

Abstract
The induction of a long-term hyperexcitability (LTH) in vertebrate nociceptive sensory neurons (SNs) after nerve injury is an important contributor to neuropathic pain in humans, but the signaling cascades that induce this LTH have not been identified. In particular, it is not known how injuring an axon far from the cell soma elicits changes in gene expression in the nucleus that underlie LTH. The nociceptive SNs of Aplysia (ap) develop an LTH with electrophysiological properties after axotomy similar to those of mammalian neurons and are an experimentally useful model to examine these issues. We cloned an Aplysia PKG (cGMP-dependent protein kinase; protein kinase G) that is homologous to vertebrate type-I PKGs and found that apPKG is activated at the site of injury in the axon after peripheral nerve crush. The active apPKG is subsequently retrogradely transported to the somata of the SNs, but apPKG activity does not appear in other neurons whose axons are injured. In the soma, apPKG phosphorylates apMAPK (Aplysia mitogen-activated protein kinase), resulting in its entry into the nucleus. Surprisingly, studies using recombinant proteins in vivo and in vitro indicate that apPKG directly phosphorylates the threonine moiety in the T-E-Y activation site of apMAPK when the -Y- site contains a phosphate. We used inhibitors of nitric oxide synthase, soluble guanyl cyclase, or PKG after nerve injury, and found that each prevented the appearance of the LTH. Moreover, blocking apPKG activation prevented the nuclear import of apMAPK. Consequently, the nitric oxide-PKG-MAPK pathway is a potential target for treatment of neuropathic pain.
Introduction
Persistent neuropathic pain is a major clinical problem that has mostly resisted effective treatment. In humans (Gracely et al., 1992) and mammalian model systems (Millan, 1999), persistent pain after nerve injury is associated with a long-term hyperexcitability (LTH) of sensory neurons (SNs) having axons in the injured nerve. LTH is manifested as increased sensitivity to electrical stimuli in the SN cell body and axon at the injury site (Wall and Devor, 1983; Study and Kral, 1996; Zhang et al., 1997; Chen and Devor, 1998; Kim et al., 1998; Abdulla and Smith, 2001). These changes result in discharge of SNs at rest or during innocuous stimulation, leading to continuing excitation of higher order neurons in the CNS and to persistent pain.
Because the appearance of LTH involves alterations in gene expression (Waxman et al., 1994; Wang et al., 2002; Park et al., 2003), a central question is how such changes in the nucleus are induced by an injury that occurs far from the cell body? Answering this question has been extremely difficult using the complex mammalian nervous system. A experimentally favorable alternative is the homogeneous cluster of SNs that reside in the bilateral pleural ganglia of the mollusk Aplysia californica (Walters et al., 2004). Noxious mechanical stimulation of the body wall (Walters et al., 1983a) or crushing SN axons in vivo or in vitro, elicits an LTH with electrophysiological properties similar to those seen after axotomy of mammalian SNs (Walters et al., 1991; Walters, 1994; Ambron et al., 1996; Bedi et al., 1998; Ungless et al., 2002; Sung and Ambron, 2004). The LTH appears after a delay, suggesting that its induction after nerve crush is attributable to a positive molecular injury signal (Walters et al., 1991; Ambron and Walters, 1996; Lin et al., 2003). Two studies support this idea. First, blocking axonal transport after nerve injury in excised nervous systems prevented the appearance of LTH (Gunstream et al., 1995). Second, LTH was induced in noninjured SNs by injecting axoplasm from injured axons (Ambron et al., 1995). LTH was also elicited in the SNs after intrasomatic injection of an ERK (extracellular signal-regulated kinase) member of the MAPK (mitogen-activated protein kinase) family (Sung et al., 2000), and cGMP and PKG (cGMP-dependent protein kinase; protein kinase G) are probably involved (Lewin and Walters, 1999). Nevertheless, we still do not know the identity of the signal from the axon, how PKG and the ERK are activated, or how these kinases might interact. Moreover, LTH was also reported to be induced by cAMP acting on PKA (protein kinase A) in a learning paradigm (Dale et al., 1988; Scholz and Byrne, 1988), and because PKG and PKA have common properties, overlap in these pathways was a possibility. To resolve these gaps in our knowledge, we cloned an Aplysia (ap) PKG and found that the induction of LTH after nerve crush requires its activation. Activated apPKG is retrogradely transported to SN cell bodies, where it phosphorylates apMAPK directly, resulting in its translocation into the nucleus. This is the first report of a direct activation of apMAPK by PKG, and we speculate that this discovery will have broad implications for other long-term changes associated with axon injury.
Materials and Methods
In vivo nerve crush. Aplysia (100-150 gm) were anesthetized with isotonic MgCl2, and a small incision was made on one side of the body wall. Pedal nerves 5-9 were crushed 2 cm from the pedal-pleural ganglia on one side. The wound was sutured, and the animal was returned to its tank. The crush-ligation protocol followed was as described (Ambron et al., 1995).
Cloning. Degenerate oligonucleotide primers 5′-tayaaytgyacnmgiacngc and 5′-ccrcaraangtccangtytt were used to amplify an apPKG cDNA fragment from Aplysia CNS cDNA. The resulting PCR product from this amplification was cloned into pCR-II (Invitrogen, Carlsbad, CA) and subsequently sequenced by the core facility at Columbia University. The 5′ end and 3′ end of the cDNA were cloned using 5′-rapid amplification of cDNA ends (RACE) and 3′-RACE, respectively. A Marathon cDNA Amplification kit (BD Clontech, Palo Alto, CA) was used to generate cDNA from Aplysia CNS poly(A+) RNA according to the manufacturer's instructions. For the 5′-RACE reaction, a specific primer, 5′-cgcctgtccagcacccatagcg, was used. The product of this PCR reaction was then confirmed by a second amplification using a nested, specific 5′ primer, 5′-gggtgaccgctttcacggagg. For the 3′-RACE reaction, a specific primer, 5′-cggcaaggttctgcgtcgcc, was used. The PCR product was then subjected to a second amplification using a nested, 3′ primer, 5′-ggacgcgaggggatacgtc. Both 5′- and 3′-RACE products were subcloned into pCR-II and sequenced. To obtain the full-length cDNA in one piece, another PCR was performed with oligonucleotides 5′-ggtggaggagatagcggcggttctgtgaacgcc and 5′-ggaggagtgagggtcagatcc, corresponding to the 5′ and 3′ ends of 5′- and 3′-RACE products, respectively. The PCR product was sequenced and designated apPKG and was deposited in the GenBank database under accession number AY362340.
Sequence analysis. Sequence alignment of various PKGs and identification of conserved residues was performed using the Clustal W and box-shade algorithms provided in the suite of programs available from Biology Workbench (http://workbench.sdsc.edu/).
