

Abstract
Reduced glutamate-mediated synaptic transmission has been implicated in the pathophysiology of schizophrenia. Because antipsychotic agents might exert their beneficial effects against schizophrenic symptoms by strengthening excitatory transmission in critical dopaminoceptive brain areas, in the present study we have studied the effects of acute and chronic haloperidol treatment on striatal synaptic plasticity. Repetitive stimulation of corticostriatal terminals in slices induced either long-term depression or long-term potentiation (LTP) of excitatory transmission in control rats, whereas it invariably induced NMDA receptor-dependent LTP in animals treated chronically with haloperidol. Haloperidol effects were mimicked and occluded in mice lacking both D2L and D2S isoforms of dopamine D2 receptors (D2R-/-), in mice lacking D2L receptors and expressing normal levels of D2S receptors (D2R-/-;D2L-/-), and in mice lacking D2L receptors and overexpressing D2S receptors (D2L-/-). These data indicate that the blockade of D2L receptors was responsible for the LTP-favoring action of haloperidol in the striatum. In contrast, overexpression of D2S receptors uncovered a facilitatory role of this receptor isoform in LTP formation because LTP recorded from D2L-/- mice, but not those recorded from wild-type, D2R-/-, and D2R-/-;D2L-/- mice, was insensitive to the pharmacological blockade of D1 receptors.
The identification of the cellular, molecular, and receptor mechanisms involved in the action of haloperidol in the brain is essential to understand how antipsychotic agents exert their beneficial and side effects.
Introduction
Dopamine (DA) receptors are implicated in the control of motor activity, reward, and cognition, and alterations of the DA receptor signaling play a prominent role in a variety of neurological and psychiatric disorders, such as Parkinson's disease, Tourette's syndrome, drug addiction, and schizophrenia (Grace, 1991; Nestler and Aghajanian, 1997; Koob et al., 1998; Leckman and Riddle, 2000; Obeso et al., 2000; Carlsson et al., 2001).
Antipsychotic drugs convey their therapeutic effects in the treatment of schizophrenia by binding DA receptors, and, in particular, the clinical potency of these agents is correlated directly with their ability to inhibit the D2-like family (D2, D3, and D4) of DA receptors (Creese et al., 1976; Seeman et al., 1976). Although blockade of DA receptors is achieved almost instantaneously after the administration of antipsychotic drugs, the therapeutic action of these compounds takes time to appear. Based on this observation, it has recently been proposed that the promotion of plastic, slowly developing rearrangements of neuronal connectivity is essential for the mode of action of antipsychotics (Konradi and Heckers, 2001). In line with this idea, chronic but not acute haloperidol treatment causes complex ultrastructural changes of glutamatergic synapses in the striatum (Meshul and Casey, 1989; Uranova et al., 1991; Kerns et al., 1992; See et al., 1992), indicative of its ability to promote long-lasting potentiation of excitatory transmission (Eastwood et al., 1997; Toni et al., 1999; Konradi and Heckers, 2001; Steward and Worley, 2002). Such an effect might play a crucial role in the therapeutic effects of this drug because a reduced glutamate-mediated transmission has been recognized to mediate, at least in part, the symptoms of schizophrenia (Carlsson et al., 2001; Goff and Coyle, 2001), and the striatum serves not only motor, but also critical, cognitive and motivational functions altered in schizophrenia. Accordingly, deficits in striatal function are thought to produce motivational deficits and to disrupt goal-directed behavior in schizophrenia patients (Swerdlow and Koob, 1987; Goldman-Rakic and Selemon, 1990; Graybiel, 1995,1997; Calabresi et al., 1997a; Canales and Graybiel, 2000). In addition, never-medicated subjects with schizophrenia sometimes exhibit motor deficits resembling those of patients with primary striatal dysfunction (Caliguiri et al., 1993; Chatterjee et al., 1995), and clinical improvement of schizophrenic symptoms treated with neuroleptics has been found to be associated with increased basal ganglia metabolism, possibly reflecting neuroadaptation processes in the striatum (Miller et al., 1997; Westmoreland Corson et al., 2002).
The mechanisms by which chronic blockade of D2-like receptors by haloperidol affects glutamate transmission in the striatum are, however, unknown. To answer this question, in the present study we evaluated the effects of acute and chronic haloperidol treatment on corticostriatal synaptic plasticity. We found that chronic but not acute haloperidol treatment facilitates the emergence of long-term potentiation (LTP) versus long-term depression (LTD) at corticostriatal excitatory synapses. In addition, in the attempt to identify the receptor subtype and isoform involved in this effect, we also studied striatal synaptic plasticity in three distinct mutant mice, in which the expression of the long (D2L) and short (D2S) isoforms of D2 receptors were variably altered. Our data allowed concluding that the D2L isoform of D2 receptors is responsible for the facilitatory action of classical antipsychotics on excitatory transmission in the striatum.
Materials and Methods
Haloperidol treatment and behavioral testing. Adult male Wistar rats and wild-type (WT) and transgenic mice (2-3 months of age) were divided into groups that received 1 or 20 daily injections of either haloperidol (1 mg/kg) or saline (0.9% NaCl). Animals were housed with food and water ad libitum in a room with a 12 hr light/dark cycle and controlled temperature (22-23°C).
