

Abstract
Nicotine addiction is initiated by its binding to high-affinity nicotinic receptors in brain composed primarily of α4 and β2 subunits. For nicotinic receptors expressed in vivo or heterologously, nicotine exposure over hours to days increases or “upregulates” high-affinity nicotine binding to receptors through a posttranslational mechanism thought to increase receptor numbers. Using heterologous expression, we find nicotine exposure causes a fourfold to sixfold higher binding to α4β2 receptors that does not correspond with any significant change in the number of surface receptors or a change in the assembly, trafficking, or cell-surface turnover of the receptors. However, upregulation does alter the functional state of the receptor, slowing desensitization and enhancing sensitivity to acetylcholine. Based on these findings, we propose an alternative mechanism to explain nicotine-induced upregulation in which nicotine exposure slowly stabilizes α4β2 receptors in a high-affinity state that is more easily activated, thereby providing a memory for nicotine exposure.
Introduction
Although it is established that neuronal nicotinic acetylcholine receptors are the primary target for nicotine, the mechanisms that cause nicotine addiction are poorly understood. Neuronal nicotinic receptors are members of the family of neurotransmitter-gated ion channels that includes GABAA receptors, glycine receptors, and 5-HT3 receptors (Karlin, 2002). Neuronal nicotinic receptor subtypes arise from different subunits. Eight subunits (α2-α4, α6-α10) contribute to the acetylcholine binding site, and four subunits (α5, β2-β4) have a structural function but contribute to the binding site. Knock-out mice without β2 subunits lack high-affinity nicotine binding sites, fail to self-administer nicotine, and no longer show nicotine-stimulated dopamine release from mesolimbic neurons (Picciotto et al., 1998). In rodents, ∼90% of the high-affinity nicotine binding sites in the brain contain α4 and β2 subunits (Whiting and Lindstrom, 1988; Flores et al., 1992; Conroy and Berg, 1998; Zoli et al., 1998). Hence, α4β2 receptors are the neuronal nicotinic receptor subtype most likely to participate in nicotine addiction, although other receptors that contain α6 or α3 subunits may also be involved (Champtiaux et al., 2003; Parker et al., 2004).
Prolonged exposure to nicotine increases nicotine binding in rat and human brain, a process termed “upregulation,” which has been linked with aspects of nicotine addiction (Schwartz and Kellar, 1985; Perry et al., 1999). Upregulation occurs in cells heterologously expressing α4β2 receptors (Wonnacott, 1990; Dani and Heinemann, 1996; Buisson and Bertrand, 2002) and has been further characterized in a variety of expression systems (Peng et al., 1994; Bencherif et al., 1995; Hsu et al., 1996; Eilers et al., 1997; Warpman et al., 1998; Whiteaker et al., 1998; Fenster et al., 1999a; Harkness and Millar, 2002; Sallette et al., 2004). Based on these and other studies, it was deduced that α4β2 receptor upregulation results from an increase in receptor number caused by posttranslational mechanisms (Marks et al., 1992). Specifically, α4β2 receptor upregulation is attributed to decreased surface turnover (Peng et al., 1994), increased receptor assembly (Bencherif et al., 1995; Whiteaker et al., 1998; Harkness and Millar, 2002; Nashmi et al., 2003; Sallette et al., 2004), or increased receptor trafficking to the surface (Harkness and Millar, 2002). Furthermore, the functional response of α4β2 receptors expressed in Xenopus oocytes decreased after long exposure to nicotine (Peng et al., 1994; Hsu et al., 1996; Fenster et al., 1999b). This effect was initially interpreted as a loss of receptor function compensated in the brain by an increase in the number of receptors (Wonnacott, 1990; Dani and Heinemann, 1996). However, recent studies also reported that functional responses of α4β2 receptors expressed in mammalian cells (Buisson and Bertrand, 2002) and midbrain neurons (Nashmi et al., 2003) is increased after long-term exposure to nicotine. In this study, using heterologous expression, we examine mechanisms underlying α4β2 receptor upregulation by nicotine and conclude that upregulation is not caused by changes in assembly, trafficking, cell-surface turnover, or by any process that increases the number of surface receptors. Instead, we propose that nicotine exposure slowly stabilizes α4β2 receptors in a high-affinity state that increases both radio-labeled agonist binding to the receptor and the response of the receptor to agonist.
Materials and Methods
cDNA constructs, cell culture, and gene transfer. Rat α4 and β2 subunit cDNAs (gift from Dr. J. Boulter, University of California, Los Angeles, Los Angeles, CA) were subcloned into pCB7, which contains a cytomegalovirus promoter and the hygromycin resistance gene used to select for cells stably expressing α4β2 hemagglutinin (HA) receptors. The HA epitope (Wilson et al., 1984), YPYDVPDYA, and a stop codon were inserted after the last codon of the 3′ translated region of the subunit DNA using the extension overlap method (Ho et al., 1989). The human embryonic kidney (HEK) cell line tsA201 was maintained in DMEM supplemented with 10,000 U/ml penicillin and streptomycin (Invitrogen, Gaithersburg, MD) and 10% calf serum (Hyclone, Logan, UT) at 37°C in the presence of 5% CO2. α4, β2, and β2HA subunits were expressed by transient transfection into tsA201 cells using a calcium phosphate method (Eertmoed et al., 1998). The α4β2HA stable cell line was established from tsA201 cells using calcium phosphate transfection and hygromycin B selection (Calbiochem, La Jolla, CA) at 0.42 mg/ml.
Metabolic labeling, immunoprecipitation, and affinity purification. Newly synthesized subunits were metabolically labeled with 35S-methionine and cysteine (1175 Ci/mmol; EXPRE35S35S Protein Labeling Mix; NEN, Boston, MA). Cultures were starved at 37°C in methionine-free DMEM for 15 min, incubated in 5% CO2, and labeled in 1 ml of methionine-free DMEM, supplemented with 111 μCi of the 35S-mix for 1 h. After labeling, cultures were rinsed with ice-cold PBS, scraped, pelleted at 5000 × g for 3 min, and resuspended in lysis buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris, pH 7.4, 0.02% NaN3, plus 1% Triton X-100) containing protease inhibitors (2 mm phenylmethylsulfonyl fluoride, 2 mm N-ethylmaleimide, and chymostatin, pepstatin, leupeptin, and tosyllysine chloromethyl ketone at 10 μg/ml). 35S-labeled lysates were incubated with anti-HA monoclonal antibody (mAb) 12CA5 or α4 subunit-specific mAb 299 (Research Biochemicals, Natick, MA) overnight at 4°C. Antibody-antigen complexes were precipitated with protein G-Sepharose (Amersham Biosciences, Arlington Heights, IL), and purified subunits were analyzed on a 4-8% gradient acrylamide-SDS gel.
