

Abstract
The development of hyperpolarizing inhibition is an important step in the maturation of neuronal networks. Hyperpolarizing inhibition requires Cl− outward transport that is accomplished by KCC2, a K+/Cl− cotransporter. We show that cultured hippocampal neurons initially contain an inactive form of the KCC2 protein, which becomes activated during subsequent maturation of the neurons. We also show that this process is accelerated by transient stimulation of IGF-1 receptors. Because the transporter can be rapidly activated by coapplication of IGF-1 and an Src kinase and can be deactivated by membrane-permeable protein tyrosine kinase inhibitors, we suggest that activation of K+/Cl− cotransporter function by endogenous protein tyrosine kinases mediates the developmental switch of GABAergic responses to hyperpolarizing inhibition.
In the developing rat hippocampus, fast GABAergic transmission is depolarizing early in development and becomes hyperpolarizing and strictly inhibitory only by the end of the first postnatal week (Cherubini et al., 1991). The direction of Cl− currents flowing through GABAA receptors depends on the transmembranal gradient for Cl− currents. Cl− influx requires low levels of intracellular [Cl−], which is achieved by extrusion of Cl− by a furosemide-sensitive transporter (Misgeld et al., 1986; Thompson et al., 1988; Jarolimek et al., 1999; Kakazu et al., 1999) whose driving force is determined by extracellular [K+] (Payne, 1997; Jarolimek et al., 1999). There is considerable evidence suggesting that the recently cloned neuronal K+/Cl−cotransporter, KCC2, accomplishes this transport (Payne et al., 1996). A reduction in the expression of KCC2 by antisense nucleotides was shown to decrease the driving force for hyperpolarizing GABA currents in hippocampal CA1 neurons (Rivera et al., 1999). KCC2 expression increases in forebrain regions during the first postnatal weeks (Clayton et al., 1998; Lu et al., 1999; Rivera et al., 1999). At approximately this time, GABAA and glycine receptor responses become hyperpolarizing (Ehrlich et al., 1999; Kakazu et al., 1999; Rivera et al., 1999; DeFazio et al., 2000), and giant depolarizing potentials that are blocked by GABAAreceptor antagonists disappear in the hippocampus (Ben-Ari et al., 1989). That KCC2 is the dominant neuronal Cl− extruding molecule, is important for synaptic inhibition by GABA or glycine, and is supported by the abnormalities observed in the developing spinal cord and brainstem of KCC2 knock-out mice (Hübner et al., 2001).
Protein tyrosine kinase (PTK) phosphorylation is considered a key biochemical event in numerous cellular processes, including proliferation, growth, and differentiation, and has also been implicated in synaptogenesis (Catarsi and Drapeau, 1993). Protein tyrosine kinases are subdivided into the cytosolic nonreceptor family and the transmembrane growth factor receptor family, which includes receptors for insulin and insulin-like growth factor (IGF-1). The maturation of postsynaptic inhibition may require both a cytoplasmic PTK, which increases GABAA receptor-mediated currents (Moss et al., 1995), and insulin, which was shown to induce a rapid translocation of GABAA receptors from intracellular compartments to the plasma membrane (Wan et al., 1997b). KCC2 is also known to have a C-terminal PTK consensus site (Payne, 1997). Therefore, the maturation of postsynaptic inhibition may, in addition to other mechanisms, also involve the effects of PTK and insulin acting on KCC2.
Previously, we presented an assay that allows one to determine the direction of KCC2 transport as a function of extracellular K+ concentration (Jarolimek et al., 1999). A major advantage of the assay is that the driving force for the transporter is set by defined intracellular and extracellular ion concentrations and that GABAA currents are not confounded by the influence of HCO3− ions (Kaila et al., 1993). In this study, we used the assay and show that PTK inhibitors reduce KCC2-mediated transport. Furthermore, we provide evidence that insulin and IGF-1 in conjunction with a cytosolic PTK, c-Src, rapidly activate KCC2.
MATERIALS AND METHODS
Cell culture. Pregnant Wistar rats were anesthetized with ether and killed by decapitation. The embryos were removed, placed in sterile, ice-cold Gey's buffered salt solution [containing (in mm): 137 NaCl, 5 KCl, 0.3 MgSO4, 1 NaH2PO4, 1.5 CaCl2, 2.7 NaHCO3, 0.2 KH2PO4, 1 MgCl2, and 5 glucose, at pH 7.4], and immediately decapitated. Hippocampal anlage from 14-d-old embryos was mechanically dissociated and plated on a primary culture of glial cells from the same area. Cell culture conditions were the same as described previously (Jarolimek and Misgeld, 1992). For the incubation experiments, neurons were raised in medium containing either bovine insulin (30 μg/ml; Life Technologies, Paisley, UK) or IGF-1 (50 ng/ml; Sigma, Deisenhofen, Germany) from 2–5 or 2–8 d in culture (DIC), during which the medium was replaced two to three times. Afterward, half of the medium was exchanged twice per week with medium lacking insulin or IGF-1. Sister control cultures, raised in the absence of insulin, were obtained from the same embryos and recorded on the same days. Cultures raised with insulin contained considerably larger cell numbers than their counterparts (data not shown).