Protein expression and purification. A His tag was added to the N terminus of the apPKG-coding region by PCR amplification from Aplysia CNS cDNA with the following primers: 5′-tggcggccgctcatgagaggatcgcatcaccatcaccatcacggcaacggtgccagttcgaacacgcacttc and 5′-gcaggctctagagaaatcaatgtcccagccggataactcgtc. The PCR product was subcloned into the NotI and XbaI sites of pFasBac-1 (Invitrogen) and was subsequently confirmed by sequencing. The resulting construct pFB1apcGK contains an N-terminal histidine epitope tag. Transformation of pFB1apcGK into Max Efficiency DH10Bac cells (Invitrogen), identification of recombinant clones, and isolation of the recombinant baculovirus shuttle vector DNA (bacmid) were performed according to the manufacturer's instructions (Invitrogen). Recombinant baculovirus was obtained by transfecting Sf9 cells (Spodoptera frugiperda), which were propagated as monolayers at 27°C in Sf-900 IISFM medium (Invitrogen) containing 100 U/ml penicillin (Invitrogen) and 0.1 mg/ml streptomycin (Invitrogen). Transfection with recombinant bacmid DNA was performed using CellFectin (Invitrogen) according to the instructions of the manufacturer. Positive viral clones were identified by their ability to direct the expression of the appropriate protein as revealed by immunoblotting of whole-cell extracts of transfected Sf9 cells harvested 3 d posttransfection using an antibody to the His tag of the protein. For apPKG protein expression, Sf9 cells were infected with the recombinant baculovirus at a multiplicity of infection of >10. After 72 hr, cells were harvested and recombinant His-apPKG was purified on nickel nitriloacetic acid resin (Qiagen, Valencia, CA) according to the manufacturer's instructions. To express VASP (vasodilator-stimulated phosphoprotein), the coding region of VASP was first obtained by PCR amplification from mouse brain cDNA with the following primers: 5′-gtcgtgggatccccatcgatagcgagacggtcatctgt and 5′-atcttgaattcctcgagggtcaaggagaaccccgctt. The PCR product was subcloned into the EcoRI and BamHI sites of pGEX3X (Amersham Biosciences, Arlington Heights, IL) and was subsequently confirmed by sequencing. The resulting construct pGEXVASP contains a C-terminal GST (glutathione S-transferase) epitope tag. VASP-GST, Elk1-GST, MEKK1C (MAP kinase kinase kinase 1C)-GST, MEK1 (MAP kinase kinase 1)-GST, and ERK1-GST fusion proteins were expressed in Escherichia coli DH5α and purified as described (Sung et al., 1996).
Northern blots. Total RNA was extracted from various tissues and resolved by denaturing agarose gel electrophoresis; the gel was then transferred to a nylon membrane. The resulting blot was hybridized with radioactively labeled apPKG and 5S ribosomal cDNA as described previously (Alberini et al., 1994; Sung et al., 2001).
Single-cell RT-PCR. Single SNs were transferred to 500 μl of Tri Reagent (Molecular Research Center, Cincinnati, OH), and total RNA was isolated according to the manufacturer's instructions. cDNA from each sample was synthesized using random hexamers as primers and reverse transcriptase (SuperScript II). Aliquots (2 μl) from each sample were used to amplify specific fragments by PCR (40 cycles), using specific primer sets for the following: (1) neuronal nitric oxide synthase (NOS) (GenBank accession number AAK83069), 5′-gtaccctcacaggacgagtc and 5′-tccttggacctctcttggtg (nt 3610-4049); (2) SN-specific neuropeptide sensorin A (GenBank accession number X56770), 5′-aacagaaacagtctttcccc and 5′-tcttgactcaccaactgcc (nt 43-331); and (3) neuron-specific actin (GenBank accession number U01352), 5′-cagagagaagatgacccag and 5′-gggtaagagaagcaagaaag (nt 416-1298).
Kinase assays. In vitro PKG activity was measured as described (Pohler et al., 1995). Briefly, 100 ng of His-apPKG was incubated with 5 μg of various peptides in a buffer containing the following (in mm): 25 Tris-HCl, pH 7.5, 5 β-glycerol phosphate, 2 DTT, 0.1 Na3VO4, and 10 MgCl2. The reaction was initiated by adding 10 μm [γ-32P]ATP. The incubation was allowed to proceed for 20 min at room temperature and terminated with 50 mm EDTA (final concentration). Labeled peptides were captured on P81 filters (Whatman, Maidstone, UK). The filters were washed with 0.5% phosphoric acid and dried, and the bound 32P-phosphopeptide was detected by liquid scintillation counting. All of the values were corrected for background counts per minute obtained without the substrate. To evaluate endogenous apPKG activity, 5 μg of ganglia extract or axoplasm was used in the kinase buffer (above) with 5 μg of PKA inhibitor and in the presence or absence of 1 μm cGMP.
ERK activity was assayed as described (Sung et al., 2001). Briefly, proteins and 500 μm ATP were incubated in kinase buffer for 20 min at room temperature. The reaction mixtures were resolved on 10% SDS polyacrylamide gels and subjected to Western blotting with antibody (Ab)pTpYmapk, AbpYmapk,AbpTmapk,orAbpElk1.
In situ hybridization. Ganglia were first isolated from animals, desheathed, and fixed in 4% paraformaldehyde in PBS, pH 7.4, for 3 hr. The ganglia were then washed several times in 1× PBS and then digested with 80 μg/ml proteinase K (Ambion, Austin, TX) in 1× PBS for 30 min at room temperature (RT). After several washes in 1× PBS, the ganglia were fixed again for 20 min with 4% paraformaldehyde and then washed several more times in 1× PBS. After treatment with 1.32% triethanolamine HCl, pH 8.0 (10 min at RT), and 0.24% acetic anhydride in 1.32% triethanolamine HCl, pH 8.0 (20 min at RT), and several washes with 1×PBS, the ganglia were prehybridized in Hyb buffer (50% formamide, 5× SSC, 5× Denhardt's solution, 0.25 mg/ml yeast tRNA, and 0.5 mg/ml salmon sperm DNA) at 60°C for 2 hr, and then hybridized overnight at 60°C with fresh Hyb buffer containing either antisense or sense digoxigenin (DIG)-labeled cRNA (1 μg/ml). After hybridization, ganglia were first washed for 30 min in fresh Hyb buffer at 68°C, and then in 0.2× SSC at 68°C for 1 hr. After equilibration in PBST (1× PBS and 0.1% Triton X-100), ganglia were blocked with 10% sheep serum in PBST for 30 min at RT, and then incubated with anti-DIG antibody (1:5000) coupled to alkaline phosphatase (Roche, Indianapolis, IN) in 1× PBST containing 1% sheep serum overnight at 4°C. Hybridization signals were visualized with nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate (Roche).
Western blotting. Protein samples were resolved on 10% SDS polyacrylamide gels and subsequently transferred onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH); the blots were probed with various gene-specific primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoreactivity was detected using the Pico-tag chemiluminescence system (Pierce, Rockford, IL).
Immunocytochemistry. Ganglia, nerve, or cultured cells were fixed with 4% paraformaldehyde in PBS, pH 7.4. The primary antibody was diluted in TBS supplemented with 0.1-0.5% Triton X-100 in TBS and 5% goat serum, and incubated overnight at 4°C. After several washes, an Alexa Fluor 594- or 488-conjugated secondary antibody (Molecular Probes, Eugene, OR) was applied for 1 hr at room temperature. Subsequently, the cells were visualized by confocal fluorescence microscopy (LSM510 confocal microscope; Zeiss, Oberkochen, Germany), and images were collected.
Cell culture. SNs were isolated from the pleural ganglia of 50-80 gm animals and were plated on poly-l-lysine-coated dishes containing L15 medium and 50% hemolymph (Dagan and Levitan, 1981; Glanzman et al., 1989). Cultures were maintained at 18°C for up to 7 d. Medium was changed every 2 d. Drugs were washed out 1 hr before electrophysiological tests.
Electrophysiology. Before the start of each recording, the hemolymph was replaced with a 1:1 mixture of artificial seawater and culture medium (without hemolymph; pH 7.6). Standard techniques were used for intracellular stimulation and recording (Ambron et al., 1996). The soma spike threshold was measured with a standard series of 20 msec depolarizing pulses. Repetitive firing was quantified by counting the number of spikes evoked by a series of 2 sec depolarizing pulses at 1, 2, 3, 4, and 5 nA, or 1 sec depolarizing pulses at 2.5 times the current for the 20 msec threshold. Spike amplitude was measured from baseline to the peak of the action potential, and spike duration was the breadth of the action potential at one-half of its maximal height.
Fluorescence protein labeling. BSA, apPKG, and ERK1 protein were labeled using an Alexa Fluor 546 Protein Labeling kit (Molecular Probes) according to the manufacturer's instructions.
SN microinjection. Microinjection pipettes were prepared with a Sutter programmable puller. Alexa Fluor 546-labeled protein (0.75 μg/μl in 10 mm Tris-HCl, pH 7.3, 100 mm NaCl, and 0.05% fast green dye) was microinjected into cultured SNs by applying positive air pressure under defined conditions (pounds per square inch and duration) using a picospritzer (Sung et al., 2000).