Nicotine-induced upregulation and 125I-epibatidine binding. Upregulation was achieved, unless otherwise noted, by incubating cells in 10 μm nicotine for 17-18 h at 37°C in medium. Transiently transfected cells were treated 24 h after transfection, and stably expressing cells were treated once confluent. In general, cells were washed four to five times and collected by gentle agitation with PBS, followed by incubation in 4-5 nm 125I-epibatidine. To prepare membranes, cultures were rinsed two to three times with PBS and harvested by gentle agitation. Cells were resuspended in lysis buffer containing protease inhibitors and sonicated on ice. Membranes were pelleted at 10,000 × g for 30 min at 4°C, washed twice with PBS, and resuspended in lysis buffer containing protease inhibitors via sonication. For competition with ACh (see Fig. 5 A, B), intact cells and membrane preparations were preincubated in the indicated concentrations of ACh for 5 min, followed by the addition of 1 nm 125I-epibatidine for 15 min. For measuring ACh competition as a function of time, longer preincubation times were used (5 min, 30 min, 1 h, and 3 h). All binding was terminated by vacuum filtration through Whatman (Clifton, NJ) GF/B filters presoaked in 0.5% polyethyleneimine using a Brandel (Gaithersburg, MD) 24 channel cell harvester. Bound 125I-epibatidine (2200 Ci/mmol) was determined by γ counting (PerkinElmer Wallac, Gaithersburg, MD) with nonspecific binding estimated by 125I-epibatidine binding to untransfected cells. There was no significant 125I-epibatidine binding to the parent HEK cell line in the absence and presence of nicotine treatment, to cells transiently transfected with the α4 or β2 subunit cDNAs alone, or to cells transfected with the β2 subunit cDNA alone in the absence and presence of nicotine treatment. Cultures were treated with Brefeldin A (10 μg/ml) 30 min before nicotine treatment. Sample sizes are expressed relative to one well from a 24-well plate containing 0.12 mg of protein.
Cell surface Ab binding. Cells were incubated with anti-HA mAb 12CA5 or anti-α4 mAb 299 for 2 h at room temperature, washed four times with PBS, and fixed for 15 min with 2% paraformaldehyde as described previously (Harkness and Millar, 2002). After a 10 min incubation in Tris-buffered saline (TBS) and two washes with PBS, cells were incubated in 0.5 pmol of 125I-protein A (9.11 μCi/μg; NEN) in PBS for 2 h at room temperature. For assays performed on live cells, cells were incubated with the antibodies in PBS-Ca2+ overnight at 4°C, washed three times with ice-cold PBS to remove unbound antibody, and incubated in 125I-protein A in PBS for 3 h at 4°C. Unbound radioactivity was removed by four washes in PBS, and binding was assayed by γ counting.
Methanethiosulfonate ethylammonium-biotinylation assays. To biotinylate surface receptors, intact, confluent cultures in poly-d-lysine-coated plates (VWR, West Chester, PA) were treated with 50 μm methanethiosulfonate ethylammonium (MTSEA)-biotin (Toronto Research Chemicals, Toronto, Canada) for 20-30 min at room temperature. To biotinylate surface plus intracellular receptors, cells were incubated in 10 mm phosphate buffer containing 0.5% saponin, 0.1% bovine serum albumin (BSA), and 10 mm EDTA for 5 min, washed twice with PBS, and treated with MTSEA-biotin as before. After MTSEA-biotin treatment, cells were washed with PBS and solubilized at 4°C in lysis buffer containing 1% Triton X-100 and protease inhibitors. Lysates with 75 μl of streptavidin immobilized on 4% beaded agarose were rotated overnight at 4°C. Afterward, biotinylated subunits were subjected to 125I-epibatidine binding.
Endoglycosidase H analysis. Biotinylated receptors were treated with endoglycosidase H (Endo H) (Calbiochem) per the protocol of the manufacturer for 36-48 h at 37°C without the 100°C heating step. Subunits were separated on SDS-PAGE, transferred to Immobilon-P membranes (Millipore, Billerica, MA), and incubated in wash buffer (10 mm Tris, pH 7.4, 0.05% Tween 20, and 150 mm NaCl) containing 3% nonfat dry milk for 1 h at room temperature. Afterward, membranes were immersed in wash buffer containing α4 subunit-specific polyclonal Ab 6964 (gift from Dr. S. Rogers, University of Utah, Salt Lake City, UT) and 1% nonfat dry milk for 1 h, washed three times, and incubated in 125I-protein A for 3 h at room temperature, followed by four washes. Membranes were dried, and proteins were visualized by exposure to X-Omat XK-1 film (Eastman Kodak, Rochester, NY).
Immunofluorescence staining. Biotinylated, intact cells were stained with a mouse monoclonal HA.11 antibody (Covance Research Products, Berkeley, CA), followed by a goat anti-mouse cyanine 5 (Cy5) secondary antibody (Jackson ImmunoResearch, West Grove, PA), and biotinylation was detected with streptavidin conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, OR). Permeabilized cells were labeled with the latter. Cells were grown on poly-d-lysine-coated coverslips (VWR), fixed with 2% paraformaldehyde (Sigma, St. Louis, MO), and preblocked with TBS supplemented with 2 mg/ml bovine serum albumin (Sigma) (TBS/BSA). Antibody and streptavidin were diluted in TBS/BSA and allowed to react with cells for 1 h at room temperature. Cells were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and analyzed using an IX70 Olympus Optical (Tokyo, Japan) Fluoview 200 laser-scanning confocal microscope using a 60× planApo (numerical aperture 1.4 oil) objective. The following negative controls were performed: (1) intact parent tsA201 cells were stained with fluorescent secondary antibody alone or secondary antibody plus the primary anti-HA antibody; (2) intact and permeabilized tsA201 cells were stained with the Alexa Fluor streptavidin; (3) intact cells stably transfected with α4β2HA receptors were stained with fluorescent secondary antibody alone or the Alexa Fluor streptavidin; and (4) permeabilized cells were stained with the Alexa Fluor streptavidin. In all cases, the fluorescent signals from the negative controls were extremely weak relative to the images in Figures 1 B, 2 A, and supplemental figures.
Electrophysiology. Cells from 35 mm cultures were recorded 2-3 d later as described previously (Buisson et al., 1998). Cells were impaled and maintained in voltage clamp in the whole-cell configuration technique using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). For nicotine treatment, cells were incubated for the indicated times and thoroughly washed three times before placing the dish in the recording setup. Cells were then washed with control solution throughout the experiment. Because impaling required at least 1 min, minimal washing time after nicotine treatment was at least 2 min. No difference in current amplitude or time course could be observed between the first and subsequent cells impaled. Currents were filtered at 1 kHz and digitized at 5 kHz by a PCI card (National Instruments, Austin, TX). Values were stored on the hard disk of a Macintosh (Apple Computers, Cupertino, CA) and analyzed using MacDatac (personal program). All recordings were done at room temperature in the following extracellular medium (in mm): 130 NaCl, 5 NaCl, 2 CaCl2, 2 MgCl2, and 10 HEPES, pH 7.4 with NaOH. Patch electrodes made of borosilicate electrodes (3-8 MΩ) were filled with the following (in mm): 130 K-gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, and 5 EGTA, pH 7.4 with KOH. The use of a liquid filament for the drug application is one of the fastest currently available systems, with an exchange time of <20 ms. Thus, for the fastest response decay time observed in this work (180 ms), a putative error at the initial onset of the response should not exceed 11%, and therefore no attempt were made for correction. ACh concentration-response curves were best fitted by the sum of two empirical Hill equations (see Fig. 4) that were adjusted using a least-squares curve-fitting procedure (Simplex). Net charge analysis was performed as follows: (1) data were first adjusted to remove the baseline offset that corresponds to the holding current in absence of ligand; and (2) net charge was calculated by integrating the current during the data epoch using a cumulative trapezoidal numerical integration algorithm (cumtrapz) built in Matlab (MathWorks, Gümligen, Switzerland). Significance of electrophysiological data were evaluated using a Student's t test for two samples assuming unequal variance and two-tailed distribution (Excel; Microsoft, Seattle, WA). p values of significant results are indicated in the figure legends.