In rats, motor abilities were measured 20-26 hr after the last injection and just before the electrophysiological experiments. Behavioral effects were first examined during a 10 min test session in a novel open field (60 × 60 × 40 cm, with lines dividing the floor into 3 × 3 squares) (Steiner and Kitai, 2001). The behavior was videotaped and assessed from tapes by an experimenter who was unaware of the treatment. The behavioral analysis started 30 sec after the animal was placed into the center of the open field. The following behavioral items were measured: number of peripheral squares crossed with all four feet, mobility (seconds), number of central crossed squares, and number of rearing. A RotaRod system constant and accelerating treadmills (Technical & Scientific Equipment GmbH, Bad Homburg, Germany) were then used to study coordinated locomotor activity. All animals were trained during at least one session before starting the saline or haloperidol treatment. The rats were placed on the rod and tested sequentially at 4, 12, 20, 28, 36, and 40 rotations per minute for a maximum of 300 sec at each speed. Overall rod performance was expressed as the integral of time spent on the rod versus turning speeds (Rozas et al., 1997; Picconi et al., 2003).
Mutant mice. Male mice lacking DA D2 (D2R-/-) (Baik et al., 1995) and D2L (Usiello et al., 2000) receptors were used along with their WT counterparts. Two distinct genotypes of D2L-lacking mice were used. In particular, D2L-/- mice were obtained by substitution of exon 6 of the D2 receptor gene with the neomycin cassette (Usiello et al., 2000). Therefore, in D2L-/- mice, D2 transcripts are all translated into D2S receptors. This leads to a higher proportion of D2S-specific mRNAs in vivo compared with the WT condition (Usiello et al., 2000; Centonze et al., 2003a). To exclude the possibility that the effects we observed were dependent on D2S overexpression and not on the specific lack of the D2L isoform, we aimed at reducing the expression of D2S mRNA and proteins to values that were closer to the WT situation. Thus, we mated D2L-/- mice with D2R-/-. Because in the latter no D2 isoforms were present, the mating with D2L-/- would result in a reduction of D2S expression to ∼50%. This is exactly what we obtained both at the mRNA and protein levels (Centonze et al., 2003a).
The D2 receptor ligand-binding assays on striatal membranes were performed as described previously (Baik et al., 1995). Briefly, striata were homogenized with a Polytron in ice-cold buffer containing 10 mm Tris-HCl, pH 7.5, and 5 mm EDTA. The homogenates were centrifuged at 19,000 × g for 40 min at 4°C, then the supernatants were collected and centrifuged again. Ligand-binding assays were performed with 15 μg of striatal membranes using [3H]spiperone (specific activity, 84 Ci mmol; Amersham Biosciences, Les Ulis, France) at concentrations of 0.01-0.6 nm; nonspecific binding was determined in the presence of 1 μm (+)-butaclamol. Binding data were analyzed with the Prism program (Graph Pad, San Diego, CA). Experiments were performed three times in triplicate.
Electrophysiology. Male rats and mice (2-3 months of age) were used for all the electrophysiological experiments. In particular, haloperidol-treated animals (1 and 20 d), and D2R-/-, D2L-/-, and D2R-/-; D2L-/- mice were used along with their respective counterparts (sham-treated rats or WT mice). Corticostriatal coronal slices (200-300 μm) were prepared from tissue blocks of the brain with the use of a vibratome (Calabresi et al., 1992a, 1997b, 2000; Centonze et al., 1999). A single slice was then transferred to a recording chamber and submerged in a continuously flowing Krebs solution (35°C; 2-3 ml/min) gassed with 95% O2-5% CO2. The composition of the control solution was (in mm) 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3.
For intracellular recordings, sharp electrodes were used. They were filled with 2 m KCl (30-60 MΩ), and an Axoclamp 2A amplifier was used for recordings in current-clamp mode.
To evoke glutamatergic corticostriatal synaptic potentials (EPSPs), bipolar electrodes were used. In both rat and mouse slices, the stimulating electrode was located in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum to activate corticostriatal fibers. In contrast, the recording electrodes were invariably placed in the striatum. As the conditioning high-frequency stimulation (HFS), we used three trains (3 sec duration, 100 Hz frequency, at 20 sec intervals) of synaptic stimulation. In ∼50% of the recordings, HFS was delivered in the presence of 10 μm bicuculline to block GABAA receptors. Consistently with previous reports (Calabresi et al., 2000; Gubellini et al., 2003), these experiments gave similar results to those obtained in the absence of this drug, and therefore all the data were pooled together.
For data presented as the mean ± SEM, statistical analysis was performed using a paired or unpaired Student's t test or Wilcoxon's test. When comparing differences between two cumulative distributions, the Kolmogorov-Smirnov test was used. The significance level was established at p < 0.05. The comparisons were made between EPSP amplitudes recorded 5-10 min before and 30-40 min after HFS of corticostriatal terminals.
Drugs were applied by dissolving them to the desired final concentration in saline. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), (+)-MK 801 maleate (MK-801), and RO 32-043 were from Tocris (Bristol, UK). Bicuculline and SCH 23390 were from RBI (Research Biochemicals, Natick, MA).
Results
Motor effects of acute and chronic haloperidol treatment
One day after the last injection of either saline or haloperidol, motor activity and coordination were measured. In an open field, locomotor activity (peripheral crossed squares and mobility) was similar in saline-injected (1 or 20 d treatment; n = 14; data pooled together) and in haloperidol-treated (1 or 20 d treatment; n = 6 for both conditions) rats. Moreover, both rearing behavior and central crossed squares were not altered by acute or chronic haloperidol treatment, as well as motor coordination measured in the RotaRod system (Fig. 1).