Materials. Recombinant bovine PKG 1α, guanosine 3′,5′-cyclic monophosphorothioate, 8-(4-chlorophenylthio)-, Rp isomer (Rp-8-pCPT-cGMPS), adenosine 3′,5′-cyclic monophosphorothioate, Rp isomer (Rp-cAMPS), 1-H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), l-thiocitrulline, NG-nitro-l-arginine methyl ester hydochloride (l-NAME), protein kinase A inhibitor 6-22 amide, and PKG substrate BPDEtide were purchased from Calbiochem (La Jolla, CA). Peptides A, C, and D, and MAPK p42 protein were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The following antibodies were obtained and used according to the manufacturer's instructions: anti-phospho-VASP (Ser239) from Upstate Cell Signaling Solutions (Lake Placid, NY), polyclonal antibodies to phosphorylated MAPK (AbpTpYmapk) and phospho-Elk1 (Ser383;AbpElk1) from Cell Signaling Technology (Beverly, MA), and anti-pY MAPK (AbpYmapk), anti-pT MAPK (AbpTmapk), and α-actin from Sigma (St. Louis, MO).
Results
Aplysia SNs contain a neuronal type-I PKG
To investigate the role of the PKG pathway in the induction of LTH, we first cloned an Aplysia PKG (GenBank accession number AY362340). ApPKG cDNA contains an open reading frame encoding a putative 733 aa protein. In concordance with all of the known PKGs, the Aplysia kinase contains two tandem cyclic nucleotide-binding domains and a C-terminal catalytic domain (Fig. 1A, top). The predicted protein encoded by the apPKG sequence is highly similar to known cGMP-dependent protein kinases with >50% amino acid identity to Drosophila PKGs and to the mammalian type-I and -II PKG isoforms (Fig. 1A, bottom). However, it appears to be most closely related to Drosophila DG1 (Fig. 1B).

A, Top, A schematic diagram of apPKG showing the position of the conserved tandem cGMP binding domains, the ATP binding and catalytic sites, and the position of an autoinhibitory sequence. Bottom, Clustal W sequence alignment of the predicted apPKG amino acid sequence with Drosophila DG1 (GenBank accession number AAB03405) and DG2T3a (AAA28459), human Iα (BAA08297) and II (CAA64318), mouse Iβ (AAD16044) and II (AAA02572), and rat II (CAA85284) PKGs. Conserved amino acids are shaded in black; similar amino acids are shaded in light gray. B, Phylogenetic analysis of the PKG family. C, Expression of apPKG in neurons. A multiple-tissue Northern blot was hybridized with a 32P-labeled N-terminal apPKG cDNA fragment. The arrow indicates apPKG mRNA. The sizes of RNA standards are indicated to the left of the figure. A 32P-labeled probe to 5S ribosomal RNA (arrow) was used to ensure loading uniformity. D, apPKG mRNA localization in pleural and pedal ganglia by in situ hybridization with a digoxigenin-labeled antisense RNA (left) or sense-RNA (right). apPKG mRNA is expressed in the pleural sensory cluster (arrow). Scale bar, 200 μm.
To determine the pattern of apPKG expression, we probed a Northern blot of total RNA from various tissues from the adult animals with a 32P-labeled 283 bp fragment corresponding to bases 209-492 of the cDNA. The probe detected a single 3.0 kb transcript that was expressed in ganglia, but not in muscle or the genital organs (Fig. 1C). A 32P-labeled probe to 5S ribosomal RNA detected a 0.19 kb transcript in all of the tissues (Fig. 1C).
To localize the source of the message, the apPKG probe was used to generate sense and antisense riboprobes for in situ hybridization. The antisense probe detected high levels of apPKG mRNA expression in the SN cluster and in most of the neurons in the pedal and pleural ganglia (Fig. 1D, left panel). Negligible labeling resulted from the sense probe (Fig. 1D, right panel).
We then investigated the catalytic properties of apPKG by first expressing apPKG cDNA in a baculovirus/Sf9 system. An inactive His-tagged recombinant apPKG was produced when the cells were grown in the presence of serum, and a constitutively active apPKG was made when the cells were deprived of serum. Both recombinant apPKG forms were purified via affinity chromatography.
Inactive apPKG was activated by 100 nm 8-Br-cGMP and readily phosphorylated BPDEtide, a peptide substrate for all of the type-I PKGs (Glass and Krebs, 1982) (Fig. 2A). Comparison of active apPKG with recombinant bovine type-Iα soluble PKG showed that both kinases readily phosphorylated several PKG peptide substrates (Fig. 2B). Significantly, neither kinase phosphorylated peptide C, which is the preferred substrate for membrane-bound type-II PKG (Hall et al., 1999) (Fig. 2B). We also examined the protein VASP, whose serine at position 239 is recognized specifically by type-I soluble PKGs (Smolenski et al., 1998), and found that both the bovine and Aplysia kinase phosphorylated this site (Fig. 2C). These studies establish apPKG as a member of the soluble type-I family of PKGs.

apPKG is a type-I PKG. A, The kinase activity of purified inactive recombinant apPKG protein (100 ng) was measured by the transfer of 32P from [32P]ATP to BPDEtide in the presence (+) or absence (-) of 100 nm 8-Br-cGMP. Activity caused by autophosphorylation was subtracted using a peptide control reaction (see Materials and Methods). Note that the kinase activity was inhibited in the presence of 10 μm PKG inhibitor Rp-8-pCPT-cGMPS (RP-G). B, Kinase activity of active recombinant apPKG (25 ng) and bovine PKG type-Iα (bPKG1α) (50 ng) in the presence of four type-I PKG peptide substrates: peptide A, RKISASGP; B, RKISASEFDRPLR (BPDEtide); and D, RKRSRAE (H2Btide). Peptide C, QKRPRRKDTP, is a type-II PKG substrate. C, apPKG phosphorylates recombinant VASP at serine-239. Purified recombinant VASP-GST (0.5 μg) was incubated with active apPKG (100 ng) or the recombinant bovine PKG (50 ng), or in the kinase buffer alone, at room temperature for 20 min. After SDS-PAGE, a Western blot was probed with an anti-phospho-VASP (Ser239) (p-VASPS239) antibody.
ApPKG is located in axons where it is activated by nerve injury and retrogradely transported to the cell body of the SNs
Aplysia peripheral nerves p5-p9 innervate the mid-body wall and tail region and contain axons of the SNs (Walters et al., 1983a,b; Billy and Walters, 1989). To determine whether apPKG is in axons, we first raised a rabbit polyclonal antibody, AbapPKG, against a peptide located at the N terminus (amino acids 18-128) of the protein. AbapPKG was affinity purified and used to probe Western blots. AbapPKG recognized an 80 and 100 kDa polypeptide in pedal and pleural ganglion extracts, but not in muscle or genital tissue as well as the affinity-purified 80 kDa recombinant protein (Fig. 3A). We believe that the 100 kDa band is the dominant form of apPKG, because it is most abundant and has a greater affinity for cGMP than the 80 kDa constituent (data not shown). Both bands were also recognized by a commercial antibody that was generated against human type-Iα PKG (amino acid residues 657-671: 50% identity to apPKG) (data not shown). apPKG contains consensus sequences for several kinases and other enzymes and the 100 kDa constituent might contain one or more posttranslational modifications.