Results
To study the nicotine-induced upregulation of α4β2 receptors, we expressed rat α4β2 receptors in the HEK cell line. An HA epitope tag was fused to the C terminus of the rat β2 subunit (β2HA) to allow detection of surface membrane receptor characterization. As illustrated in Figure 1A, the HA epitope was expected to have an extracellular location given the predicted membrane topology of the subunit. Intact cells transiently transfected with α4 and β2HA subunits were immunostained with anti-HA mAb (Fig. 1B) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), confirming that the HA epitope on the β2HA subunit C terminus is located on the extracellular domain of the subunit. α4 and β2HA subunits were metabolically labeled and immunoprecipitated by either an α4-specific mAb or an anti-HA mAb, respectively (Fig. 1C). The β2HA subunits migrated at ∼51 kDa and the α4 subunits at ∼75 kDa, similar to previous estimates of their molecular weights (Whiting and Lindstrom, 1987). Approximately 30% of newly synthesized α4 and β2HA subunits coprecipitated when its complementary subunit was immunoprecipitated with subunit-specific antibodies. Additional experiments were performed using HEK cells stably expressing α4 and β2HA subunits, and we found that 30% of α4 subunits coprecipitated with β2HA subunits and 56% of β2HA subunits coprecipitated with α4 subunits, indicating that a significant percentage of the subunits assembled into α4β2 receptors. To assess whether the HA tag on the β2 subunit perturbs receptor function, we conducted electrophysiological recordings on HEK cells stably expressing α4β2HA receptors. ACh evoked large current responses (see Fig. 4). Despite the fact that a better fit of the data were obtained with a double sigmoidal rather than a single sigmoidal, the sensitivity of the α4β2HA-expressing cells is in close correlation to that reported previously for the rat α4β2 receptors (Sabey et al., 1999). Similarly, the decay time of the ACh-evoked current closely resembles the fast decay time reported by these authors.

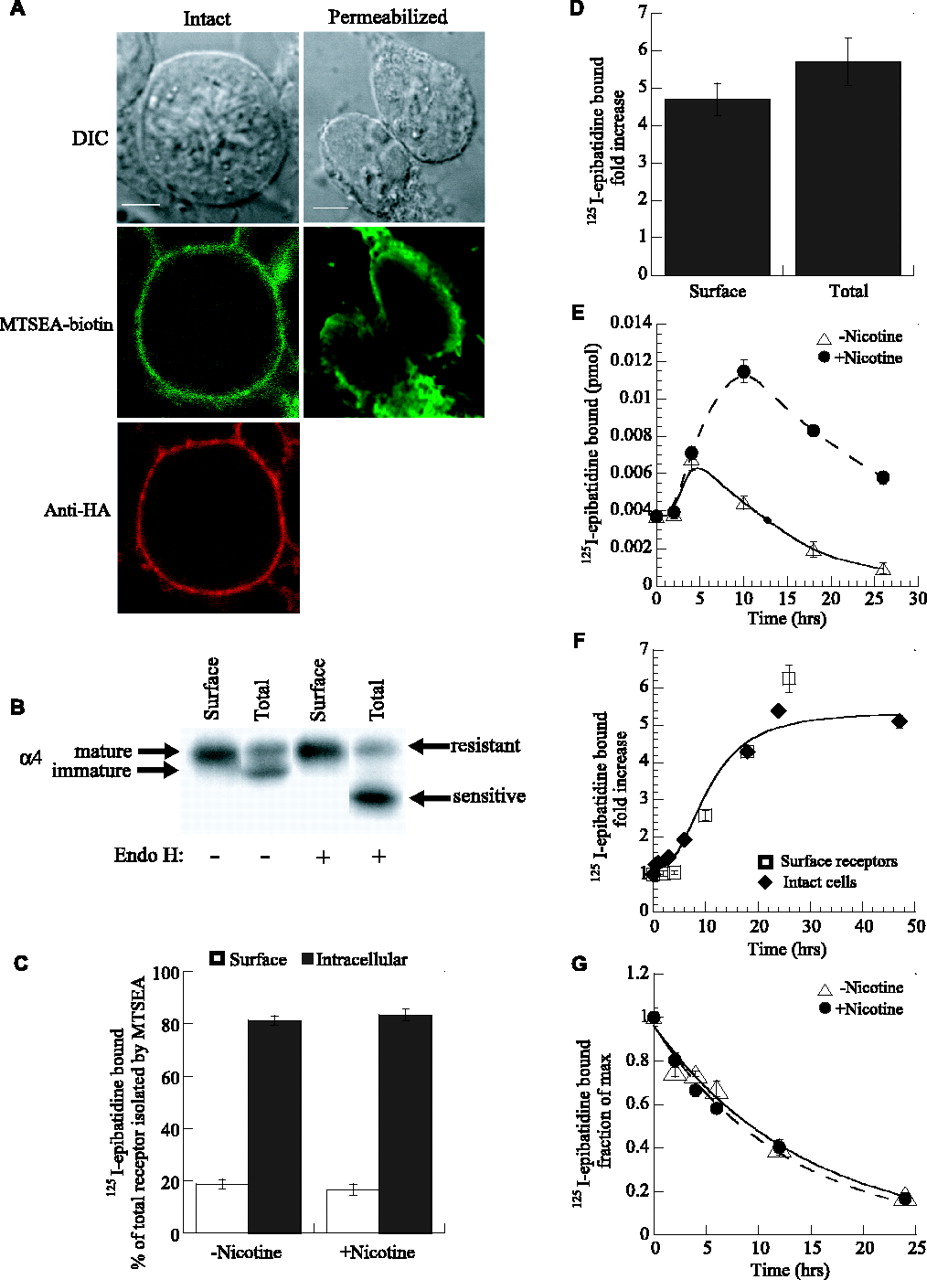
Nicotine-induced upregulation of nicotinic receptors composed of α4 and epitope-tagged β2 subunits. A, The α4 and HA epitope-tagged β2 subunits are diagrammed. Based on consensus membrane topology of the subunits shown at the bottom, the HA epitope tag on the C terminus is located within the extracellular domain of the receptors. B, Cell-surface immunostaining of α4β2HA-expressing cells. Intact cells were stained with anti-HA mAb HA.11 (bottom). A differential interference contrast (DIC) image is shown above. Scale bar, 20 μm. C, α4 and β2HA subunits were metabolically labeled and immunoprecipitated with anti-HA mAb 12CA5 (middle lane) or anti-α4 mAb 299 (right lane). Both subunits assemble into complexes, as shown by the coprecipitation of the two subunits. No subunits were precipitated in sham-transfected cells (left). D, Nicotine treatment of cells (+Nicotine) resulted in a 3.7-fold increase in 125I-epibatidine binding compared with untreated controls (-Nicotine). Sample size was one well from a 24-well plate, in which 125I-epibatidine binding was equivalent to 2.3 ± 0.20 pmol/mg (no nicotine) and 8.4 ± 0.66 pmol/mg (nicotine treated); n = 15. E, Concentration dependence of nicotine-induced upregulation for cells stably expressing α4β2HA receptors or transiently expressing α4β2 receptors lacking the HA epitope. Each point for ▵ represents the mean ± SD of three samples, each being one-third of a well from a 24-well plate. The lines through the data represent a least-squares fit of the Hill equation to the data for the α4β2HA receptors: fold increase = 1 + Max/(1 + EC50/[nicotine])n, where 1 + Max, the maximum fold increase, was 3.3; n, the Hill coefficient, was 1; and the EC50 value was 250 ± 30 nm. F, α4β2HA stably expressing cells were incubated in 10 μm nicotine for the indicated times, and 125I-epibatidine binding was determined. Each point is the mean ± SD of three samples, each being one-fourth of a well from a 24-well plate.