Lack of gross motor abnormalities in rats treated with haloperidol. A-D, Both acute (1 d) and chronic (20 d) haloperidol treatment (1 mg/kg) failed to alter horizontal (A, B, D) and vertical (C) motor activity measured in an open field. E, Overall rotarod performance was also unaffected by the two regimens of treatment.
Corticostriatal LTD and LTP
As described previously (Calabresi et al., 2000; Centonze et al., 2001), HFS of corticostriatal terminals induced either LTD (n = 6; p < 0.001) or LTP (n = 8; p < 0.001) of excitatory transmission in control rats. Both forms of activity-dependent synaptic plasticity have been found in the striatum in vivo (Charpier and Deniau, 1997; Reynolds and Wickens, 2000; Reynolds et al., 2001) and in vitro (Akopian et al., 2000; Partridge et al., 2000) in the absence of any pharmacological manipulation, indicating that they represent alternative physiological responses to HFS of corticostriatal synapses. Unless specified otherwise in the text or in the figure legends, however, in our in vitro experiments on both rats and mice, HFS was delivered in the presence of 1.2 mm external magnesium to optimize the appearance of LTD (LTD protocol), whereas LTP was studied after the removal of this ion from the bathing solution, a procedure that prevents the voltage-dependent blockade of NMDA receptors (LTP protocol). As reported previously (Calabresi et al., 2000; Centonze et al., 2003b), this experimental approach was aimed at minimizing the substantially unpredictable appearance of LTP over LTD after HFS (Akopian et al., 2000; Partridge et al., 2000).
Corticostriatal synaptic plasticity after haloperidol treatament
In rats exposed to a single dose of haloperidol, both LTD and LTP were present at corticostriatal synapses (n = 5 and p < 0.01 for both LTD and LTP) and did not differ in amplitude and duration from LTD and LTP recorded in control conditions (n = 7 and p > 0.05 at 10, 20, and 30 min after HFS for both forms of synaptic plasticity). In contrast, in chronic haloperidol-treated rats, HFS of corticostriatal fibers invariably induced LTP, either in the presence (n = 7; p < 0.01) or in the absence (n = 7; p < 0.001) of external magnesium (Fig. 2).

Chronic but not acute haloperidol treatment alters corticostriatal synaptic plasticity. A, The graph shows that in the presence of 1.2 mm external magnesium (LTD protocol), HFS (arrow) of corticostriatal fibers induced LTD of glutamatergic transmission in striatal neurons recorded from sham-treated (control) and 1-d-treated (acute haloperidol) rats. Conversely, the same stimulation protocol induced LTP in rats treated chronically (20 d) with haloperidol. B, HFS delivered in the absence of magnesium ions from the bathing solution (LTP protocol) induced LTP in sham-treated, acute, and chronic haloperidol-treated rats. Traces in the bottom part of the figure are intracellular recordings from single experiments showing EPSPs 10 min before (pre) and 30 min after (post) HFS in control and chronic haloperidol-treated rats, in the presence (A) or in the absence (B) of external magnesium. In this figure and in the following ones, the resting membrane potential and input resistance of the recorded neurons were constant and ranged between -83 ± 5 mV and 48 ± 16 MΩ.
We have measured the amplitude, the half-decay time, and the duration of the membrane depolarizations induced by HFS in control rats and in rats chronically treated with haloperidol to investigate whether differential characteristics of these events could account for the different forms of synaptic plasticity observed in the two groups. These parameters, however, did not significantly differ in the two classes of animals. Accordingly, in the presence of external magnesium, the amplitude of HFS-induced depolarization was 46 ± 5 mV in control rats (n = 7) and 43 ± 8 mV in rats chronically treated with haloperidol (n = 7; p > 0.05). The half-decay time was 3.1 ± 0.4 sec in controls (n = 7) and 3.0 ± 0.6 sec in treated rats (n = 7; p > 0.05). The duration was 8.8 ± 3 sec in controls (n = 7) and 8.8 ± 5 sec in treated rats (n = 7; p > 0.05). In contrast, in the absence of magnesium, the amplitude was 47 ± 6 mV in controls (n = 7) and 45 ± 7 mV in rats chronically treated with haloperidol (n = 7; p > 0.05). The half-decay time was 3.5 ± 0.6 sec in controls (n = 7) and 3.5 ± 0.4 sec in treated rats (n = 7; p > 0.05). The duration was 11.9 ± 2 sec in controls (n = 7) and 12.8 ± 4 sec in treated rats (n = 7; p > 0.05; data not shown). As reported previously (Calabresi et al., 1992b), the intrinsic membrane properties of these neurons were also unaltered by the treatment, as well as the pharmacology of cortically evoked EPSPs. In fact, the AMPA glutamate receptor antagonist CNQX (10 μm) fully blocked the EPSPs in the two groups of animals in the presence of external magnesium (n = 6 for both groups), whereas in magnesium-free solution, the suppression of cortically evoked EPSPs was achieved after coapplication of 10 μm CNQX plus the NMDA receptor antagonist MK-801 (30 μm) (n = 5 for both groups; data not shown).
Intrinsic and synaptic properties of striatal neurons in WT and D2 receptor mutant mice
Intracellular sharp microelectrode recordings were performed in corticostriatal slices obtained from WT, D2R-/-, D2L-/-, and D2R-/-;D2L-/- mice. In the present study, we included 49 striatal spiny neurons recorded from WT mice, 51 from D2R-/- mice, 45 from D2L-/- mice, and 44 from D2R-/-; D2L-/- animals. In these four groups, resting membrane potential and input resistance of striatal neurons were similar (p > 0.05) and ranged between -80 and -90 mV and 39 and 60 MΩ, respectively. Striatal cells were silent at rest and showed membrane rectification and tonic firing activity during depolarizing current pulses. These features closely resembled the electrical activity described previously for rat and mouse spiny neurons (Calabresi et al., 1997b, 2000; Centonze et al., 1999; Picconi et al., 2003) (Fig. 3A,B).