Electrophysiological recordings. Recordings were performed at room temperature (22–25°C) in the whole-cell voltage-clamp configuration with a patch-clamp amplifier Axopatch 200 B (Axon Instruments, Foster City, CA). The composition of the extracellular solution was (in mm): 156 NaCl, 1 CsCl, 2 KCl, 2 CaCl2, 1 MgCl2, 15 glucose, and 10 HEPES, pH 7.3. Composition of the patch pipette solution was (in mm): 3.5 NaCl, 5 KCl, 130 K-glucuronate, 0.25 CaCl2, 0.5 MgCl2, 10 glucose, 10 HEPES, 5 5-N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide (QX314), and 0.1 or 2 Mg-ATP, pH 7.3. For a more detailed description of the assay, see Jarolimek et al. (1999). Extracellular solutions were applied by a multibarreled perfusion system that was positioned ∼250 μm apart from the soma of the recorded cell. All experiments were performed with 10 μm6,7-dinitroquinoxaline-2,3-dione and 1 μmdl-2-amino-4-methyl-5-phosphono-3-pentenoic acid in the extracellular solution to block glutamatergic synaptic currents. The remaining currents could be completely blocked by application of 20 μm bicuculline (Jarolimek et al., 1999). Drugs were from Sigma, except lavendustin A (Calbiochem, Schwalbach, Germany). Insulin or IGF-1 were added to the extracellular solution. c-Src (recombinant pp60c-Src; Upstate Biotechnology, from Biomol, Hamburg, Germany) was added to the patch pipette solution directly before the experiment. For control experiments, c-Src was heat-inactivated at 60°C for 15 min. For all experiments with acute application of insulin, IGF-1, and/or c-Src, patch pipette solution containing 2 mm Mg-ATP was used.
Patch pipettes were fabricated from borosilicate glass (Hilgenberg, Malsfeld, Germany), and their resistances to bath ranged from 2.5 to 4.5 MΩ. The access resistance was estimated from the amplitude of the capacitive current evoked by a 5 mV square pulse. Only access resistances between 10 and 20 MΩ were accepted and routinely checked during the recording. For a determination of the liquid junction potential between the patch pipette and the extracellular solution, seeJarolimek et al. (1999). All values were corrected by −14 mV. Recordings were started >5 min after the whole-cell configuration was established to allow adequate time for QX314 to take effect and for anions to equilibrate. After 5 min, no additional change in dendritic or somatic EGABA was observed for the duration of recording (up to 2 hr). GABA (1 mm; Sigma) was applied by pressure ejection (1–20 kPa; 20–40 msec) every 15 sec from a pipette with a <1 μm opening.IGABA was measured in the presence of TTX (0.3 μm; Alomone Labs, Jerusalem, Israel) to avoid superposition of IGABA and action potential-dependent spontaneous IPSCs (sIPSCs). Currents were recorded at holding potentials (VH) near EGABA. After 15–45 sec at a newVH, the current amplitudes induced by three consecutive applications were averaged. Despite the rapid exchange of the extracellular solution (∼0.5 sec) around the neuron, effects of drugs were determined >1 min after start of the application. During this time, VH was set to EGABA. For the analysis of sIPSCs, we first determined the reversal potential, and, from the reversal potential, VH was changed in small steps to determine the voltage dependence of synaptic currents.
Data analysis. Data were filtered at 1.3 kHz with a four-pole Bessel filter and were acquired and analyzed with pClamp6 (Axon Instruments) and Igor Pro (WaveMetrics Inc., Eugene, OR). The amplitudes of sIPSCs were analyzed with a program written in our laboratory (Jarolimek and Misgeld, 1997). The reversal potential ofIGABA was determined by fitting the current–voltage plot by a linear regression (r ≥ 0.95). Somatic sIPSCs and all sIPSCs recorded in the presence of a blocker for cation–anion transporters were fitted together only ifr ≥ 0.75. The 10 largest inwardly and outwardly directed sIPSCs, respectively, were averaged for each data point of the current–voltage relationship. Statistic analysis of data are reported as mean ± SEM.
Reverse transcription-PCR. To evaluate levels of KCC2 mRNA in neurons, a semiquantitative reverse transcription (RT)-PCR approach was undertaken in which fragments corresponding to the KCC2 and the internal standard, neurofilament light chain (NFL) (Moshnyakov et al., 1996), transcripts were coamplified. The specificity of the reaction conditions was determined by amplifying the cDNA with either the KCC2 or NFL primer pair (see below) individually and then sequencing the reaction products. Fragments corresponding to the KCC2 and NFL transcripts so amplified were purified and32P-dCTP labeled to generate specific probes for the Southern blot analysis (see below). Conditions for the cDNA synthesis and PCR (given below) were chosen such that the reaction products were hardly visible after 20 amplification cycles but were abundant after 30 cycles (see Fig. 6C1). Because the reaction products were well within the exponential phase of the PCR, all quantitations were done on reaction products amplified for only 20 cycles. Reaction products were electrophoresed on a 2% agarose gel, subjected to Southern blot analysis, and quantified with a bio-imaging analyzer (FUJIX BAS1000; Fujifilm, Tokyo, Japan). To maximize accuracy, the same blots were stripped and then probed individually with either KCC2- or NFL-labeled fragments (see Fig. 6C2). After correcting for the amount of NFL amplified, the relative levels of KCC2 expressed by neurons after treatment with insulin to sister controls was determined for three independent experiments and expressed as a ratio (see Fig. 6D). For each cDNA, the amplifications, Southern blots, and quantitations were repeated four times.
cDNA reactions were conducted on RNA prepared from sister control and insulin-treated cultures, using Moloney murine leukemia virus reverse transcriptase and random hexanucleotide primers, and had a final volume of 30 μl. For PCR reactions, 0.5 μl of the cDNA reaction was used along with 0.5 μm of each of the four primers (given below), 0.25 mm of each dNTP, 10 μl of 10× buffer, 0.5 μl of Taq polymerase, and 1 mmMgCl2. The following primers were used: 5′-GCAGCCCCTTCA-TCAACAGCAC-3′ and 5′-CATCGCTGGGAAGAGGTAAGC-3′, which amplified a 559 bp fragment of the KCC2 transcript, and 5′-TGCACGAGGAAGAGATCGCCGAGCT-3′ and 5′-CTGTAAGCT-GCAATCTCAATGT-3′, which amplified a 495 bp fragment of the NFL transcript.