Nicotine-induced upregulation of α4β2HA receptors
Chronic exposure of α4β2HA-expressing cells to nicotine resulted, as expected, in upregulation of agonist binding sites. After a 17 h incubation of the cells at 37°C with 10 μm nicotine, we observed threefold to sixfold increases in 125I-epibatidine binding (Fig. 1D). Epibatidine is an α4β2 receptor agonist that binds to the same site as nicotine (Badio and Daly, 1994; Gerzanich et al., 1995) but has the advantages of higher affinity and offers the possibility to be iodinated (Davila-Garcia et al., 1997). The concentration dependence of the nicotine-induced increase in 125I-epibatidine binding was measured for HEK cells stably expressing α4β2HA receptors or transiently expressing α4β2 receptors lacking the HA epitope tag (Fig. 1E). The upregulation saturated at 1-10 μm, was half-maximal at 250 ± 30 nm for α4β2HA receptors, and was similar for untagged receptors. Notably, the serum concentration range achieved after smoking is 100-200 nm nicotine (Henningfield et al., 1993; Benowitz, 1996), a range expected to cause an approximate twofold increase in binding according to our data. At a saturating nicotine concentration (1-10 μm), upregulation was not observed until 2-3 h after exposure and reached a steady state after 20 h (Fig. 1F). The concentration dependence and time course of the nicotine-induced upregulation were essentially identical to those reported previously for α4β2 receptors expressed in mammalian cells (Eilers et al., 1997; Gopalakrishnan et al., 1997; Whiteaker et al., 1998).
Biotinylation of surface α4β2 receptors
Although previous studies have concluded that nicotine-induced upregulation results from increases in receptor levels, quantitative analysis of the different cellular pools of α4β2 receptors has proven difficult. 125I-epibatidine readily diffuses into cells and binds without distinction to cell-surface and intracellular receptors. To distinguish the effects of nicotine treatment on surface and intracellular receptors, we used biotinylation of surface receptors as an approach for separating surface and intracellular 125I-epibatidine binding sites. Care was taken to choose an appropriate reagent and ensure that minimal biotinylation of intracellular receptors occurred when applied to intact cells. After experimental trials, MTSEA-biotin, which cross-links to cysteinyl sulfhydryls (Akabas et al., 1992; Stauffer and Karlin, 1994), was chosen. Its biotinylation of the cells saturated with increasing concentrations, whereas the concentration dependence of other biotinylation reagents did not behave as well (data not shown). Although MTSEA is membrane permeable (Holmgren et al., 1996), MTSEA-biotin biotinylates only surface receptors significantly and appears not to permeate the membrane in our experimental conditions. Evidence that MTSEA-biotin only labeled surface receptors was as follows. (1) Fluorescent-labeled streptavidin decorates only the cell surface after biotinylation of intact cells with MTSEA-biotin, similar to the staining of the intact cells with anti-HA mAb (Fig. 2A, left) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This is different from the staining pattern detected in biotinylated permeabilized cells (Fig. 2A, right) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), in which intracellular biotinylation occurred. (2) Virtually all α4 subunits biotinylated in intact cells were insensitive to Endo H cleavage, whereas α4 subunits biotinylated in permeabilized cells had a large Endo H-sensitive component (Fig. 2B), consistent with biotinylation of predominantly fully mature subunits. (3) 125I-epibatidine binding sites purified by streptavidin-agarose after biotinylation migrated as a single 10 S peak when analyzed by sucrose gradient centrifugation (data not shown) as opposed to multiple peaks observed when intracellular plus 10 S surface receptors are analyzed on sucrose gradients (Nelson et al., 2003).
To estimate the percentage of α4β2 receptors on the cell surface, intact cells were exposed to MTSEA-biotin. Alternatively, for determination of the total receptor population, cells were treated with saponin to allow MTSEA-biotin permeation and biotinylation of intracellular as well as surface receptors. Using this technique, we observed that 20% of 125I-epibatidine binding sites are located on the surface and 80% were intracellular (Fig. 2C). Importantly, we noticed that nicotine treatment did not significantly alter these percentages, and the nicotine-induced upregulation caused no significant changes of the ratio between surface and intracellular receptors (Fig. 2D). These results are contrary to studies suggesting that nicotine upregulation results from increased receptor assembly, trafficking to the plasma membrane, and/or a reduction in the turnover of surface receptors. In studies in which an increase in receptor assembly appear to be the primary cause of upregulation, a larger increase in the number of intracellular receptors relative to surface was expected (Whiteaker et al., 1998). When increases in trafficking to the plasma membrane predominate, larger increases in the number of surface receptors than intracellular receptors are expected (Harkness and Millar, 2002). Similarly, a decrease in the turnover of surface receptors should result in larger increases in the number of surface receptors relative to intracellular receptors (Peng et al., 1994). A combination of these different mechanisms could, of course, compensate and result in no change in the percentage of surface and intracellular receptors with upregulation, as we observed.
Effects of long-term nicotine exposure on cell-surface α4β2 receptors
To further examine the mechanisms of nicotine-induced upregulation, we tested whether nicotine exposure could alter 125I-epibatidine binding to α4β2 receptors already on the cell surface. To perform these experiments, surface receptors were first biotinylated and cells were then exposed to nicotine or left untreated. At the indicated times, surface receptors previously biotinylated were purified, and 125I-epibatidine binding to receptors was measured (Fig. 2E). Over the first 5 h, 125I-epibatidine binding to both the nicotine-treated and untreated receptors increased equally. This increase is observed independent of the presence of nicotine when the culture medium is replaced by fresh serum-containing medium. After this initial increase, 125I-epibatidine binding to the nicotine-treated surface receptors continued to increase, whereas 125I-epibatidine binding to the untreated surface receptors decayed at a rate comparable with that measured for the turnover of the surface receptors (Fig. 2G). In Figure 2F, we plotted the ratio of 125I-epibatidine binding to biotinylated surface receptors: nicotine treated to untreated. The values and time course of the ratio for the biotinylated surface receptors are similar to that measured for the intact cells (Figs. 1F, 2F). A possible explanation for this observation is that nicotine treatment induces clustering of receptors so that new receptors are associated with the already biotinylated surface receptors. However, previous studies have shown by sucrose gradient analysis that the migration of nicotine-treated α4β2 receptors is indistinguishable from untreated receptors (Nelson et al., 2003). We also performed sucrose gradient analysis and confirmed that no apparent change in size can be detected in sucrose gradient sedimentation after nicotine upregulation (data not shown).