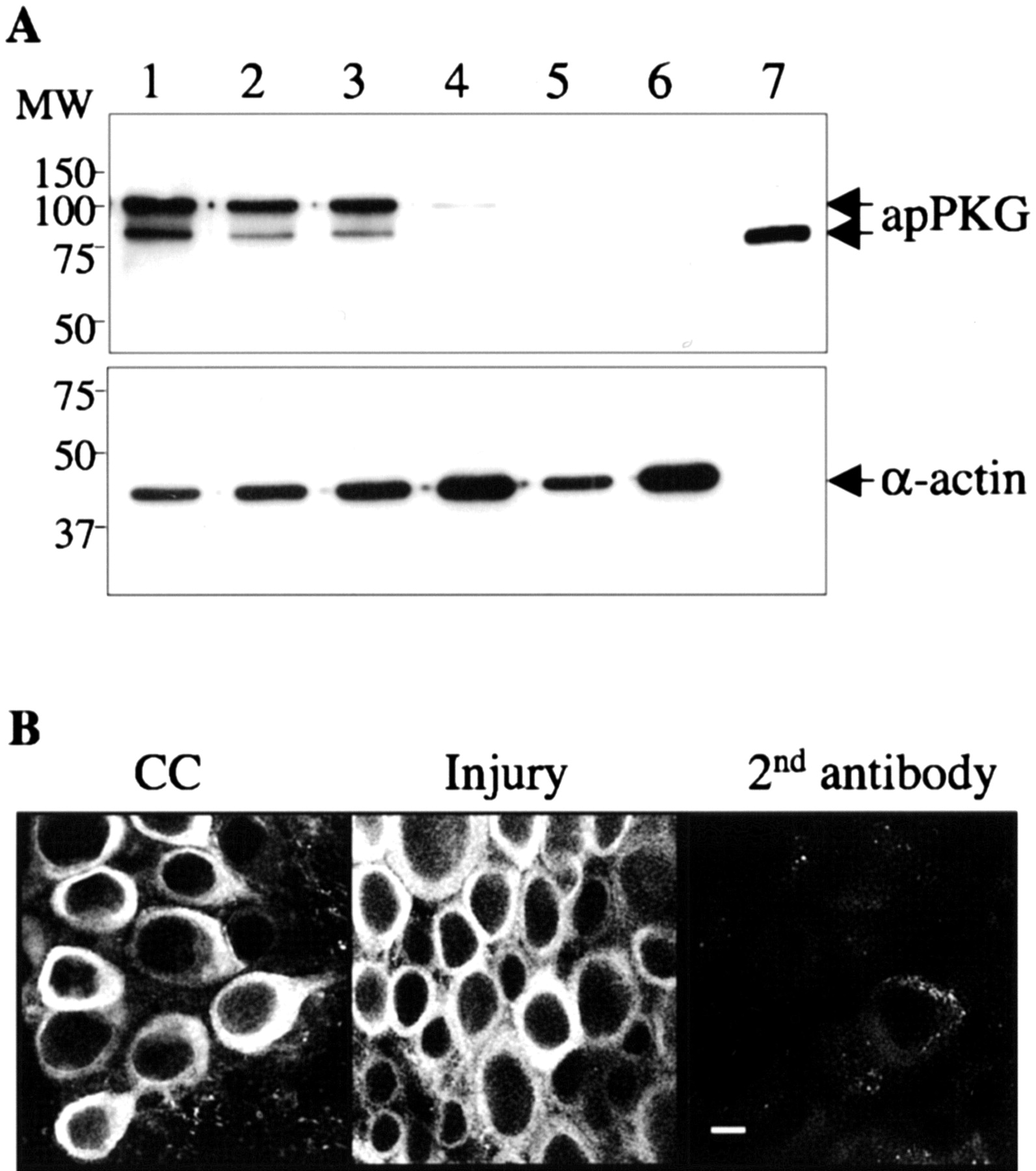
A, apPKG protein expression in the nervous system. Top, A Western blot (10 μg of protein per lane) was probed with antibody AbapPKG raised against an N-terminal peptide of apPKG protein. Lane 1, Pedal ganglia; lane 2, pleural ganglia; lane 3, axoplasm extruded from peripheral nerves; lane 4, body wall muscle; lane 5, buccal mass; lane 6, genitalia; lane 7, recombinant apPKG. The two specific apPKG signals are indicated by arrows. Bottom, The blot was stripped and reprobed with an antibody against α-actin to indicate protein load. Positions of molecular mass markers in kilodaltons are indicated on the left. B, apPKG is expressed in the Aplysia SNs. Confocal microscopy of a 2 μm optical section taken from a Z-series through the pleural sensory cluster exposed to AbapPKG 24 hr after nerve crush in vivo. Shown are representative images of contralateral uninjured (CC) and injured sensory clusters from sections in the middle of the neuron to show the nucleus. An injured sensory cluster stained in the presence of second Ab only shows the background staining. Scale bar, 20 μm. Although the staining is primarily in cytoplasm, the nucleus of some neurons is also stained. Note that the apPKG-staining pattern is essentially identical between injured and contralateral uninjured sensory clusters.
To determine whether apPKG is present in axons, we extruded axoplasm from nerves p5-p9. Figure 3A shows that both polypeptides were present. The SNs have axons in nerves p5-p9, and immunocytochemistry with AbapPKG stained essentially all of the SNs in the cluster (Fig. 3B).
To determine the effects of nerve injury on axonal apPKG, we crushed nerves p5-p9 on one side, thereby axotomizing the axons in each nerve. At various times afterward, we assayed apPKG activity in the ipsilateral and contralateral pedal and pleural ganglia, which included the cluster of SN cell bodies. As shown in Figure 4A, there was a delay of ∼16 hr after nerve crush before significant apPKG activity appeared in the ipsilateral pleural ganglion. The activity then increased for at least 24 hr, but was not significantly different from baseline at 48 hr. Little or no apPKG activity was detected in the contralateral control pleural ganglion (Fig. 4A). Significantly, apPKG activity in the cell bodies of the ipsilateral pedal ganglion neurons remained at basal level during the 48 hr after axotomy (Fig. 4A), indicating that the activation of apPKG is selective for neurons in the pleural ganglion.

ApPKG activity appears in the pleural ganglion after a delay after nerve crush. A, Peripheral nerves p5-p9 were crushed on one side. At the indicated times, pleural (white/gray squares) and pedal (dotted/black squares) ganglia were collected from the injured (black/gray) and contralateral control (CC) (white/dotted) sides and assayed for apPKG activity using BPDEtide as substrate in the presence of the PKA-specific inhibitor 6-22 amide. ApPKG activity at each point was corrected for autophosphorylation and was normalized to total apPKG activity elicited by adding 1 μm 8-Br-cGMP to a duplicate sample. Relative apPKG activity was the ratio of the apPKG activity of each sample to the basal apPKG activity in a sample from a naive animal. Six animals were examined independently at each time point. Two-way ANOVA with repeated measures showed significant effects of axotomy, time, and their interaction in the pleural ganglia (p < 0.001 in each case). B, apPKG is activated and retrogradely transported after injury. p5-p9 nerves were crushed and ligated. Twenty-four hours later, axoplasm was extruded from the crush (Cr) site, from the crush/ligation (Cr/Lig) site, and from the ligation (Lig site) on the control nerves, as indicated by brackets in the schematic. Axoplasm containing equal amounts of protein from each segment was assayed for apPKG activity as in A. The line indicates the level of basal apPKG activity, determined by assaying axoplasm collected from noninjured nerves. Error bars represent ± SEM. An asterisk indicates significant difference compared with all of the other groups (p < 0.05; ANOVA and Newman-Keuls tests). An enrichment of active apPKG at the Cr/Lig site is characteristic of positive molecular injury signals. C, ApPKG protein is retrogradely transported after injury. Peripheral nerves were crushed and ligated as in B. Twenty-four hours later, injured and control nerves were fixed, exposed to AbapPKG, and processed for immunohistochemistry. Optical sections (2 μm) through each nerve were examined by confocal microscopy. All of the images were at the same magnification and were captured after identical exposures to the confocal beam. Each image is aligned in the same direction; the central somata are to the left of the segment shown. 1, Segment of a nerve from a non injured animal. 2, Segment containing the Lig site (arrow) on a nerve contralateral to the injury. 3, Segment of nerve containing the Cr site (arrow), which has expanded over the 24 hr. 4, Segment of nerve containing the Cr/Lig site (arrow). 5, Segment of nerve exposed to the second antibody only. Scale bar, 20 μm for all of the images.