Immunocytochemistry. The abundance of KCC2 protein in neurons treated with insulin and in untreated controls was visualized with an antibody whose specificity and utility has been described previously (Williams et al., 1999). Neurons cultured on coverslips were fixed for 10 min at room temperature with 4% paraformaldehyde prepared in PBS. After brief washes with PBS, they were permeabilized by incubation for 5 min at room temperature in a 0.2% Triton X-100 (Tr)–PBS solution, rinsed again with PBS, and then incubated in a solution containing 0.2% Tr and 4% goat serum (GS)–PBS for 30 min at room temperature. The coverslips were incubated in the primary antibody (1:200 dilution; 0.1% Tr and 2% GS–PBS) overnight at 4°C. After rinses in 1% GS–PBS, the FITC-conjugated anti-rabbit secondary antibody was added (1:200 dilution; Jackson ImmunoResearch, West Grove, PA), and the coverslips were incubated for 2 hr at room temperature. The coverslips were then rinsed, mounted on slides, visualized, and photographed with an Axio Plan 2 microscope (Zeiss, Oberkochen, Germany).
RESULTS
KCC2 activity distinguishes between two types of neurons
Whole-cell recordings from embryonic (E14) hippocampal neurons 27–31 DIC were performed under conditions of 15 mm (10 mm Cl− and 5 mmBr−) (cf. Jarolimek et al., 1999) permeant anions in the patch pipette and 2 mmK+ in the extracellular solution ([K+]extra).Only cells with membrane potentials more negative than −55 mV were included in the analysis. We applied GABA (1 mm) focally to either the soma or a dendrite of a single cell. The distance between the somatic and dendritic applications was at least 100 μm. Responses to GABA applications allowed us to discriminate two types of cells, which we termed type 1 and type 2. In type 1 neurons, the reversal potential (EGABA) for dendritic currents was more negative than EGABAfor currents induced by somatic applications (−81.3 ± 2.9 and −59.5 ± 1.6 mV, respectively; p ≤ 0.001;n = 4). Furosemide (100 μm) changed the driving force of the currents induced by focal dendritic applications (Fig.1A1). Plots of current amplitudes versus holding potential (VH) revealed a positive shift of dendritic EGABA in the presence of furosemide (Fig. 1A2). In the same cells, currents induced by focal somatic applications were not altered (Fig.1A1). In type 2 neurons (Fig. 1B), on the other hand, there was no difference betweenEGABA obtained with either dendritic or somatic applications (−57 ± 0 and −56 ± 1.3 mV, respectively; n = 4), and furosemide had no effect onEGABA for dendritic currents.
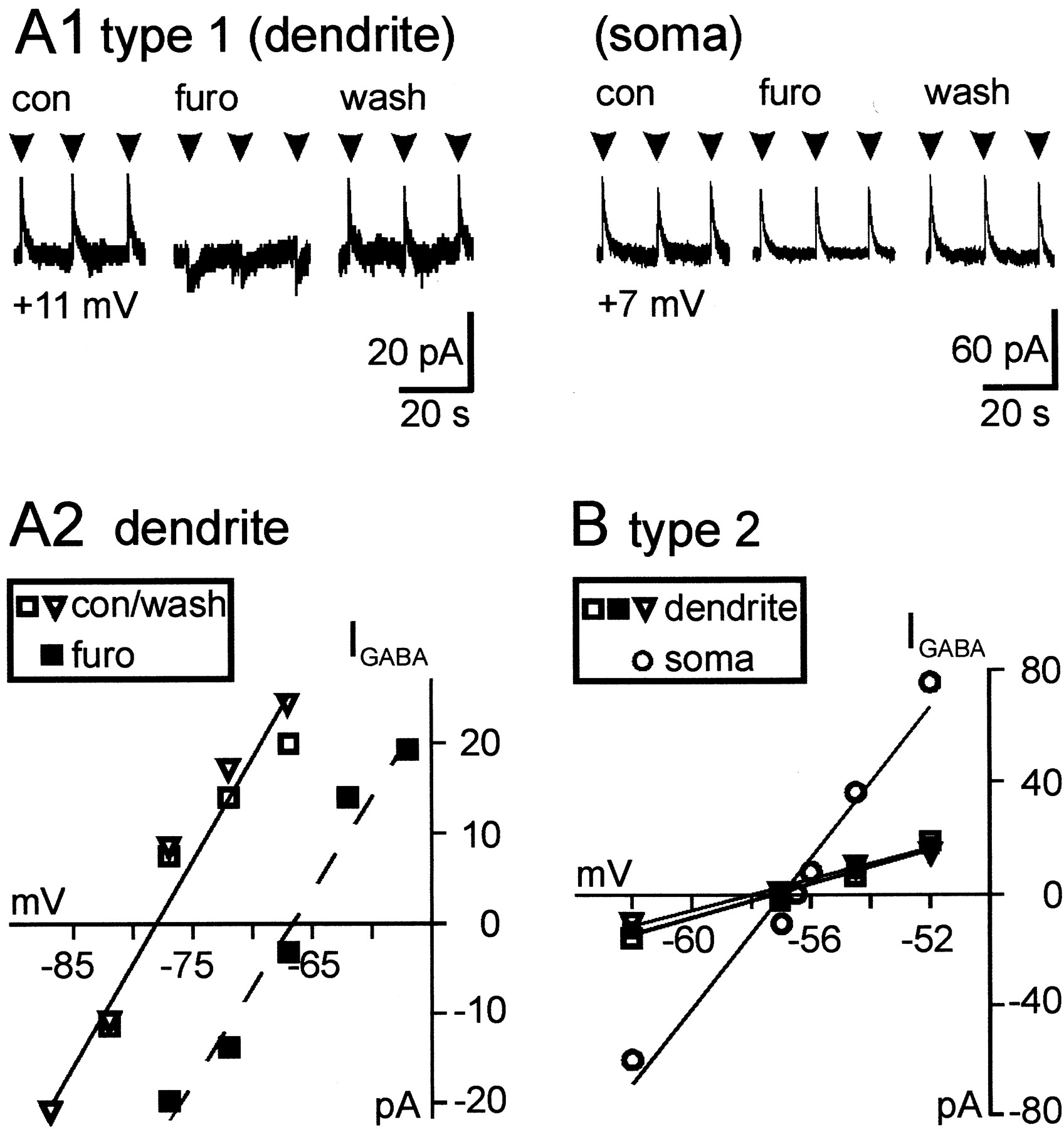
Somatodendritic gradient forEGABA in type 1 but not type 2 neurons.A, Focal application of GABA (1 mm) to a type 1 neuron. A1, Furosemide (100 μm) reversed the direction of GABA currents at the dendrite but not at the soma of the same cell. Below the traces, the driving force (VH-EGABA) is indicated. Somatic EGABA was −64 mV, and dendritic EGABA was −78 mV.con, Control; furo, furosemide;wash, washout. A2, Plots of GABA current amplitudes versus VH revealed a positive shift of dendritic EGABA in furosemide (100 μm; ▪) compared with control (■) and washout (▿).B, Focal application of GABA (1 mm) to a type 2 neuron. There was no difference betweenEGABA in the dendrite (■) and in the soma (○) of the same cell. Furosemide (100 μm; ▪) did not alter EGABA of dendritic currents. Control, ■; washout, ▿.