Another possible explanation for the results in Figure 2, E and F, is that the increase in 125I-epibatidine binding was caused by a decrease in the cell-surface turnover rate of the receptors, as suggested previously (Peng et al., 1994). Cultures expressing α4β2 receptors were exposed to nicotine for 12 h, after which intact cells, both exposed and unexposed, were treated with MTSEA-biotin to biotinylate surface receptors present at that time. The turnover of surface receptors was then monitored at the indicated times after label in Figure 2G by purification and binding with 125I-epibatidine. The rate of turnover was well fit by single-exponential decay (line through the points), and the rates obtained were similar to those measured for extrasynaptic muscle nicotinic receptors using 125I-α-bungarotoxin binding (Pumplin and Fambrough, 1982). No change in the rate of turnover for surface receptors was observed with exposure to nicotine (Fig. 2G). These data indicate that the increase in 125I-epibatidine binding observed during nicotine-induced upregulation cannot be attributed to a decrease in the rate of surface turnover.

Biotinylation of α4β2HA receptors. A, Staining of α4β2HA-expressing cells after biotinylation with MTSEA-biotin. Intact cells (left row) were biotinylated with MTSEA-biotin and stained with Alexa Fluor streptavidin after permeabilization (middle). To image the plasma membrane in parallel, surface α4β2HA receptors were bound with anti-HA mAb HA.11 and stained with goat anti-mouse Cy5 secondary Ab after permeabilization (bottom). Alternatively, cells were first permeabilized (right row) and biotinylated with MTSEA-biotin and then stained with Alexa Fluor streptavidin (middle). DIC images are shown above. Scale bars, 5 μm. B, Endo H analysis of biotinylated α4 subunits. Intact or permeabilized cells were biotinylated with MTSEA-biotin, and α4 subunits were purified using streptavidin-agarose and analyzed on immunoblots to determine the effects of Endo H. In permeabilized cells (Total), α4 subunits migrated as a doublet, consistent with a mixture of fully processed (mature) and partially processed (immature) subunits, whereas only mature subunits were biotinylated in intact cells. Only immature subunits were sensitive to Endo H cleavage, and all subunits biotinylated in intact cells were insensitive. C, Subcellular distribution of epibatidine-bound α4β2 receptors. 125I-epibatidine binding to surface and total receptor pools were obtained by biotinylating intact or permeabilized cells, purifying receptors with streptavidin-agarose, and measuring 125I-epibatidine binding. Intracellular 125I-epibatidine binding was determined as the difference between binding to the surface and total pools. Data are means ± SEM of 13 independent experiments performed in triplicate. Sample size was one well from a 24-well plate. D, Nicotine-induced upregulation of 125I-epibatidine binding was determined for cell-surface receptors and the total population of receptors by biotinylating intact or permeabilized cells as in C. Cells were treated with 10 μm nicotine for 17 h. Data are means ± SEM of 13 independent experiments performed in triplicate. E, 125I-epibatidine binding to surface receptors is upregulated by nicotine treatment. Intact α4β2HA-expressing cells were biotinylated, after which cells were incubated with or without 10 μm nicotine for the indicated times. Biotinylated receptors were precipitated, and 125I-epibatidine binding was determined. Each point is the mean ± SD of three samples, each being equivalent to three wells from a 24-well plate. Similar results were obtained in three separate experiments. F, Biotinylated surface receptors are upregulated to the same degree and with the same time course as the total cell receptor population. Data in E have been replotted with the data from Figure 1 E to show the extent of nicotine-induced upregulation of preexisting surface receptors and to compare the time courses of upregulation. G, Cell-surface turnover of α4β2HA receptors. Intact cells expressing α4β2HA receptors were maintained in the presence or absence of 10 μm nicotine for 12 h and biotinylated, medium was replaced, and the cells were cultured at 37°C for the indicated times. Biotinylated surface α4β2HA receptors were purified with streptavidin-agarose and bound with 125I-epibatidine. The rate of turnover for the surface receptors was determined from the decay in 125I-epibatidine bound to the receptors with increasing time. The amount of 125I-epibatidine bound is displayed as 125I-epibatidine bound divided by the maximum bound (fraction of maximum), which occurred immediately after biotinylation of the cells (time 0). Each point is the mean ± SD of three samples. Sample size is the same as in Figure 3E. Similar results were obtained in three separate experiments. The lines through the points are a least-squares fit of the data to the following equation: fraction of maximum bound = exp (-t/τ), in which τ is the time constant of the surface turnover. Maximal binding for untreated samples was 0.036 ± 0.001 and 0.14 ± 0.006 pmol for nicotine-treated samples, and the τ values are 14.3 and 12.8 h for untreated and nicotine-treated samples, respectively.
Effects of long-term nicotine exposure after block of receptor surface delivery
We next addressed whether receptor assembly and trafficking to the plasma membrane could be contributing to upregulation. Cells expressing α4β2 receptors were exposed to Brefeldin A, a fungal metabolite that disrupts the structure and function of the Golgi apparatus, thereby inhibiting intracellular trafficking of proteins to the cell surface (Klausner et al., 1992). As shown in Figure 3A, after Brefeldin A treatment for 1.5 h, formation of new 125I-epibatidine binding sites was blocked, indicating that the maturation of the receptors as well as surface trafficking was inhibited by Brefeldin A treatment. If Brefeldin A was applied before and during nicotine treatment for 18 h, the number of 125I-epibatidine binding sites was reduced (Fig. 3B). Despite disruption of the Golgi apparatus, the number of 125I-epibatidine binding sites was still upregulated by nicotine to approximately the same magnitude as in control cells (Fig. 3C). From these data, we conclude that the increase in 125I-epibatidine binding observed during nicotine-induced upregulation is not caused by an increase in receptor maturation or trafficking to the plasma membrane.

Effects of long-term nicotine exposure after block of receptor surface delivery. A, Effect of Brefeldin A treatment on formation of 125I-epibatidine-binding receptors. α4β2HA-expressing cells were incubated in the presence or absence of Brefeldin A (10 μg/ml) for the indicated times, and 125I-epibatidine binding was determined. Data are plotted as the fraction of initial amount of 125I-epibatidine bound, and each point is the mean of two experiments performed in triplicate. B, Effect of Brefeldin A treatment on nicotine-induced upregulation. 125I-epibatidine binding was performed on α4β2HA-expressing cells after exposure to Brefeldin A (right set of bars, 10 μg/ml Brefeldin A; left set of bars, unexposed) and nicotine treatment (filled bars, 10 μm nicotine for 18 h; open bars, untreated). C, Brefeldin A does not significantly affect nicotine-induced upregulation. Data in B have been replotted as a nicotine-induced fold increase in 125I-epibatidine binding for cells unexposed and exposed to Brefeldin A. Samples were equivalent to one-fourth of a well from a 24-well plate.
