent component, caused by n-sybΔF33Bmutation (Yoshihara et al., 1999).
Finally, we examined the effect of Co2+ on the forskolin-induced mSC frequency change in the double mutant. After the mSC frequency increase was induced by forskolin, 2 mmCo2+ was perfused in the bath. The mSC frequency dropped immediately to almost zero in all five preparations examined (Fig. 5; data from one cell are depicted because the timing of Co2+application was not the same among cells examined). Thus, we concluded that the increase in mSC frequency in the double mutant was solely attributable to influx of Ca2+ induced by forskolin. Therefore, the Co2+-insensitive minor component of the forskolin effect on mSC frequency observed inMhc1 was external Ca2+-independent and n-syb-dependent and was the same component as we had identified previously (Yoshihara et al., 1999).

Effects of Co2+ on the forskolin-induced mSC frequency increase in the double mutantMhc1; n-sybΔF33B, in the presence of external Ca2+ (1 mm). Co2+ (2 mm) was perfused to block voltage-gated Ca2+ channels during the period in which forskolin (500 μm) was applied. Forskolin and Co2+ were applied during the time indicated by thehorizontal bars below the abscissa. A representative example is shown here. All five samples in the same series of experiments showed complete disappearance of mSCs after application of Co2+. Because the timing of Co2+application was not the same in all cases, only one example is depicted here.
Synaptic transmission in a double mutant,Mhc1DC0B3
So far we have shown that there are two pathways, n-syb-dependent and n-syb-independent, for forskolin–cAMP to increase the mSC frequency. In most cases cAMP exerts effects through activating PKA (Coffino et al., 1976). We next examined whether PKA was involved in these pathways. In Drosophila, the DC0 locus codes the sole or major catalytic subunit of PKA, andDC0B3 is a null allele (Lane and Kalderon, 1993).
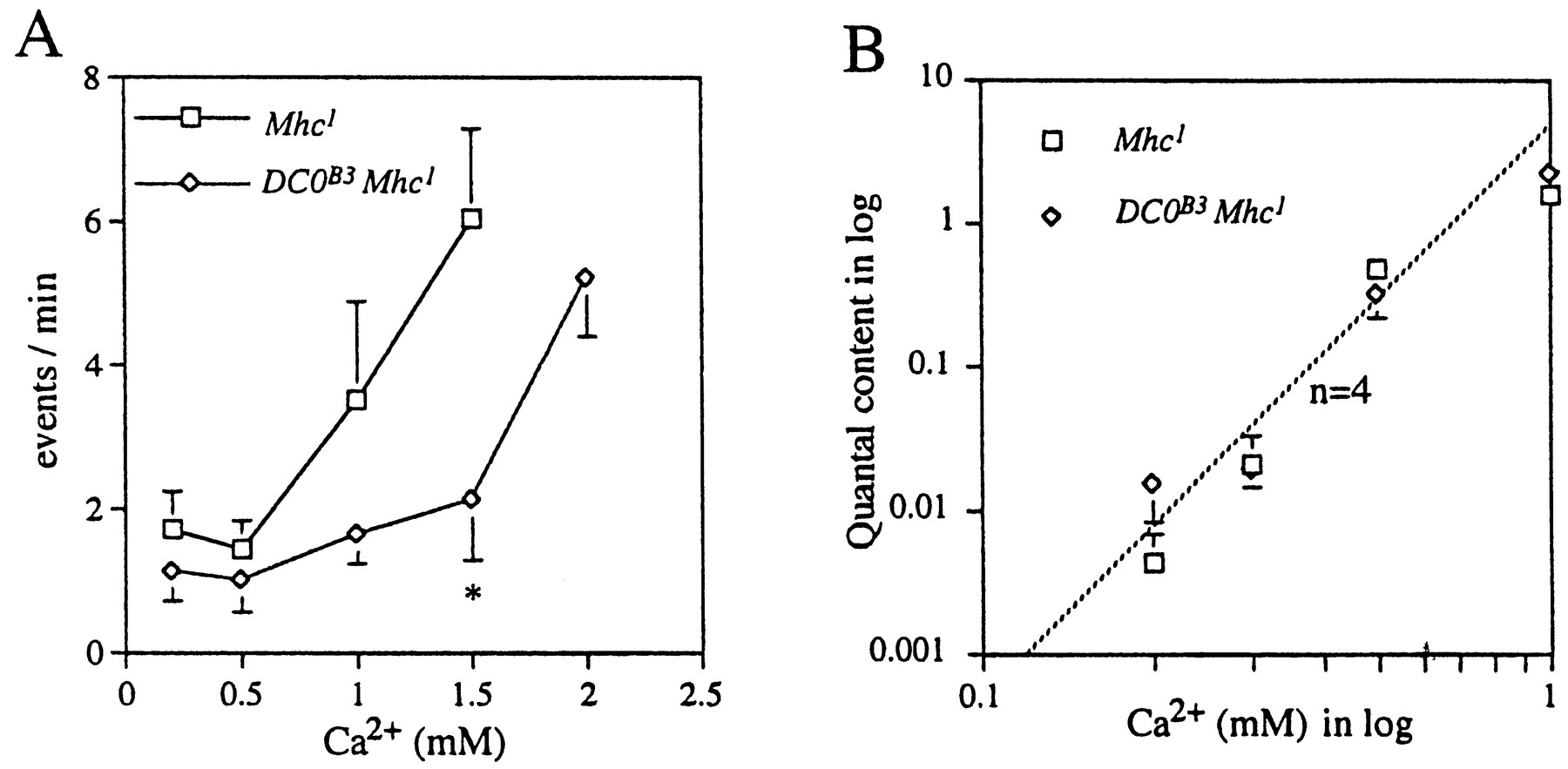
Generally the mSC frequency in the double mutantMhc1DC0B3 was lower than that inMhc1. At normal K+ (2 mm) and 0.5 mm Ca2+, the frequency was 1.1 ± 0.7/min (n = 14) in the double mutant, whereas it was 3.7 ± 2.2/min (n = 28) in Mhc1 (these two means are significantly different at p = 0.01; Student'st test). In 20 mmK+ solutions the mSC frequency also tended to be lower in the double mutant compared withMhc1 (Fig.6A). The mean amplitude of mSCs was 144 ± 28 pA (n = 5), which is similar to that in