The long delay before apPKG activity appeared in the cell bodies is consistent with the idea that the kinase is a positive injury signal. Moreover, apPKG contains a nuclear localization sequence (NLS) that can provide access to the retrograde transport system (Ambron and Walters, 1996; Schmied and Ambron, 1997; Hanz et al., 2003). To investigate this possibility, we used a standard crush-ligation protocol (Ambron et al., 1995; Johanson et al., 1995) (Fig. 4B). Nerves p5-p9 were crushed unilaterally, and a ligation was placed on each nerve proximal to the crush site. Proteins that are transported away from the crush site (toward the cell bodies) accumulate in the axoplasm behind the ligation where they can be collected. 24 hr later we removed 0.5 cm nerve segments: (1) proximal to the crush (Cr) site, (2) distal to the ligation on the crushed nerves (the Cr/Lig site), and (3) distal to the ligation on the control side (the Lig site) (Fig. 4B, top). Axoplasm was then extruded from each segment, as well as from segments of nerves p5-p9 from an animal that did not receive a nerve injury.
When equal amounts of each axoplasm were screened for apPKG activity, there was a 10-fold increase in apPKG activity in axoplasm from the Cr/Lig segment relative to basal activity in axoplasm from noninjured animals (Fig. 4B, bottom). In contrast, apPKG activity at the Cr segment was only threefold greater than the basal activity, and that at the Lig segment was at the basal level.
The accumulation of apPKG activity at the Cr/Lig site was supported by immunocytochemical studies in which we used AbapPKG to examine the distribution of apPKG protein in noninjured and injured nerves. apPKG staining was uniformly distributed along axons in naive nerves (Fig. 4C, panel 1). After nerve crush, however, there was a decrease in the staining at the Cr site (Fig. 4C, panel 3) and a significant increase at the terminations of axons at the Cr/Lig site (panel 4). There was no increase in staining at the Lig site (Fig. 4C, panel 2). The accumulation of both apPKG protein and activity at the Cr/Lig site relative to the Cr and Lig sites is strong evidence that apPKG is a positive injury signal (Ambron et al., 1995).
The nitric oxide-cGMP-PKG pathway regulates the induction of LTH in SNs in vitro
When Aplysia SNs are axotomized by removal from the ganglion and individually placed in culture, they regenerate their axons and exhibit a decrease in spike threshold and an increase in repetitive firing, which are two characteristics of LTH (Ambron et al., 1996; Bedi et al., 1998; Sung and Ambron, 2004). To examine the appearance of this LTH in greater detail, we recorded from the SNs in vitro on the second to the seventh day and compared their electrical properties with those from SNs within the cluster in situ. Approximately 10% of the cells tested in vitro did not have a sufficient resting potential or were refractory to firing and were not included in the study. We detected the same decrease in threshold and increase in repetitive firing as reported previously, and also found that the axotomized SNs exhibited significant spike broadening and an increase in spike amplitude relative to the controls (Fig. 5). The changes were significant on the second day and persisted until at least the seventh day. The fact that these same four changes in electrical properties occur in hyperexcitable rat DRG SNs after axotomy (Abdulla and Smith, 2001), affirms the use of dissociated Aplysia SNs as a model system for studies of sensory alterations that may contribute to neuropathic pain.

SNs develop an LTH in vitro. The electrical properties of SNs after time in vitro were compared with those in control SNs in the sensory cluster in vivo (d 0). A, Top, A representative single action potential elicited in response to a 20 msec depolarizing pulse showing the increase in spike amplitude after 7 d in vitro. Bottom, Action potential discharge in response to a normalized 1 sec intracellular test pulse. Note the repetitive firing in the neurons after 7 d in vitro. B, Data comparing spike duration, spike amplitude, spike threshold, and repetitive firing of control SNs (gray bars) with those after 2-7 d in vitro (open bars). Each bar contains the number of cells examined. Error bars represent ± SEM. An asterisk indicates significant difference from the in vivo value (p < 0.01; ANOVA and Newman-Keuls test).
To investigate whether the appearance of the LTH requires apPKG activity, we removed the SNs from the ganglion in the presence of either the soluble guanylyl cyclase (sGC) inhibitor ODQ, the PKG inhibitor Rp-8-pCPT-cGMPS, or the PKA inhibitor Rp-cAMPS. We used the latter because PKA has properties in common with PKG (Scott, 1991; Francis and Corbin, 1994) and has been implicated in various forms of synaptic plasticity in Aplysia SNs (Ghirardi et al., 1992; Byrne and Kandel, 1996; Bedi et al., 1998; Muller and Carew, 1998; Chain et al., 1999; Liao et al., 1999; Sutton and Carew, 2000; Antonov et al., 2003). The cells were subsequently allowed to regenerate in the presence of the inhibitors in vitro, and on the third day, their electrophysiological properties were compared with those of SNs that were removed from the same ganglia at the same time, but that had not been exposed to a drug. We found that both Rp-8-pCPT-cGMPS and ODQ prevented the increase in repetitive firing (Fig. 6A) and the decrease in spike threshold (B). In contrast, the PKA inhibitor had no significant effect on either parameter. Neither Rp-8-pCPT-cGMPS nor ODQ inhibited the appearance of excitability when added to 2-d-cultured SN (data not shown), confirming the data in Figure 4A showing that apPKG activation is transient. We did not examine spike broadening or amplitude in these experiments. None of these treatments, which used similar or even lower concentrations of the drugs than that reported for mammalian (Monfort et al., 2002) and Aplysia neurons (Lewin and Walters, 1999), affected the resting membrane potential. Significantly, they also did not alter the extent or pattern of neurite growth. In contrast, exposing SNs to U0126, a selective MEK inhibitor, produced severe growth defects (data not shown). We did not examine these cells further.

Inhibiting NOS, sGC, or apPKG prevents the induction of LTH in SNs in vitro. SNs were removed from the cluster and grown in vitro in the presence of Rp-8-pCPT-cGMPS (Rp-cGMPS), Rp-cAMPS, or ODQ (all 10 μm). Other SNs removed at the same time were not exposed to any inhibitors as controls (C). A, B, On the third day in vitro, 12 SNs exposed to Rp-cGMPS, Rp-cAMPS, or ODQ, and 12 control SNs were impaled with a microelectrode to assess repetitive firing in response to stimulation at three test currents (A) and to determine spike threshold (B). We examined the cells on the third day, and not later, to avoid more prolonged exposure to the drugs. Two-way ANOVA with repeated measures showed that both Rp-cGMPS and ODQ significantly reduced repetitive firing elicited by test currents of 2 and 3 nA relative to controls. Error bars indicate SEM, and the asterisks indicate significance (p < 0.0001 in each case). Similarly, Rp-cGMPS and ODQ significantly prevented the injury-induced decrease in threshold compared with C cells (ANOVA and Fisher's PLSD tests; p < 0.05). There was a considerable variability in threshold in the presence of the Rp-cAMPS, and the mean difference from controls was not significant. C, Detection of nNOS mRNA in single SNs by RT-PCR. Fragments of appropriate lengths were amplified with primer sets for apnNOS, sensorin A, and the neuron-specific isoform of actin from five separate sets of samples from SNs in vivo (0) or after 16 hr in vitro. The size of the synthesized fragments detected by ethidium bromide staining on 2% agarose gels were identical with those predicted from the known sequences in the database. In addition, the PCR products were verified by DNA sequence analysis. Finally, there was no amplification in the absence of reverse transcriptase, indicating the RNA preparations were not contaminated by genomic DNA (bottom panel). Positions of molecular markers are indicated on the left. D, Effects of the NOS inhibitors on LTH. l-Thiocitrulline (50 μm) and l-NAME (1 mm) were used as described above. n, Number of SNs. The data were normalized to the average excitability of control cells in the same preparation. The asterisk indicates significance (p < 0.001), comparing LTH with and without inhibitor by ANOVA and Dunnett's test. Error bars indicate SEM.