Nicotine exposure alters the functional state of α4β2 receptors
Electrophysiological recordings were obtained from cells stably expressing α4β2HA receptors to characterize functional changes that accompany nicotine-induced upregulation. Currents evoked by a series of different ACh concentrations were significantly different if cells were treated with 1 μm nicotine for at least 19 h before the recordings (Fig. 4A, C), conditions in which upregulation was ∼90% of the maximum value (Fig. 1E). At each ACh concentration that elicited a response, the currents were larger for the nicotine-treated cells (Fig. 4A, bottom) compared with the untreated cells (Fig. 4A, top). More than a twofold increase in the ACh-evoked currents was observed for nicotine-treated cells at the highest ACh concentrations (Fig. 4C). Note that the ACh concentration-response curve could not be fitted by a single Hill equation but were adequately described by the sum of two Hill equations (Fig. 4, continuous lines and legend). In addition to the increase in magnitude of the response, the profile of the concentration-response relationship changed after nicotine treatment. Moreover, ACh concentration-response before and after nicotine treatment can be fitted using the same apparent low and high affinities and the same Hill coefficient; a marked change in the fraction of high versus low affinity is observed. This is best visualized in Figure 4D, which illustrates the change in the fraction of the high- and low-affinity fractions. An important statistical difference is observed for the high-affinity component, with a p value <0.006, whereas no statistical difference could be observed for the low-affinity component. In addition, upregulation is accompanied by a significant slowing of the response fast desensitization (Fig. 4B). As shown in this figure, a single exponential plus a constant adequately describe responses evoked by brief ACh test pulse (200 ms). Comparison of mean currents recorded in control conditions (Fig. 4B, top traces) or after nicotine exposure (Fig. 4B, bottom traces) illustrate that functional upregulation is accompanied by a slowing down of the response time constant. Significant differences in the decay time were observed at the four concentration tested (see figure legend). Similar results were reported previously for human α4β2 receptors expressed in HEK cells (Buisson and Bertrand, 2001). Nicotine upregulation is further demonstrated when computing the net charge carried through the activated α4β2 receptors. Net charge analysis was performed as described previously (Mike et al., 2000; Papke and Porter Papke, 2002) and yielded values of 34 ± 2.4, 48 ± 3.1, 92 ± 11, and 109 ± 12 pC for responses evoked by, respectively, 10, 30, 100, and 300 μm ACh in control conditions (n = 5) and 131 ± 24, 168 ± 36, 265 ± 57, and 344 ± 84 pC for responses evoked by the same ACh concentrations in nicotine-treated cells (n = 5). Net charges were significantly increased in nicotine-treated cells for the four ACh concentrations tested (p < 0.05).

Nicotine exposure modified the functional properties of α4β2 receptors. A, Typical currents evoked by five ACh concentrations recorded in control conditions (top traces) and in another cell that has been treated for 20 h with 1 μm nicotine. Horizontal bars above the traces indicate the timing of the ACh application. Cells were maintained in control medium throughout the experiments and held at -100 mV. B, Mean currents recorded in control or after nicotine treatment (as in A). Response decay time was fitted with a single exponential. Mean values for the exponentials (n = 5) are for the control conditions (304 ± 37, 252 ± 15, 219 ± 22, and 194 ± 24 ms) and after nicotine treatment (514 ± 17 ms, p = 0.003; 484 ± 33 ms, p = 0.001; 450 ± 64 ms, p = 0.02; and 412 ± 53 ms, p = 0.01). A significant slowing of the response in nicotine-treated cells was observed for all concentrations tested. C, ACh concentration-response curves recorded in control (open circles; n = 7) and nicotine-treated (filled squares; n = 7) cells. Continuous lines are the best fit obtained with the sum of two empirical Hill equations in the form y = Imax × [a × (1/1 + (EC50H/ACh)nH) + (1 - a) × (1/1 + (EC50L/ACh)nL)], where y is the peak current (in picoamperes), Imax is the maximal evoked current recorded at 1 mm ACh, a is fraction of high-affinity current, and EC50H, nH, EC50L, nL are the half-activation and Hill coefficients for the high- and low-affinity components. Values for the control were as follows: Imax = 360 ± 76 pA, a = 0.2 ± 0.02, EC50H = 2.1 ± 0.3 μm, nH = 1.1 ± 0.03, EC50L = 92 ± 13 μm, nL = 1.3 ± 0.04; nicotine-treated Imax = 1131 ± 279 pA, a = 0.44 ± 5.2, EC50H = 3.2 ± 0.3 μm, nH = 1 ± 0.04, EC50L = 90 ± 5 μm, nL = 1.7 ± 0.07. The difference in maximal evoked current was statistically significant, with p = 0.03. D, Plot of the fraction of high-affinity (filled bars) and low-affinity (open bars) currents measured in control and nicotine-treated cells. High-affinity fraction of current measured in seven cells was significantly increased (p = 0.004) from 77 ± 19 to 427 ± 79 pA, whereas the low-affinity current was increased from 283 ± 58 to 710 ± 208 pA (p = 0.09). E, Typical currents evoked by three epibatidine pulses (400 ms) are superimposed. Left traces illustrate currents recorded in control conditions, and right traces illustrate currents recorded in nicotine-treated cells. F, The amplitude of current evoked by three epibatidine concentrations in control (n = 4) and after nicotine treatment (n = 6) is plotted. Currents ranged from 6.7 ± 1.8, 22.7 ± 5.2, and 64.4 ± 12 pA in control to 23.4 ± 4.8 (p = 0.01), 73.3 ± 12 (p = 0.006), and 173 ± 35 (0.02) in nicotine-treated cells. Values are indicated with their SEM, and asterisks indicate the values that were statistically significant (see Materials and Methods).
Because binding data have been performed using labeled epibatidine, we examined if a comparable functional upregulation could be observed for this agonist. It should be remembered, however, that, although rat α4β2HA receptor display a high affinity for this ligand, epibatidine evokes only a fraction of the ACh response and behaves as a partial agonist (Buisson et al., 2000). In agreement with this previous observation, currents of smaller amplitude were recorded in response to epibatidine test pulses (Fig. 4E). Despite the low current amplitude, a marked increase in current was, however, measured after nicotine treatment (Fig. 4E, right trace). This current increase is illustrated in Figure 4F, in which responses evoked by three epibatidine concentrations are compared. These data demonstrate that functional upregulation is not specific for ACh but also applies to other ligands, such as epibatidine, and further correlates with binding data.
Other assays of surface α4β2 receptors
Previously, the binding of epibatidine to surface and intracellular α4β2 receptors was distinguished using cell-impermeant competitive agonists (Whiteaker et al., 1998; Harkness and Millar, 2002). We attempted a similar approach with ACh to block surface 125I-epibatidine binding. When increasing concentrations of ACh were applied to α4β2 receptors in a broken cell membrane preparation, ACh blocked 100% of 125I-epibatidine binding (Fig. 5A). We expected ACh to block only surface receptors in intact cells, which should consist of ∼20% of the total binding (Fig. 2C), and that the block would occur with a similar concentration dependence as with the α4β2 receptors in the membrane preparation (Fig. 5A, dotted line). Instead, ACh blocked 100% of 125I-epibatidine binding in intact cells, with an altered concentration dependence shifted toward higher concentrations. As shown in Figure 5B, the slope and midpoint of the concentration dependence of the ACh block changed when the incubation time of ACh was increased in increments from 5 min to 3 h and began to approach values observed with membranes. These data indicate that ACh is slowly entering the cells by an unknown mechanism and blocking 125I-epibatidine binding to intracellular receptors as well as to surface receptors.