Intrinsic and synaptic properties of striatal spiny neurons of WT and D2 receptor mutants. A, The electrophysiological traces show that in WT and D2L-/- neurons, the membrane responses to hyperpolarizing and depolarizing current steps were similar. B, This graph shows that the current-voltage relationships of striatal neurons recorded from WT, D2R-/-, D2L-/-, and D2R-/-;D2L-/- mice were virtually coincident (n = 15 for each genotype). C, The electrophysiological traces are cortically evoked EPSPs recorded from WT, D2R-/-, D2L-/-, and D2R-/-;D2L-/- striatal neurons in the presence and in the absence of magnesium ions and in the presence of magnesium plus 10 μm CNQX, antagonist of non-NMDA glutamate receptors. Note that in the absence of external magnesium, EPSP amplitude and duration increased in the four genotypes, as an expression of the contribution of glutamate NMDA receptors in corticostriatal transmission.
The pharmacology of EPSPs was also similar in WT (n = 5), D2R-/- (n = 6), D2L-/- (n = 6), and D2R-/-;D2L-/- (n = 5 for each genotype) mice. In fact, similar to the results obtained in control and chronic haloperidol-treated rats, in the presence of physiological concentrations of external magnesium (1.2 mm), CNQX (10 μm) fully suppressed the EPSPs in the four groups of animals, whereas 10 μm CNQX plus 30 μm MK-801 were needed to block corticostriatal EPSPs recorded after removal of external magnesium (Fig. 3C).
D2 receptor ligand-binding sites on striatal membranes of D2R-/-;D2L-/- mice
Increased expression of D2S receptors has been described in D2L-/- mice (Usiello et al., 2000). D2L-/- mice therefore were mated with D2R-/- mice in an attempt to bring the amount of D2S sites to levels similar to those of WT animals. The pharmacological features of D2S receptor-binding sites in D2R-/-;D2L-/- mice in comparison with D2L-/- mice were assessed by binding assays using [3H]spiperone, as described previously (Baik et al., 1995; Usiello et al., 2000). The results of these experiments showed that in D2R-/-;D2L-/- mice, the maximum binding capacities (Bmax = 237 + 49 fmol/mg protein) were strongly reduced compared with D2L-/- (Bmax = 497 + 56 fmol/mg protein) and WT (Bmax = 416 + 51 fmol/mg protein) mice (Centonze et al., 2003a).
Synaptic plasticity in WT and D2 receptor mutant mice
To identify the DA receptor subtype and isoform involved in the electrophysiological effects of haloperidol in the striatum, we measured corticostriatal synaptic plasticity in WT, D2R-/-, D2L-/-, and D2R-/-;D2L-/- striatal neurons. As reported previously (Calabresi et al., 1997b, 2000), both forms of synaptic plasticity were present in WT mice (n = 9 for LTD and n = 10 for LTP; p < 0.001), whereas LTD was absent in D2R-/- mice, in which HFS of corticostriatal fibers produced a robust LTP either in the presence (n = 8; p < 0.001) or in the absence (n = 5; p < 0.001) of external magnesium ions. We also investigated the effects of HFS in D2L-/- and D2R-/-;D2L-/- mice. Also in these mutants, HFS failed to induce LTD but invariably caused LTP in the presence (n = 5 and p < 0.001 for both genotypes) or in the absence (n = 5 and p < 0.001 for both genotypes) of magnesium ions (Fig. 4A,B). As reported previously and in line with the experiments on rats chronically treated with haloperidol, the amplitude, half-decay time, and duration of HFS-induced membrane depolarizations of D2 receptor-lacking neurons were similar to those recorded in WT mice, either in the presence (amplitude, 39 ± 4 mV; half-decay time, 3.1 ± 0.2 sec; duration, 8.1 ± 3 sec) or in the absence (amplitude, 44 ± 6 mV; half-decay time, 3.4 ± 0.5 sec; duration, 12.1 ± 3 sec) of external magnesium (Calabresi et al., 1997b).

DA-regulated synaptic plasticity in WT and mutant mice. A, The graph shows that HFS (arrow) of corticostriatal fibers induced LTD of glutamatergic transmission in WT neurons recorded in the presence of 1.2 mm external magnesium (LTD protocol). In contrast, the same stimulation protocol induces LTP in D2R-/-, D2L-/-, and D2R-/-;D2L-/- striatal neurons. Superimposed traces in the bottom part of the figure are intracellular recordings from single experiments showing EPSPs 10 min before (pre) and 30 min after (post) HFS in WT, D2R-/-, D2L-/-, and D2R-/-;D2L-/- striatal neurons. B, HFS delivered in the absence of magnesium ions from the bathing solution (LTP protocol) induced LTP in WT, D2R-/-, D2L-/-, and D2R-/-;D2L-/- striatal cells. Note that LTPs recorded from the three mutants had higher amplitude than the one recorded from WTs. C, The histogram shows that chronic haloperidol treatment enhanced the amplitude of corticostriatal LTP in WT mice but not in the three D2 receptor mutants. All these experiments were performed in the absence of magnesium ions. *p < 0.01 at 30 min after HFS. D, This graph shows that preincubation with the D1-like receptor antagonist SCH 23390 (10 μm) prevented LTP in WT, D2R-/-, D2R-/-;D2L-/- neurons but not in D2L-/- cells. In this graph, LTP-inducing protocol in WT cells consisted in the application of HFS in the absence of magnesium ions. In contrast, in the three mutants, HFS was delivered either in the absence (at least 3 cells per group) or in the presence (at least 3 cells per group) of magnesium ions. Because the experiments gave similar results, the data were pooled together for each genotype.