Pharmacological blockade of ionotropic glutamate receptors allowed us to measure sIPSCs mediated by GABAA receptors. Cells exhibiting little spontaneous synaptic activity were not considered in this study. In half of the hippocampal neurons (type 1), the simultaneous occurrence of inward and outward sIPSCs (Fig.2A1) prevented the determination of a single reversal potential (Erev). The other half (type 2) was characterized by sIPSCs that were either outward or inward depending on the respective VH (Fig.2B1). Only in type 1 neurons was it possible to alter the driving force for sIPSCs with the cation–anion transport blocker furosemide.

Comparison of furosemide effects on spontaneous IPSCs in type 1 and type 2 neurons. A1, Simultaneous outward and inward sIPSCs characterize type 1 neurons. Application of furosemide (100 μm) reversed the direction of sIPSCs.A2, In the same neuron, dendritic and somatic sIPSCs could be fitted separately by linear regression lines (■). In furosemide (100 μm), all sIPSCs could be fitted with the somatic regression line (▾). B1, B2, Type 2 neuron. Measurement of sIPSCs at varying VH allowed to determine a reversal potential by linear regression (r > 0.95; ■), and furosemide (100 μm; ▾) had no effect. Con, Control;furo, furosemide.
Figure 2A1 shows a type 1 hippocampal neuron. Application of furosemide (100 μm) reversed the direction of synaptic currents in <1 min. This effect was reversible during washout of furosemide. When amplitudes of the sIPSCs of this neuron were measured at varying VH(−69 to −85 mV), the data points could be fitted by two linear regression lines (Fig. 2A2), and two different reversal potentials could be extrapolated. According to results obtained with focal GABA applications, data points yielding the more negative reversal potential are from dendritic sIPSCs. The other line connected values for somatic sIPSCs. Data points obtained in the presence of the transport blocker furosemide could be fitted with the somatic currents (Fig. 2A2). In contrast, for a type 2 neuron (Fig. 2B), measurement of sIPSC amplitudes at varying VH allowed determination of a single reversal potential. In this cell, furosemide had no effect.
According to the data reported above, in type 1 neurons, dendritic sIPSCs can be discriminated from somatic sIPSCs by their respective driving forces. Driving forces for dendritic sIPSCs approached those of somatic sIPSCs during blockade of Cl−transport by furosemide. Bumetanide mimicked the effects of furosemide (Fig. 3A) but was somewhat less effective than furosemide. At high concentration (100 μm), bumetanide as well as furosemide reversed the direction of sIPSCs (n = 3) (Fig. 3A). At low concentration, bumetanide (25 μm) had no effect on spontaneous synaptic currents, whereas application of 25 μm furosemide to the same type 1 neuron increased the amplitude of inward sIPSCs and decreased those of outward sIPSCs (n = 4). The stilbene derivative 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) was more effective than furosemide, but recovery was not always complete (Fig.3B,C). These pharmacological characteristics match those of KCC2 expressed in human embryonic kidney cells (Payne, 1997).