Additional measurements of surface α4β2 receptors. A, Concentration-dependent block of 125I-epibatidine binding by ACh. 125I-epibatidine binding to intact cells or membrane preparations was determined after incubation with the indicated concentrations of ACh for 5 min. Data are plotted as the fraction of the maximum 125I-epibatidine bound. Each point is the mean ± SD of three samples. The line through the data represents a least-squares fit to the following Hill equation: fraction of max = Max/(1 + IC50/[ligand])n, where Max was 1; n, the Hill coefficients, were 0.35 ± 0.2 and 0.87 ± 0.04; and the IC50 values were 5.3 ± 0.88 × 10-5 m and 3.9 ± 0.24 × 10-7 m for intact cells and membranes, respectively. Samples were one-sixth of a well from a 24-well plate for intact cells and one-fourth of a well for membranes. B, Concentration-dependent block of 125I-epibatidine binding by ACh changes with increased ACh incubation time. 125I-epibatidine binding to intact cells was determined after incubation in the indicated concentrations of ACh for 5 min, 30 min, 1 h, or 3 h. The data were fit to the equation in A, and the IC50 values and Hill coefficients obtained for increasing lengths of ACh exposure were 6.3 ± 2.6 × 10-4 and 0.33 ± 0.05, 1.3 × 10-4 ± 5.1 × 10-5 and 0.36 ± 0.05, 6.2 ± 1.7 × 10-5 and 0.35 ± 0.03, and 8.0 ± 2.8 × 10-6 and 0.38 ± 0.04, respectively. Samples were one-sixth of a well from a 24-well plate. C, The fold increase of 125I-epibatidine binding. Specific 125I-epibatidine binding was determined for α4β2HA stably expressing cells that were grown in the presence or absence of 10 μm nicotine for 18 h at 37°C. Binding was performed on both live cells and cells that had first been fixed with 2% paraformaldehyde as described previously (Harkness and Millar, 2002). D, Antibody binding to surface α4β2 receptors on cells fixed with 2% paraformaldehyde. Nicotine-treated or nontreated cells were incubated in anti-β2HA mAb 12CA5 (left) or anti-α4 mAb 299 (right). Afterward, cells were fixed with 2% paraformaldehyde, and specific 125I-protein A binding was determined. E, Antibody binding to surface α4β2 receptors on live cells. Cells were treated as in D except that 125I-protein A binding was performed on live cells. Statistical significance of the results, determined by two-tailed Student's t test, were p = 0.023 for 12CA5 and p = 0.99 for mAb 299. Data in C-E are the mean ± SD of experiments done in triplicate. Each sample was equivalent to five wells from a 24-well plate.
A recent publication concluded that nicotine-induced upregulation of α4β2 receptors is caused by an increase in receptor numbers (Harkness and Millar, 2002). The evidence was based on measurements of the binding of subunit-specific Abs to intact cells expressing α4β2 receptors. We repeated these experiments with the same protocol except substituting 125I-protein A for HRP-labeled secondary Abs to quantify Ab binding. 125I-protein A allowed us to measure the amount of Abs bound. Using this protocol, we found that there was a twofold increase in the binding of the α4-specific mAb 299 with nicotine treatment and 1.7-fold increase in the anti-HA mAb to the β2 subunit (Fig. 5D). These increases are smaller than those measured previously (3.3-fold for mAb 299) (Harkness and Millar, 2002) and do not correspond to the fivefold increase in 125I-epibatidine binding observed in unfixed cells or to the fourfold increase observed in fixed cells (Fig. 5C). Surprisingly, we found that very little of the Ab was bound to the cells compared with the number of 125I-epibatidine binding sites. There were 1.8 pmol of 125I-epibatidine bound to nicotine-treated cells (Fig. 5C). Assuming that 20% of the 125I-epibatidine bound to surface receptors, there are 0.36 pmol of surface receptors (Fig. 2C). Only 0.003 pmol of either Ab bound to the surface of nicotine-treated cells (Fig. 5B), which is 100-fold less than the amount of 125I-epibatidine bound.
In the protocol used previously (Harkness and Millar, 2002), cells were fixed after Ab binding. The results of these experiments are questionable given how little Ab binds to the receptors (Fig. 5D). Levels of Ab binding consistent with 125I-epibatidine binding were obtained when cells were not fixed (Fig. 5E). Under these conditions, 0.1 pmol of the anti-HA Ab bound to surface, which is only 3-4-fold less than the 0.36 pmol of 125I-epibatidine that appears to bind to surface receptors. The failure to saturate Ab binding may be attributable to steric hindrance between Abs binding to subunits within the same receptor. Importantly, binding of either anti-HA Abs to β2 subunits or α4-specific mAb 299 changed little with nicotine treatment of the cells with a 20% increase in anti-HA Ab binding and no detectable change in the binding of mAb 299 (Fig. 5E). The α4-specific mAb 299 bound 10-fold less than the anti-HA Ab (Fig. 5E). This difference in binding could be caused by 125I-protein A having a lower affinity for mAb 299 than the anti-HA Ab, which is expected based on the species and IgG subtype of the two Abs. However, based on the immunoprecipitation of 125I-epibatidine-bound receptors, the anti-HA Ab immunoprecipitated fivefold more of the receptors than mAb 299 (data not shown). Thus, less mAb 299 binds to the receptors compared with anti-HA Ab, perhaps because mAb 299 has a lower affinity or because its binding is dependent on the conformation of α4 subunits, as suggested by a previous study (Harkness and Millar, 2002).
Discussion
Since the initial observation that chronic nicotine exposure, at concentrations achieved with smoking, increased 3H-acetylcholine binding in rat brains (Schwartz and Kellar, 1985), the phenomenon of upregulation has been associated with nicotine addiction. The assumption has been made that nicotine-induced upregulation is a measure of an increase in the number of nicotinic receptors. The debate has been whether changes in receptor surface turnover, assembly, or trafficking to the cell surface underlie the apparent increase in receptor numbers. To assay the mechanisms underlying upregulation in detail, we and others (Peng et al., 1994; Bencherif et al., 1995; Hsu et al., 1996; Eilers et al., 1997; Warpman et al., 1998; Whiteaker et al., 1998; Fenster et al., 1999a; Harkness and Millar, 2002; Sallette et al., 2004) have resorted to heterologous expression of α4β2 receptors in mammalian cell lines because of the limitations of brain preparations and cultured neurons. Using two independent measurements, nicotine treatment of surface receptors after biotinylation (Fig. 2E, F) and Ab binding to surface α4β2 receptors (Fig. 5E), we presented evidence that nicotine induces an increase in 125I-epibatidine binding without significant changes in the number of surface receptors. We also performed experiments to directly test whether surface turnover of α4β2 receptors, receptor assembly, or their trafficking to the cell surface are altered by nicotine treatment. Previous studies implicating a decrease in surface turnover contributing to upregulation were performed with cycloheximide to block protein synthesis during the measurement (Peng et al., 1994). However, the turnover rate of surface α4β2 receptors when directly measured was unchanged by nicotine treatment (Fig. 2G).