The absence of D2L receptors in the three genotypes of mutants indicates that inactivation of this isoform of D2 receptors favors the formation of striatal LTP over LTD and therefore mimics the effects of chronic haloperidol on striatal synaptic plasticity.
To strengthen the conclusion that haloperidol effects on striatal synaptic plasticity were mediated by the blockade of D2L receptors, we performed occlusion experiments. As with rats, HFS invariably induced LTP in chronic haloperidol-treated WT mice, either in the presence (n = 5; p < 0.01 at 10, 20, 30, and 40 min after HFS) or in the absence of external magnesium. Notably, chronic haloperidol significantly enhanced LTP in WT mice recorded in magnesium-free medium (n = 6; p < 0.01 at 10, 20, 30, and 40 min after HFS), whereas it failed to affect the amplitude and duration of striatal LTPs recorded from D2R-/-, D2L-/-, and D2R-/-;D2L-/- animals (n = 5 and p > 0.05 at 10, 20, 30, and 40 min after HFS for the three genotypes) (Fig. 4C).
Role of D2S receptors in striatal synaptic plasticity
Does the D2S isoform of D2 receptors play a role in corticostriatal synaptic plasticity? On the basis of behavioral experiments, a negative interaction between D1 and D2S receptors has been identified (Usiello et al., 2000). Given the requirement of D1 receptor stimulation for LTP induction in both mice and rats (Calabresi et al., 2000; Kerr and Wickens, 2001; Picconi et al., 2003), we therefore decided to investigate the sensitivity to D1 receptor blockade of striatal LTP seen in our mutants. As with WT animals (Calabresi et al., 2000; Kerr and Wickens, 2001; Centonze et al., 2003b), the D1-like receptor antagonist SCH 23390 (10 μm; 5-7 min preincubation) fully blocked LTP in D2R-/- mice (n = 6 and p > 0.05 for the two groups) but was unable to prevent this form of synaptic plasticity in D2L-/- mice (n = 7; p < 0.001). The insensitivity of striatal LTP recorded in D2L-/- to D1 receptor inactivation was likely attributable to overexpression of D2S receptors, because mice lacking D2L receptors, but expressing normal levels of D2S receptors (D2R-/-;D2L-/-), exhibited LTP fully sensitive to SCH 23390 (n = 7; p > 0.05) (Fig. 4D). SCH 23390 (10 μm; 5-7 min preincubation) also blocked LTP in control rats (n = 5 and p > 0.05 at 10, 20, 30, and 40 min after HFS in magnesium-free experiments) and chronic haloperidol-treated rats [p > 0.05 at 10, 20, 30, and 40 min after HFS in 1.2 magnesium (n = 3) and p > 0.05 at 10, 20, 30, and 40 min after HFS in magnesium-free solution (n = 3)] (data not shown).
Role of NMDA receptor and protein kinase C in corticostriatal LTP expressed in haloperidol-treated rats and in D2 receptor mutant mice
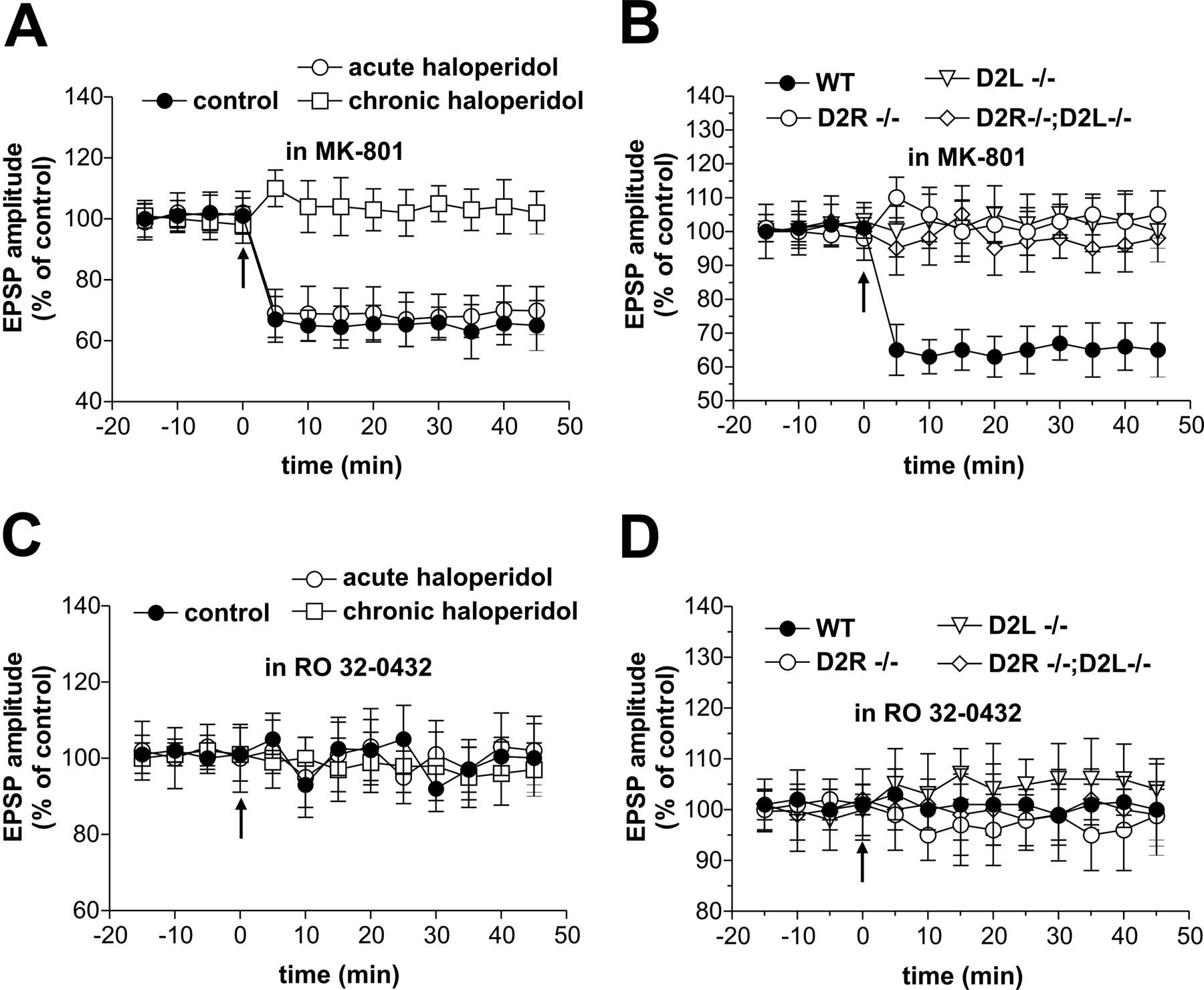
Activation of NMDA glutamate receptors is a critical requirement for the induction of LTP at corticostriatal synapses (Calabresi et al., 1992a, 1997b). To test the role of NMDA glutamate receptors in the formation of LTPs induced by HFS in rats chronically treated with haloperidol and in D2R-/-, D2L-/-, and D2R-/-;D2L-/- mice, we recorded spiny neurons in the presence of 30 μm MK-801 from the four groups of animals. As with their respective controls (n = 6 and p > 0.05 for both rats and mice), MK-801 fully prevented LTP in haloperidol-treated rats (n = 5) and D2R-/- (n = 12), D2L-/- (n = 13), and D2R-/-;D2L-/- (n = 9) slices (p > 0.05 for the four groups of animals) (Fig. 5A,B).