The inhibition of LTH by the sGC and PKG inhibitors indicated that cGMP synthesis and PKG activation occur within the sensory neurons. NO is known to elevate cGMP production via sGC (Schlossmann et al., 2003), and NO is produced by the enzyme NOS (Bredt and Snyder, 1990; Moroz et al., 1996). To investigate the likelihood that NOS activation is required for LTH, we first used RT-PCR to detect the cellular levels of apnNOS mRNA in single SNs immediately after their removal from the ganglion (0 hr) or after 16 hr in vitro. As shown in Figure 6C (top panel), four of five cells expressed significant amounts of apnNOS mRNA after 16 hr in vitro, whereas none was detected in the 0 hr cells. In contrast, the mRNA for the SN-specific neuropeptide sensorin A (Brunet et al., 1991) and the neuron-specific isoform of actin (DesGroseillers et al., 1994) was abundant in every cell (Fig. 6C, second and third panels, respectively).
l-Thiocitrulline is an effective inhibitor of apNOS, and we found that exposing the SNs to this drug in vitro as above markedly reduced the maximum firing relative to untreated controls (Fig. 6D). Another NOS inhibitor, l-NAME also reduced LTH under the same conditions, but was not as effective as l-thiocitrulline (Fig. 6D).
These data indicate that the NO-cGMP-PKG pathway is required for inducing LTH in the SNs.
Somatic apMAPK is phosphorylated at its activation site by apPKG
The induction of LTH in Aplysia SNs after peripheral injury requires gene transcription (Lewin and Walters, 1999) and could be effected directly by apPKG if the kinase translocated to the nucleus after entering the cell body. Immunostaining revealed a low constitutive level of apPKG in some SN nuclei, but there was no increase after axotomy (Fig. 3B). This suggested that apPKG contributes to the induction of LTH by activating a factor that is imported into the nucleus. An ERK member of the MAPK family is a good candidate for this factor, because LTH can be induced by injecting recombinant ERK1 into SN somata (Sung et al., 2001). To assess whether an ERK was activated after injury, we crushed p5-p9 in vivo and subsequently probed Western blots of injured and control pleural neurons with an antibody (AbpTpYmapk) that recognizes ERKs that have been activated by dual phosphorylation at the T-E-Y site. The antibody recognized a single 43 kDa polypeptide that had little activity in pleural neurons 4 or 8 hr after the injury (Fig. 7A, top panel). By 16 hr, however, there was more active kinase on the crush side relative to the contralateral control, and the level continued to increase for at least 48 hr (Fig. 7A, top panel).

A, Activation of apMAPK in pleural ganglia after nerve crush. Top, Left, Twenty-five micrograms of a pleural ganglia lysate collected at the indicated times after p5-p9 nerve crush were resolved by SDS-PAGE, and a Western blot was probed with AbpTpYmapk to detect active ERK-MAPKs. The antibody recognized a 43 kDa kinase whose activity was increased on the injured side (I) relative to the contralateral control (CC) 16 hr and later after injury. Bottom, Left, The injury-activated kinase was apMAPK. The blot was stripped and probed with the D8 antibody, which recognizes both active and inactive apMAPK. D8 recognized the same 43 kDa protein that was recognized by the pTpY antibody. Right, Relative MAPKpTpY was determined by densitometry (Sung et al., 2003). All of the values were normalized to levels of total apMAPK. The ratio of the normalized MAPKpTpY intensity at each time to the normalized naive control is presented. The value for the naive control was arbitrarily set to 1.0. The Western analysis in this figure was performed with the same material used to assess apPKG activity in Figure 4 A. The apMAPK values represent an average of six animals. The line indicates the basal level of apMAPK activity from naive animals. Each of the following experiments (B-E) was repeated twice at least, and representative results are shown. B, apPKG phosphorylates endogenous apMAPK in neurons, but not axoplasm, in vitro. Left, Pleural neurons were removed from a noninjured animal, a lysate was prepared, and 25 μg was incubated with 100 ng of active apPKG protein or with 1 μm 8-Br-cGMP in the presence or absence of 10 μm U0126. Right, Twenty-five micrograms of axoplasm was incubated with active apPKG as above. Active apMAPK was detected by immunoblotting with AbpTpYmapk. C, apPKG phosphorylates serine-383 in Elk1. One hundred nanograms of apPKG and 0.5 μg of purified recombinant Elk1 protein were incubated with either 5 μg of the pleural neuronal lysate or 0.2 μg of purified recombinant ERK2. Phosphorylated Elk1 (p-Elk1) was detected by probing a Western blot with an antibody that recognizes phosphorylated Ser383. D, Direct phosphorylation of ERK2 at T183 by apPKG. Two hundred nanograms of recombinant ERK2 was incubated with 100 ng of apPKG in the presence or absence of 1 μg of BPDEtide. The reaction mixture was divided into thirds, a Western blot of each was prepared, and ERK2 was detected with AbpTpYmapk (top), AbpYmapk (middle), and AbpTmapk (bottom), respectively. E, Relative activation of ERK2 by apPKG and MEK1. One hundred nanograms of apPKG, the catalytic subunit of MEKK1, and MEK1 were used as indicated, and the production of phospho-Elk1 was measured with AbpElk1 as shown in C. F, Nerve injury increases the level of apMAPKpY in the cell soma of injured (I) relative to contralateral (CC) neurons. A Western blot prepared using 25 μg of a pleural neuronal lysate collected at the indicated times after p5-p9 nerve crush was probed with the monospecific pY antibody, AbpYmapk, followed by D8 to detect total apMAPK as in A. The blot shows the results of two animals for each point. N, Lysate from animal without nerve crush.
Aplysia neurons contain an ERK2 homolog, apMAPK (Michael et al., 1998), that enters the nucleus after injury in vitro (Martin et al., 1997). apMAPK has the same catalytic domain and T-E-Y activation site as ERKs 1 and 2. To determine whether apMAPK was the kinase activated after injury, we probed the same blot with an antibody, D8, which specifically recognizes apMAPK (Martin et al., 1997), and found that the antibody recognized the 43 kDa injury-activated kinase (Fig. 7A, bottom panel). The antibody detects both active and inactive apMAPK, and there was little difference in the amount of total apMAPK protein among the samples.
The finding that apMAPK activity began to increase just after the arrival of active apPKG in the cell body (compare Figs. 4A, 7A) suggested a link between apPKG and apMAPK. We tested this idea by preparing a lysate of pleural ganglion neurons and using AbpTpYmapk to monitor samples for active apMAPK. Little endogenous phospho-apMAPK was detected in the lysate (Fig. 7B, lane 1), but adding active apPKG or 8-Br-cGMP markedly enhanced the level of active apMAPK (lanes 2, 3) that was recognized by antibody D8 (data not shown). Surprisingly, the activation was not blocked by U0126, a potent inhibitor of the upstream kinase MEK (Fig. 7B, lane 4). Interestingly, adding active apPKG to axoplasm did not activate apMAPK (Fig. 7B, lanes 5, 6) (see Discussion). Another study with the lysate showed that the apPKG-activated apMAPK phosphorylated its physiological substrate, Elk1, at the appropriate Ser383 (Fig. 7C, lane 3).