Pharmacological blockade of Cl− outward transport in type 1 neurons.A, Bumetanide (100 μm) and furosemide (100 μm) comparably reversed the direction of sIPSCs in the same neuron. B, Another blocker, DIDS, mimicked the effect of furosemide in a concentration-dependent manner, although, at high concentrations, the drug is known to block Cl−channels as well as Cl− transport.C, Plots of amplitudes of sIPSCs versusVH show that, in 100 μm DIDS, all values for sIPSCs could be fitted together with the values for somatic sIPSCs in control. bume, Bumetanide;con, control; furo, furosemide;wash, washout.
The data indicated that a Cl− gradient exists between soma and dendrites of type 1 neurons, because Cl− transport exceeds the Cl− load provided by the patch pipette solution in the dendrites of the neurons (Jarolimek et al., 1999). In contrast, type 2 neurons possess a uniform distribution of intracellular [Cl−], because they lack net outward Cl− transport activity. These findings are summarized in Figure 4. In type 1 neurons (Fig. 4A1),EGABA for dendritic GABA currents shifted to positive values in furosemide (ΔVrev) (Fig.4A2). For the quantification of furosemide effects on sIPSCs, we measured VH at which the first outward sIPSCs with amplitudes twice the noise appeared. Furosemide induced a significant positive shift in this value (Fig.4A3). In type 2 neurons, in which dendritic and somatic EGABA were the same (Fig.4B1), furosemide had no effect on dendriticEGABA (Fig. 4B2).VH at which the first outward sIPSCs appeared was also not changed (Fig. 4B3). In conclusion, a furosemide-sensitive KCC generates the somatodendritic [Cl−] gradient in type 1 neurons, whereas in type 2 neurons KCC activity is absent.

Somatic and dendritic [Cl−] in type 1 and type 2 neurons. A1, Schematic drawing of a type 1 neuron. Net Cl− outward transport lowers intracellular [Cl−] in dendritic regions compared with the soma, which is loaded with [Cl−] by the patch pipette solution. A2,EGABA was determined for currents induced by focal application of GABA (1 μm) to the dendrite of type 1 neurons; furosemide (100 μm) reversibly shiftedEGABA to more positive potentials (p ≤ 0.001). A3, Furosemide (100 μm) induced shifts in theVH to more positive potentials (p ≤ 0.01) at which the first sIPSCs with amplitudes twice the noise appeared. B1 displays a type 2 neuron with an even [Cl−] in the dendrites and in the soma. B2 displays focal applications of GABA (1 mm) to dendrites of type 2 neurons. No shift inEGABA occurred when furosemide (100 μm) was applied. B3, Same asA3 for type 2 neurons, but there was no effect of furosemide (100 μm). con, Control;furo, furosemide; wash, washout.
Outward transport of chloride requires tyrosine kinase activity
Neuron-specific KCC2 differs from the widely expressed KCC1 in a PTK consensus site (Payne, 1997). Hence, we tested the effect of PTK inhibitors on Cl− outward transport in type 1 neurons. Perfusion of lavendustin A (10 μm), a membrane-permeable inhibitor of PTK (O'Dell et al., 1991), like furosemide, resulted in an increase of inward and a decrease of outward sIPSCs that was fully reversible only after a single application (Fig.5A). This treatment also resulted in a decrease in the frequency of sIPSCs. To quantify the effect of lavendustin A (11 applications to five cells), we determined the amplitude ratio of outward to inward sIPSCs, which was strongly reduced during the application (0.51 ± 0.07; p ≤ 0.001) (Fig. 5B). Control experiments were performed using type 2 neurons because these cells have no KCC2-mediated transporter activity. Lavendustin A was applied at differentVH, and the resultant sIPSCs were measured (17 applications to seven type 2 neurons). Under these conditions, the frequency and amplitudes of inward (0.58 ± 0.12) and outward sIPSCs (0.6 ± 0.08) normalized to control were reduced (Fig. 5C), but their ratio did not change.

Reduction of Cl− outward transport by inhibitors of PTK. A, In a type 1 neuron, lavendustin A (10 μm) had an effect on sIPSCs comparable with furosemide. B, The ratio of outward to inward sIPSCs normalized to control was significantly reduced during application of lavendustin A (10 μm; p ≤ 0.001) or genistein (50 μm for 30 sec; p≤ 0.001) but not by daidzein (50 μm for 30 sec), the inactive analog of genistein. C, In type 2 neurons, inward currents and outward currents normalized to control at the respective VH were reduced, but the ratio did not change. D1, Currents induced by focal applications of GABA (1 mm) to a dendrite decreased or reversed in direction, respectively, when genistein (50 μm) or furosemide (100 μm) were applied to a type 1 neuron (VH of −75 mV). AtEGABA(VH of −85 mV), there was an increase in inward currents in genistein and furosemide. D2, The current–voltage relationship for the neuron shown in D1revealed a positive shift in EGABA(ΔVrev) in genistein (▪) and furosemide (●). Control, ■; washout, ▿. E, Correlation of ΔVrev induced by genistein (50 μm) and furosemide (100 μm) in 12 neurons was highly significant (r = 0.98). con, Control; daidz, daidzein; furo, furosemide; genist, genistein;lav A, lavendustin A; wash, washout.