LTPs from haloperidol-treated rats and from D2 receptor mutant mice are sensitive to NMDA receptor and PKC blockade. A, B, The graphs show that preincubation with the glutamate NMDA receptor antagonist MK-801 (30 μm) blocked LTP induction in control and chronic haloperidol-treated rats (A) as well as in WT and D2R-/-, D2L-/-, and D2R-/-;D2L-/- striatal neurons (B). Note that in control rats and in WT mice, blockade of NMDA receptors not only prevented LTP but also favored the emergence of LTD. C, D, Intracellular application of the PKC inhibitor RO 32-043 prevented LTP in control and chronic haloperidol-treated rats (C) as well as in WT and mutant mice (D). In the four graphs, the LTP-inducing protocol in WT cells consisted of the application of HFS in the absence of magnesium ions. In contrast, in the three mutants, HFS was delivered either in the absence (at least 4 cells per group and experimental condition) or in the presence (at least 4 cells per group and experimental condition) of magnesium ions. Because the experiments gave similar results, the data were pooled together for each genotype.
Activation of protein kinase C (PKC) has been implicated in the molecular steps required to induce striatal LTP (Calabresi et al., 2000; Gubellini et al., 2003). Moreover, it has been reported that this kinase is stimulated by the activation of a subclass of D2 receptor (Choi et al., 1999). Thus, to address the role of PKC in the expression of LTP induced in haloperidol-treated rats and in D2R-/-, D2L-/-, and D2R-/-;D2L-/- mice, we used intracellular electrodes filled with RO 32-043 (100 μm), a PKC inhibitor. As with their respective controls (n = 7 and p > 0.05 for both rats and mice), this experimental condition fully prevented LTP observed in neurons recorded from the four groups of animals (n = 10 and p > 0.05 for all the experimental conditions) (Fig. 5C,D).
Depotentiation of striatal LTP in haloperidol-treated rats and D2 receptor mutant mice
Sustained inactivation of D2 receptors by classical antipsychotics may lead to dyskinetic involuntary movements (Turrone et al., 2003). Drug-induced dyskinesias have been recently reported to rely on the loss of corticostriatal depotentiation [i.e., on the impossibility to erase LTP by low-frequency stimulation (LFS) because of abnormal, unbalanced stimulation of D1 receptors] (Picconi et al., 2003). Thus, to see whether LTPs recorded after chronic haloperidol and in D2 receptor-deficient mice were subject to depotentiation, we applied, 10 min after the induction of LTP, a protocol of LFS of corticostriatal afferents (10 min, 2 Hz) able to fully reverse LTP in control rats (n = 5; p > 0.05) and WT mice (n = 7; p > 0.05). LFS was equally effective in depotentiating striatal LTP in haloperidol-treated rats, as well as in D2R-/-, D2L-/-, and D2R-/-;D2L-/- mice (n = 5 and p > 0.05 for the four groups) (Fig. 6). Notably, all these experiments were performed in the absence of magnesium ions from the external solution because synaptic depotentiation is a phenomenon critically dependent on NMDA receptor activation (Bashir and Collingridge, 1994; O'Dell and Kandel, 1994; Picconi et al., 2003).