Mhc1 (179 ± 36 pA,n = 14; these two means are not significantly different at p = 0.05). Davis et al. (1998) reported that inhibition of PKA increased the mSC amplitude significantly at neuromuscular junctions in third instar larvae. Their result is seemingly in contradiction to our data inDC0B3, in which the mean amplitude of mSCs was not larger than control. However, because our experiments were performed in embryos, developmental changes to increase the mSC amplitude might not yet have happened.

Dependence of transmitter release on external Ca2+ in Mhc1 andDC0B3. A, The relation between the frequency of mSCs and external Ca2+concentration in the 20 mm K+ solution in the presence of 3 μm TTX. The frequency was plotted against the external Ca2+ concentration.Squares are for theMhc1 mutant, anddiamonds are for the double mutantDC0B3Mhc1. Error bars attached to each symbol indicate the SEM. Neighboring data points were connected bystraight lines. An asterisk indicates statistical difference between Mhc1and DC0B3Mhc1 at p = 0.05.B, The relation between the quantal content and external Ca2+ concentration. The quantal content was calculated by the following equation: m =ln (N/n0), where n0 is the number of failures, andN is the total number of stimuli. Each data point represents the mean, and the error bar indicates SEM from more than five muscle fibers. The ventral ganglion was stimulated once every 3 sec for 30–400 times to measure the failure rate. Pentobarbital (1 mm) was included in the bath solution to suppress CNS activities. MCCG-I (100 μm) was also added to the bath to block metabotropic glutamate receptors (Zhang et al., 1999). The line of slope n = 4 is drawn for reference.
The nerve-evoked synaptic currents were not different in the double mutant from those in Mhc1. The mean amplitude in 0.5 mmCa2+, excluding failures, was 205 ± 30 pA (n = 7) in the double mutant, whereas it was 289 ± 124 pA (n = 6) inMhc1 (these two means are not significantly different at p = 0.05). To determine the Ca2+ dependence of synaptic transmission, we measured the quantal content by the failure method in various external Ca2+ concentrations, assuming Poisson statistics for release of transmitter (Katz, 1969). There was no difference in the Ca2+ dependence of the double mutant and Mhc1 (Fig.6B). The slope of the dependence was ∼4. This result was in accord with a behavioral observation thatDC0B3 larvae shortly after hatching exhibited coordinated movements.