The activation of apMAPK in the presence of U0126 suggested that apPKG activates apMAPK directly, and we therefore incubated active apPKG with recombinant mammalian ERK2. ERK2 was a surrogate for apMAPK, because we do not have recombinant apMAPK. Both kinases contain the target T-E-Y site, however. As in the lysate, apPKG activated ERK2 to phosphorylate Elk1 (Fig. 7C, lane 6). When the experiment was repeated, the activated ERK2 was recognized by AbpTpYMAPK, indicating that it was doubly phosphorylated (Fig. 7D, top, lane 2). The activation was specific, because it was reduced in the presence of BPDEtide (Fig. 7D, top, lane 3). ERK2 is maximally activated when both the -T- and -Y- amino acids are phosphorylated, yet PKGs are serine/threonine kinases. It is relevant, therefore, that bacterial recombinant ERK2 has a low level of activity that is attributable to the presence of a phosphate on the -Y- moiety (Cha and Shapiro, 2001). Indeed, the antibody to ERKpY185 recognized the recombinant ERK2 substrate on a Western blot (Fig. 7D, middle). There was no increase in phosphorylation of the -Y- when ERK2 was incubated with apPKG (Fig. 7D, middle, lane 2). However, when a duplicate blot was probed with an antibody to ERKpT183, there was an increase in the phosphorylation of the -T- in the presence of apPKG (Fig. 7D, bottom, lane 2). This antibody reacts specifically with the monophosphorylated threonine and doubly phosphorylated ERKs. Our result indicates that apPKG can fully activate ERK2 by phosphorylating ERK2 that already contains a phosphate on the -Y-.
We next examined whether incubating apPKG and recombinant ERK2 produces an active ERK2 with enzymatic activity comparable with that produced by MEK1, which produces the maximally activated kinase. ERK2 activity was measured by the phosphorylation of Elk1 at Ser383, and MEK1 was activated by the catalytic subunit of MEKK1 (Xu et al., 1995). As we anticipated, apPKG produced an ERK2 with similar activity as that produced by MEK1 (Fig. 7E). This result indicates that, in addition to MEK1, PKG is an activator of ERK2.
Because PKG phosphorylates T183 on ERK2 that already has a phosphate on Y185 (Fig. 7D), an essential question is whether monophosphorylated apMAPK at -Y- is available for phosphorylation by apPKG after axotomy. We answered this question by first crushing p5-p9 in vivo. Samples were then blotted and probed with an antibody to ERKpY185. Indeed, apMAPKpY was present in pleural neurons 8 hr after the injury, and its expression was increased on the crush side relative to the contralateral control at 16 hr (Fig. 7F, top panel). ApMAPKpY was not detected in two naive animals that were analyzed (Fig. 7F, top panel). Probing the blot with an antibody that detects both active and inactive apMAPK showed that there was little difference in the amount of total apMAPK protein among the samples (Fig. 7F, bottom panel). Thus, the presence of apMAPKpY in the soma 16 hr after the injury, in conjunction with the arrival of apPKG from the crush site (Fig. 4A), should result in full activation of apMAPK.
The level of apMAPK in the nucleus of SNs in vitro is reduced when apPKG activity is inhibited
We have evidence above that apPKG does not enter the nucleus of the SNs in response to nerve crush in vivo (Fig. 3B). Nevertheless, type-I PKGs have a putative NLS (Gudi et al., 1997), and apPKG has a short stretch of positively charged amino acids (453KCLKKKHI) in the ATP-binding domain that could function as an NLS. The import of proteins into the nucleus of Aplysia neurons is readily assessed by injecting their fluorescently labeled cognates directly into the cell body (Ambron et al., 1992; Schmied et al., 1993; Gunstream et al., 1995). We therefore injected Alexa-labeled recombinant apPKG into the soma of SNs after 2 d in vitro, severed the neurites to initiate an injury response, and, 30 min later, examined the cells by fluorescence microscopy. All of the labeled protein remained in the cytoplasm (Fig. 8A). Alexa-labeled BSA also remained in the cytoplasm after injection, as expected (Ambron et al., 1992; Schmied et al., 1993; Gunstream et al., 1995). In contrast, injected Alexa-labeled active recombinant vertebrate ERK1, which is imported into the nucleus in a variety of cell types (Karin, 1994) rapidly entered the nucleus where it was distributed in discrete patches (Fig. 8A). Similar patches have been seen after the import of other proteins into Aplysia nuclei (Ambron et al., 1992; Schmied et al., 1993; Gunstream et al., 1995).

A, apPKG does not enter the nucleus of SNs. Equal amounts of Alexa Fluor 546-labeled BSA, apPKG, or ERK1 were microinjected into the cytoplasm of SNs after 2 d in vitro. The neurites were then severed with a fine needle to elicit an injury response, and 30 min later, the cells were examined by confocal microscopy. The image is a 2 μm optical section through the center of the cell that shows the nucleus. Scale bar, 20 μm. B, Rp-8-pCPT-cGMPs inhibits axotomy-induced apMAPK nuclear translocation. SNs untreated as controls, and those exposed to either 10 μm Rp-8-pCPT-cGMPs (Rp-cGMPs) or Rp-cAMPs for 2 d in vitro, were immunostained with D8 antibody to localize apMAPK. Top, Representative examples of 2 μm optical sections of control SNs and those exposed to Rp-8-pCPT-cGMPs or Rp-cAMPs. Scale bar, 20 μm. Bottom, Histogram of the mean value of nuclear MAPK immunoreactivity. The staining intensity was determined by a person who was blind to the treatment that the cells received. n, Number of cells for each treatment. Asterisk indicates significant difference from control (p < 0.05 by ANOVA and Newman-Keuls tests).
The inability of apPKG to enter the nucleus of Aplysia SNs is consistent with the idea that it contributes to the induction of LTH by promoting the nuclear import of apMAPK. If so, then inhibiting the apPKG pathway should block the axotomy-induced entry of apMAPK into the nucleus. We therefore exposed SNs in vitro either to the PKG blocker, Rp-8-pCPT-cGMPS, or the PKA blocker, Rp-cAMPS, under the conditions that induce LTH (Fig. 6A). On the third day, the cells were fixed, permeabilized, and exposed to antibody D8 to visualize apMAPK. Confocal microscopy showed that untreated cells and those exposed to Rp-cAMPS had the same levels of nuclear staining (Fig. 8B). In contrast, there was a dramatic reduction in nuclear staining in the cells treated with Rp-8-pCPT-cGMPS (Fig. 8B). Thus, both the induction of LTH and the presence of apMAPK in the nucleus depend on apPKG activity.
Discussion
Persistent neuropathic pain in humans after nerve injury is physically and psychologically debilitating. Because an important component of this pain is often the LTH that appears in primary afferent neurons, it is important to understand how axotomy induces LTH. An LTH with similar properties appears in nociceptive SNs of Aplysia after axotomy, and we exploited the experimental advantages of these neurons to define a signaling pathway responsible for the induction of LTH. One major surprise was that this pathway involves the phosphorylation of apMAPK by PKG. This is an alternative to the traditional MAP kinase cascade and suggests that PKG and MAPK have unique roles after nerve injury.
ApPKG is a positive injury signal in SNs
We cloned an Aplysia type-I PKG whose mRNA is in the SNs and other neurons in the pleural ganglion (Fig. 1D). When we crushed the peripheral nerves in vivo, apPKG activity appeared in the somata of pleural neurons, but only after a delay of ∼16 hr (Fig. 4A). A similar delay in response to nerve injury has been attributed to positive injury signals (Schmied et al., 1993; Sung et al., 2001; Lin et al., 2003), and we knew from Western blots probed with AbapPKG that apPKG is present in axoplasm extruded from the peripheral nerves (Fig. 3A). In addition, because proteins are retrogradely transported along Aplysia axons at a rate of 1.5 mm/hr (Ambron et al., 1992; Schmied et al., 1993; Gunstream et al., 1995), the delay is consistent with the transport of apPKG from the crush site, which was located 2 cm from the ganglion. We then placed a ligation proximal to the crush site and found that activated apPKG and total apPKG protein accumulated on the distal side of the ligation, relative to the crush site (Fig. 4B,C). These findings establish apPKG as a positive injury signal.