Synaptic depotentiation is unaffected by chronic haloperidol treatment or genetic manipulations of D2 receptors. A, B, After the induction of LTP by HFS, a 10 min application of LFS (2 Hz) of corticostriatal terminals reversed this form of synaptic plasticity in control and chronic haloperidol-treated animals (A) as well as in WT and in the three genotypes of D2 receptor mutant mice. These experiments were performed in the absence of magnesium ions from the external solution because synaptic depotentiation is regulated by NMDA receptors.
Discussion
In the present study, we provided evidence that antipsychotic treatment significantly influences corticostriatal excitatory transmission. In fact, after chronic but not acute blockade of D2 receptors by haloperidol, HFS of corticostriatal terminals invariably enhanced excitatory transmission in the striatum by preventing LTD induction and by favoring the emergence of LTP. Although both forms of synaptic plasticity also represent physiological responses to repetitive activation of excitatory synapses in the striatum in vivo (Charpier and Deniau, 1997; Reynolds and Wickens, 2000; Reynolds et al., 2001), their impact on striatal physiological activity is opposite. Haloperidol therefore, by blocking LTD and favoring LTP, dramatically interferes with the processing of cortical inputs to the striatum and with the final output of this nucleus.
We also demonstrated that the two isoforms of DA D2 receptors are differentially involved in the effects of haloperidol at corticostriatal synapses. Accordingly, although selective D2L receptor inactivation mimics and occludes haloperidol effects, overexpression of D2S receptors in D2L-/- mice reveals that D2S receptor stimulation facilitates LTP formation by overcoming the requirement of D1 receptor activation for its emergence. This finding is in agreement with a previous report showing that D2S receptors, expressed along with D2L receptors on striatal neurons, interfere with D1 receptor-mediated functions, presumably via a receptor-receptor interaction or an interaction at the level of the intracellular signaling (Usiello et al., 2000). D2S receptor overexpression, however, does not affect the dependency of striatal LTP after NMDA receptor and PKC stimulation.
The two isoforms of the D2 receptor display differential affinities for inhibitory G-proteins (Guiramand et al., 1995), indicating that their transduction pathways and physiological roles are not equivalent. In fact, D2L receptors act mainly at postsynaptic sites, whereas D2S receptors preferentially play autoreceptor functions and regulate DA synthesis and release (Usiello et al., 2000; Centonze et al., 2002; Lindgren et al., 2003). The location of D2L receptors at postsynaptic sites has been shown by RNase protection analyses, which is presently the only experimental tool to evaluate the absence of the D2L isoform in the striatum in the absence of either D2L-specific antibodies or ligands able to discriminate between D2L and D2S receptors. As reported, RNase protection analyses showed the specific absence of D2L transcripts in the striatum of D2L-/- mice (Usiello et al., 2000). Evidence exists, however, that D2S receptors also mediate some postsynaptic actions in the striatum: D2S receptors, in fact, act in concert with D2L receptors in modulating striatal GABA transmission, which is mainly mediated by striatal interneurons and axon collaterals of GABAergic projection cells (Centonze et al., 2004).
The promotion of LTP by haloperidol is likely to contribute to the therapeutic effects of this compound rather than to its motor side effects. Accordingly, in our experimental model, the electrophysiological effects of chronic haloperidol were not associated with gross motor abnormalities, whereas the LTP observed in treated rats, as well as those seen in the three D2 receptor mutant mice, were normally reversed by LFS. Because hypokinetic, parkinsonian-like, and dyskinetic symptoms in rats are associated with the loss of corticostriatal LTP and of its depotentiation, respectively, it is conceivable that the changes in synaptic plasticity induced by our pharmacological treatment does not reflect such motor effects. Based on the results of the present study, it is therefore conceivable that pharmacological agents able to selectively block D2L receptors, and/or to selectively stimulate D2S receptors, might prove useful to treat psychotic symptoms.
Complex regulatory role of DA receptors in corticostriatal synaptic plasticity
Long-term rearrangements of synaptic strength in the striatum have been proposed to underlie specific forms of associative learning and reward (Reynolds et al., 2001; Schultz, 2002), to play a key role in the maturation of neural circuitry during development (Choi and Lovinger, 1997), and to be involved in the drug-induced reorganization of synaptic transmission that occurs during addiction (Berke and Hyman, 2000; Hyman and Malenka, 2001; Nestler, 2001) and neuroleptic medications (Konradi and Heckers, 2001). Corticostriatal LTP may profoundly affect the pattern of firing of striatal neurons because, in the intact brain, it mainly depends on the release of glutamate from cortical terminals (Stern et al., 1998).
DA D1 and D2 receptors exert opposite roles in the formation of striatal LTP. In fact, ipsilateral nigral lesions and pharmacological or genetic inactivation of D1 receptors prevent LTP induction (Centonze et al., 1999, 2003b; Calabresi et al., 2000; Kerr and Wickens, 2001), whereas pharmacological or genetic inactivation of D2L receptors enhances this process (Calabresi et al., 1997b; present study). The differential effects of D1 and D2L (but not D2S) receptors in LTP induction might rely on their opposite actions on the phosphorylation of DARPP-32, a phosphoprotein particularly enriched in dopaminoceptive neurons of the striatum that acts as a potent inhibitor of protein phosphatase-1 (Greengard et al., 1999). In fact, corticostriatal LTP is absent in mice lacking DARPP-32 (Calabresi et al., 2000), and D1 receptor stimulation increases DARPP-32 activity, whereas D2L but not D2S receptors decrease it (Lindgren et al., 2003). Stimulation of D2 receptors has also been shown to reduce striatal neuron excitability through the activation of a phospholipase C-IP3-calcineurin signaling cascade (Hernandez-Lopez et al., 2000), raising the possibility that our electrophysiological findings reflect the dysregulation of this alternative intracellular pathway.