We then examined the effect of forskolin and cAMP on spontaneous transmitter release in the Mhc1DC0B3 in the presence of 0.5 mm Ca2+ and 3 μm TTX. Neither forskolin nor CPT-cAMP changed the mSC frequency (Fig. 7B,C). Forskolin did not alter the mSC frequency in the absence of external Ca2+, even in theDC0B3 single mutant (Fig.7A). These results indicated that neither of the forskolin–cAMP-induced pathways that increase the mSC frequency was operating in the DC0B3 embryos. Thus, the effect of forskolin–cAMP is mediated by the cAMP–PKA cascade.
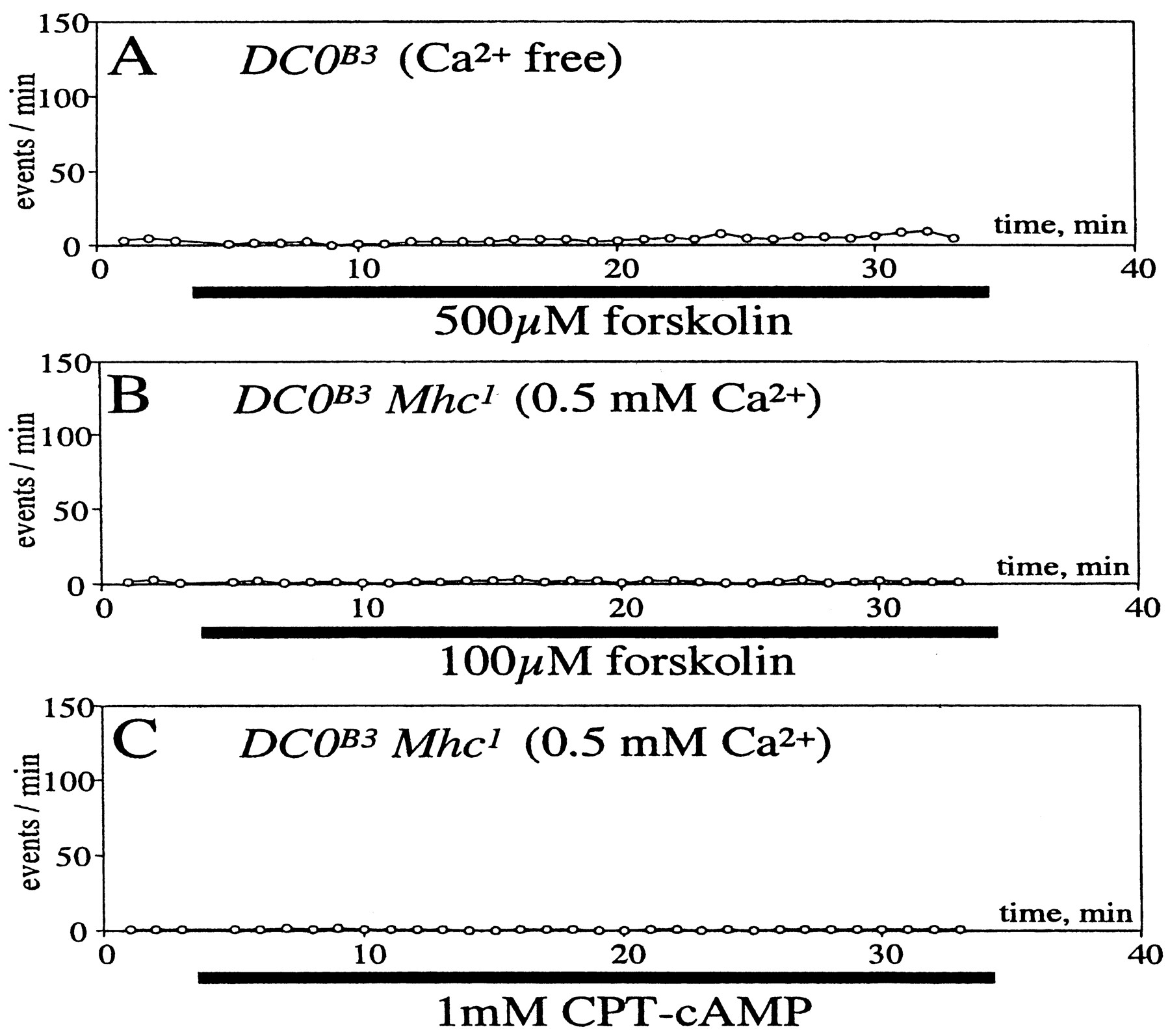
Effects of cAMP on the mSC frequency in theDC0B3Mhc1 double mutant. A,Forskolin (500 μm) was applied toDC0B3 mutant in the absence of external Ca2+ during the time indicated by the horizontal bar below the abscissa. B, Forskolin (100 μm) or an analog of cAMP, CPT-cAMP (1 mm) (C) was applied toDC0B3Mhc1 double mutant in the presence of external Ca2+ (0.5 mm) during the time indicated by the horizontal bars below the abscissa. Tetrodotoxin (3 μm) was included in external saline to prevent generation of action potentials in the nerve. Sample numbers are 3 in A, 4 in B, 5 inC, and data from each sample were averaged. All error bars are smaller than the symbol.