We do not know the mechanism responsible for the injury-induced transport. Although apPKG contains several potential myristoylation sites that could bind it to vesicles after injury, we always found apPKG to be soluble after subcellular fractionation. Hanz et al. (2003) have shown that proteins containing an NLS are retrogradely transported via dynein and importins in the rat sciatic nerve, and we are presently investigating this interesting possibility. The latter gains importance, because we recently found that a type-I PKG is activated and transported retrogradely in the sciatic nerve (Y.-J. Sung and R. T. Ambron, unpublished observations).
The appearance of the LTH in axotomized Aplysia SNs in vitro depends on NOS, sGC, apPKG, and apMAPK
The LTH that appears in both the SNs (Fig. 5) and mammalian nociceptive SNs after nerve injury (Abdulla and Smith, 2001) have similar electrophysiological properties. If, as we believe, this congruence reflects conserved mechanisms, then LTH might be induced by common pathways in both types of cells. We examined SNs in vitro and found that the axotomy-induced reduction in spike threshold and the increase in hyperexcitability were blocked by Rp-8-pCPT-cGMPS, ODQ, and l-thiocitrulline, inhibitors of apPKG, sGC, and NOS, respectively (Fig. 6 A,B,D). Moreover, Rp-8-pCPT-cGMPS also caused a nearly threefold reduction in the level of apMAPK in the nucleus of the SNs (Fig. 8B). These findings pointed to a direct relationship between the activation of NOS and apPKG, the entry of apMAPK into the nucleus, and the induction of LTH. The finding that NOS mRNA expression in the SNs was increased after injury (Fig. 6C) implies that the level of NOS protein might be the rate-limiting step in the pathway. Neuronal NOS (nNOS) mRNA and protein expression also increase in DRG neurons after severing their peripheral axons (Verge et al., 1992; Fiallos-Estrada et al., 1993; Zhang et al., 1993).
cAMP and PKA have also been implicated in the induction and maintenance of LTH (Scholz and Byrne, 1988; Goldsmith and Abrams, 1992; Bedi et al., 1998), and this was a concern here because PKA and PKG have properties in common. However, we found that exposing the SNs to Rp-cAMPS, a membrane-permeable inhibitor of PKA, neither prevented the induction of LTH nor blocked apMAPK entry into the nucleus. This confirms previous findings that PKA inhibitors failed to block LTH induced by noxious stimulation (Lewin and Walters, 1999).
The activation of both apPKG and apMAPK by injury was intriguing, because it suggested a possible link between these two kinases (Zaragoza et al., 2002). Indeed, when we added active apPKG to a neuronal lysate, it not only activated apMAPK (Fig. 7B), but did so via a pathway that did not require MEK, the ubiquitous upstream activator of the ERKs. This response was physiologically relevant, because activated apMAPK phosphorylated its nuclear substrate Elk1 at the Ser383 (Fig. 7C), which is essential for transcriptional activity (Marais et al., 1993; Whitmarsh et al., 1995). We established a direct interaction between the kinases when we incubated active apPKG with recombinant vertebrate ERK2, which has the same T-E-Y activation site as apMAPK, and found that the ERK2 was both activated (Fig. 7C) and doubly phosphorylated (D, top). The latter finding indicated that the ERK2 was fully active, and this was supported by another study in which equal amounts of apPKG or MEK1 activated ERK2 to comparable levels (Fig. 7E).
The indications that apPKG produces a maximally activated ERK2 creates a paradox, because PKGs are serine-threonine kinases that cannot phosphorylate the tyrosine. However, because the recombinant ERK2 already contains phospho-tyrosine-185 (Fig. 7D, middle), the phosphorylation of threonine-183 by apPKG (Fig. 7D, bottom) should produce a fully activated ERK2.
The phosphorylation of apMAPKpY by apPKG is attractive, because ERK2pY185 has been detected in vertebrate cells (Yao et al., 2000; Cha and Shapiro, 2001; Zhou and Zhang, 2002), and we have evidence that apMAPKpY expression increases in Aplysia neurons after nerve injury (Fig. 7F). apMAPKpY could be produced by a phosphatase that removes the phosphate from doubly phosphorylated apMAPK, or by an injury-activated tyrosine kinase that phosphorylates the tyrosine at the T-E-Y site. We are now determining which mechanism is correct. The possibility that the induction of LTH requires the convergence of apPKG and a tyrosine kinase on apMAPK would confer more control over this pathway. This is reasonable given that LTH radically alters the function of the injured neurons and leads to significant changes in the behavior of the animal.
apMAPK is also present in axoplasm, but is not activated by nerve injury (Sung et al., 2001; Lin et al., 2003), which is paradoxical, given that injury activates the axoplasmic apPKG. One explanation would be that the two kinases are located in different axons. However, when we added active apPKG to axoplasm extruded from the peripheral nerves, apMAPK was not activated under the same conditions that caused its activation in the cell soma (Fig. 7B). Evidently, there is a mechanism in the axon that shields apMAPK from apPKG. Nevertheless, these observations mean that, after its activation in the axon by injury, apPKG must be transported back to the soma to influence nuclear events via apMAPK.
A working model for the induction of LTH
We propose that nerve injury induces nNOS expression in the SNs. The nNOS is then activated in the axon, leading to NO production, resulting in the activation of sGC, the formation of cGMP, and the activation of apPKG. Activated apPKG is transported to the cell body of the SNs where it activates apMAPKpY in the cytoplasm, which then enters the nucleus. Although our findings strongly support this model, we have not completely excluded the possibility that nerve injury causes translocation of apMAPK or activation of somatic PKG by other pathways as well. An NO-cGMP-PKG-MAPK signaling pathway might also be important for LTH induced by the inflammation that develops around a nerve injury site, both in mammals (Millan, 1999; Zimmermann, 2001) and in Aplysia (Clatworthy et al., 1995; Clatworthy and Grose, 1999; Farr et al., 1999, 2001). How might nuclear apMAPK regulate LTH? apMAPK can phosphorylate CREB2 (cAMP response element-binding protein 2), a cAMP response element (CRE) site repressor (Bartsch et al., 1995; Michael et al., 1998), C/EBP (CCAAT/enhancer-binding protein), a transcription factor that binds to the estrogen response element (ERE) site (Alberini et al., 1994), and Elk1, a transcription factor that regulates the serum response element (SRE) site (Lin et al., 2003). Binding to all three sites increases after nerve injury, but with different time courses. Thus, binding of C/EBP and CREB to the ERE and CRE, respectively, is rapid, but relatively short lived (Dash et al., 1998; Sung et al., 2001), whereas the binding of Elk1 to the SRE is biphasic, with an early phase that lasts for a few hours and a second phase that persists for weeks (Lin et al., 2003). CREB is phosphorylated in DRG neurons in response to intense activity (Ji and Woolf, 2001), and the CRE site is required for the LTH response to noxious stimuli in Aplysia SNs (Lewin and Walters, 1999). Based on these considerations, we propose that CREB2 and C/EBP are the targets of apMAPK during the initial induction of the LTH in the SNs, and that the persistence of LTH for weeks is mediated by the phosphorylation of Elk1 by apMAPK. This pathway is selective, because inhibiting NOS, sGC, or PKG prevented the appearance of LTH in dissociated SNs, but did not block growth. If the link between this pathway and LTH represents a fundamental, widely conserved relationship, then therapeutic interventions that target this pathway might mitigate persistent pain after nerve injury without blocking axon regeneration.
Footnotes
This work was supported by Javits National Institutes of Health Grant NS22150 and by private funds. Y.-J.S. is supported by National Institutes of Health Grant NS 35979. We thank Dr. E. R. Kandel for providing D8 antibody, Dr. M. Karin for the pGEXElk1construct, Dr. J. Kyriakis for the pGEXMEKK1-C and pGEXMEK1 constructs, and Dr. R. B. Denman for reviewing this manuscript.
Correspondence should be addressed to Dr. Ying-Ju Sung, Department of Anatomy and Cell Biology, Columbia University, New York, NY 10032. E-mail: yjs8{at}columbia.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249583-13$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}