Another membrane-permeable inhibitor of PTK, genistein (50 μm) (Akiyama et al., 1987), produced a reversible decrease (13 applications to nine cells) in the amplitude ratio of outward to inward sIPSCs (0.25 ± 0.06; p ≤ 0.001) in type 1 neurons (Fig. 5B). In contrast, daidzein (50 μm), the inactive analog of genistein (Akiyama et al., 1987), when applied to neurons that responded to genistein (six applications to four cells), had no effect on the ratio of outward to inward currents (1.2 ± 0.14) (Fig. 5B). In type 2 neurons (18 applications to three cells), genistein did not reduce the mean amplitude of either outward (0.99 ± 0.08) or inward (0.84 ± 0.13) sIPSCs (Fig. 5C).
To circumvent effects of PTK inhibitors on spontaneous activity, we applied GABA focally to dendrites of type 1 neurons and measured ΔVrev for GABA currents. Genistein shifted EGABA to positive values (ΔVrev was +7.5 ± 1.1 mV;n = 10) and caused a new steady state, which was reached within 3 min after application and was reversible in most instances. Genistein, however, had no effect onEGABA if GABA was applied to the soma of cells (n = 3) because somatic [Cl−]intra is determined by the Cl− load through the patch pipette. We next compared ΔVrev induced by genistein or furosemide if they were applied to the same type 1 or type 2 cell (n = 12). As shown in Figure 5, D1 andD2, a given cell responded with almost the same ΔVrev to either genistein or furosemide. The correlation of ΔVrevinduced by genistein or furosemide was highly significant (r = 0.98), strongly suggesting that both drugs acted by blocking the same pathway (Fig. 5E). Furthermore, the slope of the curve revealed that the genistein effect amounted to 80% of the furosemide effect.
Neuronal chloride channels, ClC-3 and ClC-2, are known to be activated or blocked by activators of protein kinase A and protein kinase C, respectively (Smith et al., 1995). Therefore, we applied forskolin, an activator of adenylylcyclase, (20 μm; n = 5), and phorbol-12-mystirate-13-acetate, an activator of protein kinase C (1 μm; n = 3), to the bath solution. None of them had an effect on the Cl− gradient and furosemide sensitivity (data not shown), although spontaneous synaptic activity increased as described previously (Capogna et al., 1995).
Developmental activation of outward chloride transport
We determined the developmental onset of type 1 neurons in cultures derived from hippocampus. There were time points when either type 1 or type 2 neurons predominated and a transitory period wherein both types existed. Type 1 neurons were the predominant cell type only after 35–44 DIC (26 of 29 cells), whereas type 2 predominated up to 23 DIC (18 of 19 cells). The time of appearance of type 1 neurons could be shortened by incubation with a growth factor that activates PTK. In hippocampal cultures raised in insulin (30 μg/ml), type 1 neurons predominated as early as 21–24 DIC (17 of 18 cells). In contrast, very few type 1 neurons (1 of 12 cells) were found in sister cultures raised without insulin (Fig.6A). The same results were obtained with physiological concentrations of IGF-1 (50 ng/ml).

Abundance of type 1 neurons, but not expression of KCC2 mRNA and protein, is enhanced by insulin. A, The relative number of type 1 neurons (black bars) is increased when hippocampal cell cultures are incubated in insulin, whereas type 2 neurons (white bars) predominate in untreated cultures. B, KCC2 protein is abundant in all neurons, regardless of insulin pretreatment. C1, An ethidium bromide-stained agarose gel demonstrating the relative abundance of the coamplified KCC2 and NFL fragments after 10, 20, 30, or 40 (lanes 2–5, respectively; the 636, 517, and 396 bp DNA size markers are visible in lanes 1 and6) amplification cycles. C2, A representative Southern blot result illustrating the relative abundance of the KCC2 and NFL DNA fragments in insulin-treated and untreated samples. D, The relative abundance of KCC2 expression in insulin-treated samples expressed as a ratio of the amount of KCC2 amplified in insulin-treated samples to the amount in untreated samples. The results from three separate experiments (n = 4) indicate that the ratio did not deviate significantly from 1.0, suggesting that KCC2 expression was unaltered by insulin treatment.
To determine whether the difference in Cl− transport between the two groups of neurons was attributable to differential expression of KCC2, we amplified KCC2 transcript by single-cell RT-PCR from individual type 1 and type 2 neurons. Single-cell RT-PCR revealed no difference because 15 of 18 type 1 neurons and 10 of 15 type 2 neurons contained KCC2 mRNA (data not shown). To quantify the relative expression of KCC2 transcript in type 1 and type 2 neurons, we determined the abundance of KCC2 mRNA in 21–24 DIC cultures treated with insulin and in sister control cultures. Semiquantitative RT-PCR, wherein KCC2 mRNA was amplified and normalized to the expression of NFL, indicated that both cultures expressed comparable amounts of KCC2 (Fig.6D). In addition, immunocytochemical visualization of KCC2 protein revealed that it was abundant in both types of cultures (Fig. 6B). Thus, we concluded that the onset of transporter activity cannot be correlated with the amount of mRNA or with the mere presence of KCC2 protein.