Forskolin did not affect the quantal content of synaptic transmission in Mhc1DC0B3 embryos in the presence of 0.3 mm Ca2+ (Fig.8C). In contrast, the effect of forskolin in the quantal content was evident inMhc1 (Fig. 8A). As expected, ddFSK did not affect the quantal content, even inMhc1 (Fig. 8B). Thus, we concluded that null mutation at the DC0 locus completely eliminated the forskolin–cAMP effect on nerve-evoked synaptic transmission.

Effects of forskolin on the quantal content of nerve-evoked synaptic currents in 0.3 mm external Ca2+. A,Mhc1 homozygous embryos treated by forskolin (100 μm). B,Mhc1 homozygous embryos treated by ddFSK (100 μm) for control. C,DC0B3Mhc1 homozygous double mutant embryos treated by forskolin (100 μm). The ventral ganglion was stimulated once every 3 sec. A failure rate was calculated for 50 stimuli twice before application of the drug. Ten minutes after application of the drug, CNS was stimulated again once every 3 sec up to 35 min after application of the drugs, and failure rates were calculated for each of set of consecutive 50 stimuli. Pentobarbital (1 mm) was included in the bath solution to suppress activity in the ganglion. MCCG-I (100 μm) was also added to the bath to block metabotropic glutamate receptors in the terminal. Each sample number is 4 for all treatments. All error bars are smaller than the symbol.
DISCUSSION
Two pathways for cAMP-induced enhancement of spontaneous transmitter release
In the presence of external Ca2+, forskolin and CPT-cAMP increased the mSC frequency in control embryos (Mhc1) (Figs. 1,2Aa,Ba). This increase of mSC frequency was reduced to a lower level after application of a nonspecific Ca2+ channel blocker, Co2+ (Fig. 3A), suggesting that an influx of Ca2+ through voltage-gated Ca2+ channels is playing a major role in the cAMP-induced mSC frequency increase. Forskolin or CPT-cAMP also induced an increase of mSC frequency in a double mutant,Mhc1; n-sybΔF33B (Fig.4Aa,Ba). In the n-sybΔF33Bmutant no n-syb protein is synthesized (Deitcher et al., 1998). Therefore, at least a part of the cAMP-induced mSC frequency increase observed in Mhc1 did not require n-syb protein (n-syb-independent).
However, even in the presence of Co2+, a minor but significant increase of mSC frequency was induced by forskolin (Fig. 3B). This small increase may be either attributable to the external Ca2+-independent effect of cAMP as previously described in this system (n-syb-dependent; Yoshihara et al., 1999) or to Ca2+ influx that was not blocked by Co2+. To distinguish these two possibilities we used the double mutant,Mhc1; n-sybΔF33B. Inn-sybΔF33Bembryos an elevation of internal Ca2+ by various means, such as high K+ or Ca2+ ionophore in the presence of external Ca2+, does increase the mSC frequency (Yoshihara et al., 1999). Therefore, if Co2+ were not blocking all Ca2+ influx induced by cAMP, there should be an increase in the mSC frequency inMhc1; n-sybΔF33B. On the other hand, if cAMP were increasing the mSC frequency independently from Ca2+ influx inMhc1 embryos (n-syb-dependent pathway), there should be no increase in the double mutant,Mhc1; n-sybΔF33Bin the presence of Co2+. In the double mutant, Mhc1; n-sybΔF33B, Co2+ completely blocked the effect of forskolin (Fig. 5). Therefore, the remaining increase induced by cAMP in Co2+-treatedMhc1 preparations was likely caused by the external Ca2+-independent, n-syb-dependent effect on vesicle fusion. Taken together, we conclude that there are two pathways for the cAMP effect on mSC frequency, namely external Ca2+-dependent (n-syb-independent) and external Ca2+-independent (n-syb-dependent) pathways.