In addition, a presynaptic mechanism might also be implicated in the emergence of LTP over LTD in response to chronic pharmacological or genetic inactivation of D2 receptors. In fact, presynaptic D2 receptors inhibit the release of glutamate from corticostriatal terminals (Maura et al., 1988; Bamford et al., 2004), as also indicated by the increased frequency of spontaneous glutamate-mediated synaptic events in D2R-/- mice (Cepeda et al., 2001; Tang et al., 2001). Therefore, it is possible that during the HFS of corticostriatal synapses, the loss of D2-mediated inhibitory control of glutamate release might increase the membrane depolarization of striatal neurons, thereby favoring massive calcium influx and LTP. Nevertheless, we have shown that comparable membrane depolarizations are induced in control and haloperidol-treated rats as well as in WT and D2 receptor-lacking mice (Calabresi et al., 1997b; present study), making this possibility unlikely. Indeed, future studies are necessary to unravel the cellular and molecular mechanisms responsible for the effects of D2 receptor blockade on corticostriatal synaptic plasticity.
D2L receptor inactivation facilitates excitatory transmission in the striatum
Our electrophysiological data are strongly supportive of the concept that chronic inactivation of D2L receptors is responsible for the rearrangement of excitatory synapses seen in the striatum after treatment with antipsychotic agents. In line with this idea, the behavioral effects of haloperidol are absent in D2L-/- mice, indicating that this compound mediates its effects by targeting this receptor isoform (Usiello et al., 2000).
The finding that D2L receptor inactivation favors striatal LTP well correlates with the evidence that, in the striatum, haloperidol increases the size of axon terminals (Benes et al., 1985; Uranova et al., 1991; Kerns et al., 1992), the number of glutamatergic synapses (Meshul and Casey, 1989; Uranova et al., 1991; Kerns et al., 1992; See et al., 1992), and the absolute number of vescicles per synapse (Benes et al., 1985). All these morphological changes indicate hyperactivity of glutamatergic transmission and represent structural correlates of LTP (Eastwood et al., 1997; Toni et al., 1999; Konradi and Heckers, 2001; Steward and Worley, 2002). In this context, particularly interesting is the evidence that haloperidol treatment also alters postsynaptic density (Uranova et al., 1991), an electron dense area that contains NMDA glutamate receptors and scaffolding proteins that anchor receptors to the membrane and facilitate the receptor and postreceptor events required for LTP induction (Elgersma et al., 2002).
The promotion of NMDA receptor-dependent potentiation of excitatory synapses by D2L receptor blockade is likely to represent a mechanism by which antipsychotic agents exert their therapeutic effects against schizophrenic symptoms. In fact, several lines of evidence converge in indicating that hypoactivity of NMDA-mediated glutamatergic transmission is involved in the pathophysiology of schizophrenia. For example, a relative decrease of the NR1 subunit of NMDA receptors has been reported in the brains of schizophrenic patients, suggesting less than normal receptor responsiveness to endogenous glutamate (Gao et al., 2000). In addition, noncompetitive NMDA receptor antagonists such as MK-801, phencyclidine, and ketamine produce a syndrome in normal individuals that closely resembles schizophrenia and exacerbates symptoms in patients with chronic schizophrenia (Goff and Coyle, 2001). Finally, mice lacking the NR1 or NR2 subunits of NMDA receptor exhibit schizophrenia-like behavioral alterations responsive to neuroleptic treatment (Mohn et al., 1999; Miyamoto et al., 2001), which has been reported, in turn, to increase the amount of mRNA encoding both subunits of NMDA receptors in the striatum (Riva et al., 1997).
Conclusion
Although classically regarded as a brain area involved in motor planning and execution, increasing evidence indicates that the nucleus striatum is part of a complex cortico-striatal-thalamo-cortical loop involved is several association, motivational, and cognitive functions. Alterations of this circuit are therefore likely to mediate some of the symptoms of schizophrenia. The identification of the synaptic mechanisms responsible for the rewiring of neural circuits that takes place after inactivation of D2 receptors by haloperidol is essential to understand how antipsychotic agents exert their beneficial effects and side effects.
Footnotes
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Hopitaux Universitaires de Strasbourg, and Mission Interministerielle de Lutte contre les Drogues et al Toxicomanie to E.B.; by a grant from Ministero della Salute, Ricerca Finalizzata to D.C.; and by CNR Biotecnologie and Telethon grants to P.C. We thank M. Tolu for technical assistance.
Correspondence should be addressed to either of the following: Dr. Paolo Calabresi, Clinica Neurologica, Dipartimento di Neuroscienze, Università Tor Vergata, Via Montpellier 1,00133 Rome, Italy, E-mail: calabre{at}uniroma2.it; or Dr. Emiliana Borrelli, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique-Institut National de la Santé et de la Recherche Médicale-Université Louis Pasteur 1, Rue L. Fries, BP10142, 67404 Illkirch Cedex, Communauté Urbaine de Strasbourg, France, E-mail: eb{at}titus.u-strasbg.fr.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248214-09$15.00/0
↵* P.C. and E.B. should be considered equal senior authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}