The data presented above suggested that neurons with passiveECl− as well as neurons with functional transport contained KCC2 protein. Furthermore, PTK appeared to play a major role in regulating the transport. To test whether PTK could activate the transport, we applied insulin (30 μg/ml) in the bath for at least 30 min. This had no effect on its own (n = 3). However, if the application of insulin (30 μg/ml; n = 7) or IGF-1 (50–100 ng/ml;n = 3) was combined with the perfusion (15 min before application of insulin or IGF-1) of a cytoplasmic PTK (c-Src, 60 U/ml) via the recording pipette, type 2 cells could acquire type 1 activity within 5 min (Fig. 7). The change manifested itself in the appearance of inward and outward sIPSCs at a holding potential, at which the cell had shown previously inward currents only (Fig. 7B). The application of c-Src alone (30 or 60 U/ml for 30 min) was not effective (n = 6). Furthermore, whereas the cells recorded with c-Src did not respond to furosemide, furosemide induced a shift in the driving force for sIPSCs in the same cells after insulin or IGF-1 application (n= 9) (Fig. 7B). To confirm that active tyrosine kinase was required for the change from type 2 to type 1 characteristics, we used heat-inactivated c-Src, which was unable to support the insulin effect (n = 3).

Instantaneous transformation of a type 2 into a type 1 neuron. A, Only inward sIPSCs occurred at the indicated VH during recording with c-Src (60 U/ml) in the patch pipette solution. After 10 min of insulin application (30 μg/ml), simultaneous outward and inward currents were recorded at the same VH. B, In this neuron, sIPSCs were fitted by a single linear current–voltage relationship. During insulin application, sIPSCs appeared that had to be fitted by a separate line. The change reversed during application of furosemide (100 μm), which had been ineffective before insulin application (data not shown). con, Control;furo, furosemide; wash, washout.
DISCUSSION
KCC2-mediated outward transport of chloride
In a previous study (Jarolimek et al., 1999), we demonstrated that a furosemide-sensitive K+/Cl−cotransporter generates a pronounced somatodendritic [Cl−] gradient in cultured midbrain neurons provided that appropriate intracellular and extracellular K+ and Cl−concentrations are chosen. The somatodendritic gradient is built up by [Cl−] in the patch pipette solution, which serves as a Cl− source, and the K+/Cl−cotransporter, which counteracts that Cl−load. This gradient makes it possible to study Cl− regulation through K+/Cl−cotransport in neurons by measuring differences between somatic and dendritic EGABA. Using this assay, we could identify two groups of neurons in hippocampal cultures on the basis of the respective GABAA receptor-mediated synaptic currents generated by the input of spontaneously discharging inhibitory neurons. One group (type 1) comprised mature neurons in which dendritic and somatic sIPSCs could be discriminated by their different driving forces. Immature neurons constituted the second group (type 2) and revealed no difference between dendritic and somatic sIPSCs. Furosemide sensitivity ofEGABA and a difference in somatic and dendritic EGABA were found only in type 1 neurons. Based on the rank order of potencies of the blockers for KCC2 (Payne, 1997), we conclude that the somatodendritic Cl− gradient is established by KCC2. Keeping with this notion, our previous study (Jarolimek et al., 1999) showed that the direction of transport is sensitive to small changes of [K+]extra, as predicted by the thermodynamic properties of an electroneutral K+/Cl−cotransport. As expected, there was no interference with voltage-gated Cl− channels because their influence on the described Cl− homeostasis was very small and could be identified only after a complete blockade of Cl− transport (Smith et al., 1995).
KCC2 function requires tyrosine kinase activity
KCC2 is a member of the family of cation–anion cotransporters that regulate cell volume and are essential for salt and water transport across epithelia. To become activated, they are either phosphorylated or dephosphorylated. Abnormalities of erythrocyte K+/Cl−cotransport infgr−/−/hck−/−double-mutant mouse demonstrate that Src family kinases are involved in the regulation of membrane transport. In this case, the kinases are likely to negatively regulate a cotransporter-activating phosphatase (De Franceschi et al., 1997). Another member of this family, NKCC1, is regulated by protein kinases A and C, as has been shown for secretory epithelia (for a review of the literature, see Russel, 2000). Neuron-specific KCC2, in contrast to the ubiquitously expressed KCC1, has a tyrosine kinase consensus site (Payne, 1997). Recently, the inhibitory effect of phosphatase inhibitors demonstrated that serine–threonin phosphorylation events regulate KCC2 activity in oocytes (Strange et al., 2000). Thus, there are many reasons to believe that phosphorylation could be important in regulating KCC2 activity.
Genistein and lavendustin A, both membrane-permeable PTK inhibitors, had a furosemide-like effect on the somatodendritic Cl− gradient in mature neurons. The comparable strength of furosemide and genistein indicated that most of the developmental increase in the number of cultured neurons with KCC2 activity was attributable to phosphorylation. In a recent study conducted in Xenopus laevis oocytes, site-directed mutagenesis of exogenously expressed KCC2 (Strange et al., 2000) suggested that the predicted C-terminal tyrosine kinase consensus site of KCC2 (Payne, 1997) may not be phosphorylated. However, we believe that factors required in neurons for activation of KCC2 are not present in that artificial expression system because concentrations of genistein twice as high as those used in our study had no effect in the oocytes. A synergistic regulation of the efficacy of inhibitory synapses is suggested by the fact that tyrosine phosphorylation also increases amplitudes of GABAA currents (Moss et al., 1995; Wan et al., 1997a). Thus, the phosphorylation step leading to activation of KCC2 may be part of the overall maturation of GABAergic synapses.