Ca2+-independent pathway for cAMP-enhanced transmitter release
In various preparations cAMP, although facilitating nerve-evoked synaptic transmission in a prolonged time course, does not increase the Ca2+ concentration in the presynaptic terminal. In the crayfish neuromuscular junction, Wojtowicz and Atwood (1988) have shown that long-term facilitation after tetanic nerve stimulation occurs without an increase in the terminal Ca2+ concentration. This long-term facilitation is later shown to be mediated by the adenylyl cyclase system (Dixon and Atwood, 1989a). 5-HT facilitates synaptic transmission at the crayfish neuromuscular junction, and this effect is not accompanied with an increase in the Ca2+ level in the terminal (Delaney et al., 1991) and is partly mediated by cAMP (Dixon and Atwood, 1989b). Furthermore, cAMP did not increase the presynaptic Ca2+ concentration during presynaptic facilitation in a rat cerebellar synapse (Chen and Regehr, 1997), and at least some part of cAMP-induced facilitation was not mediated by Ca2+ in synapses between cultured hippocampal neurons (Trudeau et al., 1996, 1998). In Aplysiathe second component in facilitating synaptic transmission induced by 5-HT is not mediated by spike broadening. That is, when the synapse is depressed after repeated activation of the involved pathway, the presynaptic spike broadening has little effect on transmitter release. Yet 5-HT enhances synaptic transmission, suggesting another component for synaptic facilitation (Hochner et al., 1986). Injection of BAPTA in the presynaptic neuron in Aplysia neuronal cultures did not change the effect of 5-HT in enhancing the frequency of miniature synaptic potentials (Dale and Kandel, 1990). Thus, Ca2+-independent effects of cAMP on nerve-evoked transmitter release and on spontaneous release are observed in various systems.
In the n-syb-dependent, external Ca2+-independent pathway, cAMP might be directly modulating the vesicle fusion mechanism without increasing internal Ca2+, as are the cases cited above. However, we could not entirely exclude the possibility that this pathway is mediated by release of Ca2+from internal stores. If that were the case, this Ca2+ release mechanism must be dependent on n-syb. Thus, we have to assume two roles for n-syb; one is an essential element of the SNARE complex, and the other is an entirely new role in cAMP-induced Ca2+ release from internal stores.
External Ca2+-dependent, n-syb-independent pathway for cAMP-enhanced transmitter release
In Aplysia, cAMP enhances synaptic transmission through the external Ca2+-dependent pathway (Byrne and Kandel, 1996). The facilitation induced by 5-HT or by tail shocks is the result of both a broadening of the presynaptic spike and an enhancement in the membrane excitability. This effect is mediated by the cAMP–PKA cascade. In search of the substrates of PKA, a novel K+ current was found to be modulated by cAMP. Siegelbaum et al. (1982) have shown that cAMP closes the K+ channels in sensory neurons. A blockade of the K+ channels results in broadening of presynaptic action potentials and enhances an electrical excitability. Later it was found that the effect of 5-HT on the voltage-gated slow, transient K+ current also contributes more to the spike broadening (Baxter and Byrne, 1989). These effects on K+ currents are considered to be underlying mechanisms for tail shock-induced synaptic facilitation.
In larval Drosophila neurons in culture, cAMP reduces slowly inactivating or noninactivating outward K+currents (Delgado et al., 1998). Motoneurons innervating muscles may have those cAMP-sensitive currents. A blockade of these currents at the terminal by cAMP may increase membrane potential fluctuations caused by spontaneous openings of undefined ion channels, which leads to spontaneous opening of voltage-gated Ca2+channels. This chain of events may result in an increase in the mSC frequency.