Development of KCC2-mediated outward transport of chloride
We noted an increasing number of cells with transport activity when recording from mature cultures. Maturation of neurons, as determined by the relative amount of type 1 neurons, could be accelerated if cultures were transiently incubated in insulin or in physiological concentrations of IGF-1, suggesting that the effect was mediated by the IGF-1 receptor. Insulin is known to be synthesized by hippocampal neurons (Bartlett et al., 1991) and can be released in a calcium-dependent manner during depolarization (Clarke et al., 1986;Wei et al., 1990). GABAergic stimulation of immature neurons in hippocampal culture leads to a Ca2+influx, but it reduces the levels of intracellular Ca2+ in mature neurons (Berninger et al., 1995).
Responses to GABA with an elevation of [Ca2+]i have been correlated with KCC2 expression in developing hippocampal cultures. A nonlinear relationship between the mRNA of KCC2 and its functional effects was observed. Ca2+ influx during GABA application persisted at a time at which expression of mRNA for KCC2 increased. The transformation of GABA responses could be attributed to a reduced rate of Cl−uptake as a result of decreasing NKCC1 activity, as well as to increasing KCC2 activity (Ganguly et al., 2001). Other factors to consider when monitoring Ca2+ influx are developmental changes in the kinetics of Ca2+ current activation attributable to a reduced expression of L-type channels (Pravettoni et al., 2000) and increases in resting membrane potentials. Because KCC2 protein was abundant and because comparable amounts of mRNA for KCC2 were present both in insulin-treated and untreated cultures, we concluded that the presence of KCC2 protein alone is not sufficient to induce transport activity. Assuming that tyrosine kinase phosphorylation provides the key to activate KCC2, we applied insulin or IGF-1 in conjunction with intracellular perfusion of c-Src protein and found that the transport was rapidly activated. Perfusion with c-Src alone was not sufficient for activation of the transport, excluding the possibility that c-Src mediated the entire response to IGF-1. Our data, however, do not provide direct evidence for upregulation of tyrosine kinase during chronic insulin exposure. Also, an upregulation of other KCCs, which may contribute to net Cl− outward transport, was not excluded.
IGF-1 is known to rapidly activate calcium channels by phosphorylation of a subunit via a cytosolic PTK (Bence-Hanulec et al., 2000). An alternative mechanism can be inferred from studies demonstrating that insulin-induced potentiation of NMDA receptor activity does not result from direct phosphorylation of the receptor but from phosphorylation of associated C-terminal tails of anchoring or signaling proteins (Skeberdis et al., 2001). Insulin is also known to potentiate GABAA receptor currents by recruitment of functional GABAA receptors to postsynaptic CA1 synapses (Wan et al., 1997b). However, because high concentrations of insulin were necessary to potentiate the GABAAreceptor in that study, it is likely that the effect was mediated via interaction with the IGF-1 receptor that is expressed at high levels in the developing and adult hippocampus (Kar et al., 1993). Interestingly v-Src kinase has been shown to phosphorylate and activate IGF-1 receptors (Peterson et al., 1996). Therefore, endogenous c-Src, once activated, might act via both direct and indirect mechanisms, creating a positive feedback cycle to drive the phosphorylation mechanism mediating KCC2 activity to higher levels.
In conclusion, the rapid activation of KCC2-mediated transport by IGF-1–insulin and c-Src in cultured neurons indicates that development of neuronal K+/Cl−cotransport requires cooperation of a growth factor with PTK-dependent phosphorylation. In this regard, KCC2 is similar to ionotropic receptors and other ion channels whose function is also regulated by PTK-dependent phosphorylation. Transcriptional mechanisms, although known to regulate KCC2 levels in the brain, are not sufficient for the induction of KCC2 activity, at least in cultured neurons. Our studies show that tyrosine phosphorylation is important for rapidly regulating a transport in neurons, which determines the efficacy of postsynaptic inhibition. What may the role of phosphorylation of KCC2 throughout postnatal life be? The very fast and almost complete effect of PTK inhibitors suggested a high turnover rate of phosphorylation and dephosphorylation. In concert with the regulation of GABAA receptors by Src family kinases, fast regulation of KCC2 activity and, hence, postsynaptic inhibition may be important for synaptic plasticity and network excitability. Furthermore, protein tyrosine phosphorylation has been implicated in modifications of neuronal function in pathological conditions such as epilepsy (Sanna et al., 2000). A reduction of KCC2-mediated transport by furosemide promotes epileptiform activity in neuronal networks (Jarolimek et al., 1996), whereas activation of the transport might counteract an excessive enhancement of NMDA receptor-dependent excitatory transmission induced by c-Src (Yu and Salter, 1999). Given the prominent role of synaptic inhibition in mediating many brain functions and dysfunctions, modulation of KCC2 activity by PTKs may be important in a wide range of physiological and pathological processes in the CNS.
Footnotes
The study was supported by Deutsche Forschungsgemeinschaft Grant MI255/4-1 (U.M.), Sonderforschungsbereich 488 (H.M.), and the Schilling Foundation (H.M.). The excellent technical assistance of Ulla Amtmann is gratefully acknowledged. We are particularly grateful to Dr. J. A. Payne for the gift of antibody and Dr. K. Kaila for helpful discussions.
Correspondence should be addressed to Dr. Ulrich Misgeld, Institut für Physiologie und Pathophysiologie, Universität Heidelberg, Im Neuenheimer Feld 326, D-69120 Heidelberg, Germany. E-mail: ulrich.misgeld{at}pio1.uni-heidelberg.de.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}