Another possible mechanism for cAMP-induced synaptic facilitation involves a direct action of cAMP on the Ca2+ influx through voltage-gated Ca2+ channels. In bovine chromaffin cells, activation of D1 dopamine receptors enhances Ca2+ currents through the cAMP–PKA-dependent mechanism (Artalejo et al., 1990). It has also been shown that activation of a catalytic subunit of PKA enhances calcium currents in rat nodose neurons (Gross et al., 1990). A similar mechanism in the presynaptic terminal will facilitate synaptic transmission after elevation of cAMP.
Synaptic transmission in a PKA-null mutant,DC0B3
In a double mutant, Mhc1DC0B3, the amplitude and Ca2+ dependency of nerve-evoked synaptic currents were not significantly different from those in the control, Mhc1, whereas the mSC frequency was lower (Fig. 6). Conversely, in a n-syb null mutant, n-sybΔF33B, no nerve-evoked synaptic currents were detected, whereas mSCs were readily observable (Deitcher et al., 1998; Yoshihara et al., 1999). Thus, it appears that these two modes of vesicle fusion, nerve-evoked and spontaneous, seem to have distinct requirements.
Under various conditions there is a good correlation between the frequency of mSCs and the number of quanta released by nerve stimulation (Van der Kloot and Molgó, 1994). In rat cerebellar synapses, Chen and Regehr (1997) have shown a clear correlation between the frequency of mSCs and the amplitude of evoked synaptic currents in preparations treated with various concentrations of forskolin. In accordance with their report, inMhc1 embryos, forskolin increased the frequency of mSCs (Fig. 2Aa) and the quantal content (Fig. 8A) in a similar time course. Furthermore, in the in Mhc1 DC0 mutant the effect of forskolin was observed neither in spontaneous transmitter release (Fig. 7) nor in nerve-evoked release (Fig. 8). These results suggest that these two modes of transmitter release are similarly affected by cAMP–PKA.
cAMP is shown at the Drosophila neuromuscular junction in third instar larvae to increase the size of exo–endo cycling pool (readily releasable pool) of synaptic vesicles (Kuromi and Kidokoro, 1998, 1999). The size of this pool is closely correlated with the quantal content of synaptic potentials evoked by nerve stimulation at a low frequency (Kuromi and Kidokoro, 1999). Forskolin increases the mSC frequency in Mhc1 embryos (Figs. 1,2Aa) and in newly hatched wild-type larvae (Zhang et al., 1999). Thus, it is likely that this pool supplies vesicles for both modes of transmitter release. However, the two modes, namely nerve-evoked and spontaneous transmitter release, dichotomize after this step. For nerve-evoked release n-syb protein is required, whereas for spontaneous fusion this protein is not of absolute necessity, although its presence facilitates spontaneous transmitter release (Deitcher et al., 1998; Yoshihara et al., 1999). The cAMP–PKA cascade seems to influence the vesicle fusion process at multiple levels: (1) vesicle mobilization and translocation, which increase the size of exo–endo cycling pool (Kuromi and Kidokoro, 2000), (2) modification of Ca2+ influx through voltage-gated Ca2+ channels (n-syb-independent pathway; this study), and (3) modulation of transmitter vesicle fusion (n-syb-dependent pathway; Yoshihara et al., 1999). It is likely that the first mechanism affects both modes of vesicle fusion similarly. However, the second and third mechanisms may act differentially on the two modes of vesicle fusion, which may explain the phenotype of the double mutant, Mhc1DC0B3. Namely at the resting state the cAMP–PKA cascade might not be affecting the nerve-evoked transmitter release, whereas spontaneous release might be supported by the baseline activity level of the cascade.
Footnotes
This work was supported by a grant-in-aid from Ministry of Education, Science, Sports, and Culture of Japan to M.Y. and Y.K., and by the Uehara Memorial Foundation to M.Y. We thank Dr. Kaname Mogami for theMhc1 strain and his advice about the mutant, Drs. Mary B. Rheuben and Hiroshi Kuromi for critical reading of the manuscript, Dr. Sanford I. Bernstein for the MHC-β-gal transformant, Dr. Tadashi Uemura for theCyO, [y+] strain, Dr. Daniel Kalderon for the DC0B3strain, and Ms. Masako Terada, Fumiko Sekiguchi, and Nobuko Yoshihara for technical assistance.
Correspondence should be addressed to Dr. Yoshi Kidokoro, Gunma University, School of Medicine, 3-39-22 Showa-machi, Maebashi, 371–8511 Japan. E-mail: Kidokoro{at}med.gunma-u.ac.jp.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}