Nicotine-induced upregulation of α4β2 receptors. Based on the presented data, α4β2 receptors exist in at least two states: (1) a resting state that binds epibatidine with low affinity, and (2) an upregulated state, with high-affinity epibatidine binding. In the presence of nicotine, the receptors in the resting state are slowly stabilized in the upregulated state. This effect of nicotine on the receptors increases 125I-epibatidine binding fourfold, as shown without altering the number of receptors. Because nicotine readily crosses the plasma membrane, both intracellular and surface receptors undergo the same nicotine-induced changes. In addition to a difference in high-affinity epibatidine binding, nicotine also alters receptor function, with receptors in the upregulated state more active because of increased sensitivity to an agonist and a slowing of desensitization.
We tested whether receptor trafficking of newly assembled receptors to the surface contributes to upregulation by incubating cells expressing α4β2 receptors using Brefeldin A. Brefeldin A blocks receptor trafficking to the surface by disrupting the Golgi apparatus and the formation of new epibatidine-binding sites, which we assume is a direct measure of α4β2 receptor assembly (Fig. 3A). Moreover, even after extended Brefeldin A incubation, we observed no change in the upregulation (Fig. 3C). Other evidence addressing whether changes in receptor assembly underlie upregulation is that 30-56% of synthesized α4 and β2 subunits assemble into receptors. This result indicates that, even if the assembly efficiency were increased to 100%, assembly would cause at most a twofold to threefold increase in the number of assembled receptors and could only account for part of the measured fourfold to fivefold increase in ligand binding. Even if such an increase in subunit assembly were to occur, we would expect to measure a large increase in the percentage of receptor found in the intracellular pool. No such change in the percentage of intracellular receptors was observed (Fig. 2C).
What causes nicotine-induced upregulation if it cannot be attributed to changes in receptor surface turnover, assembly, or trafficking to the surface? Radiolabeled ligands specific for nicotinic receptors, such as 125I-epibatidine used in this study, have been widely used to characterize the pharmacology of α4β2 and other nicotinic receptor subtypes in brain and their regional and subcellular distribution (Sargent, 1993; McGehee and Role, 1995; Role and Berg, 1996). It has been assumed that the high-affinity binding measured using the radioligands is an immutable feature of the receptors. Such an assumption is reasonable for competitive antagonists such as 125I-α-bungarotoxin but questionable for agonists such as nicotine, cytisine, or epibatidine. Agonist binding causes nicotinic receptors to enter different functional states, and it is well established that muscle-type nicotinic receptors bind agonists with much higher affinity after entry into the desensitized state (Katz and Thesleff, 1957; Rang and Ritter, 1970; Weber et al., 1975). Thus, it is possible that long exposures of α4β2 receptors to nicotine could induce conformational changes in the receptor that alters its affinity for nicotine, cytisine, and epibatidine. Previous studies have measured the α4β2 receptor affinity for these agonists using the radiolabeled agonists and were unable to detect any change in the affinity after nicotine exposure (Peng et al., 1994; Whiteaker et al., 1997). Based on these results, nicotine would have to change α4β2 receptor conformation from a resting state in which the affinity for radiolabeled agonists was immeasurably low to the upregulated state with high-affinity binding (Fig. 6). In other words, there is no measurable binding to receptors in the resting state, and only receptors in the upregulated state bind radiolabeled agonist. In this way, radiolabeled agonist binding is a measure of the number of receptors that have entered the upregulated state, not the total number of receptors. As illustrated in Figure 6, the increase in receptor 125I-epibatidine binding occurs because nicotine treatment has shifted the equilibrium between the resting and upregulated states for surface and intracellular receptors.
The electrophysiological measurements are also consistent with a shift in equilibrium between two different receptor states after nicotine exposure. We measured changes in the concentration dependence of activation by ACh (Fig. 4A, C) and a slowing of the rate of desensitization (Fig. 4B) after nicotine treatment that is a typical signature of functionally upregulated receptors. Similar data have been obtained for human α4β2 receptors stably expressed in HEK cells (Buisson and Bertrand, 2001). Moreover, we showed that nicotine treatment upregulates both Ach- and epibatidine-evoked currents, indicating an additional correlation between binding and electrophysiological measurements. It should be remembered, however, that, because of technical limits, these two approaches are probing receptors that are probably in different states. Namely, in binding experiments, labeling is performed using long exposures that can desensitize the receptors, whereas functional test are realized with brief (200 ms) test pulses.
The two apparent affinities observed for the ACh concentration-response curves can be interpreted as the consequence of two distinct functional states of the α4β2 receptors. Although desensitization for brief pulses are well fitted with a single exponential, it should be remembered that the upregulated human receptors had a larger single-channel conductance when activated, providing even more evidence for a functional change of state with nicotine exposure. An alternative mechanism, a change in receptor α4:β2 subunit stoichiometry from 2:3 to 3:2 during assembly has been proposed to explain the change in receptor function with nicotine exposure (Nelson et al., 2003). Our findings that preexisting surface receptors can be upregulated (Fig. 2C, D) argue against such a mechanism because it is highly unlikely that receptor subunit stoichiometry would change after insertion into the plasma membrane.
Previously, the basic properties of nicotinic receptors were thought to remain unchanged with chronic nicotine exposure. Instead, our data indicate that nicotine slowly transforms α4β2 receptors into an upregulated state in which the function of the receptor differs from the original state. At nicotine levels achieved by smokers, α4β2 receptors in the brain initially cycle through one set of resting, activated, and desensitized states. With continued smoking, the same α4β2 receptors would slowly shift into the upregulated condition with different resting, activated, and desensitized states. Importantly, the characteristics of the upregulated functional states result in increased response and sensitivity to agonist. The increased response and sensitivity to agonist together with the slow entry into and exit from the upregulated state should provide a long-lasting memory of the presence of nicotine in neurons containing α4β2 receptors. Because other agonists, including the endogenous ACh, can upregulate α4β2 receptors (Gopalakrishnan et al., 1997; Whiteaker et al., 1998), it is possible that the molecular memory provided by α4β2 receptor upregulation could occur under conditions of high cholinergic synaptic activity. Previously, agonist-induced conformational changes of nicotinic receptors were thought to occur only over the timescale of milliseconds to seconds. The effects of nicotine on nicotinic receptors that occur over hours to days were attributed to effects downstream of any receptor state changes, and these downstream effects were thought to cause changes in receptor numbers. Our results suggest that the slower, longer-lasting effects of nicotine that occur during smoking or increased neurotransmitter release can result from nicotine-induced conformational changes that alter receptor function.
Footnotes
This work was supported by grants from the National Institute on Drug Abuse, National Institutes of Health, Alzheimer's Association, and Brain Research Foundation (W.N.G.), from the Swiss National Science Foundation (D.B.), and by a fellowship from Philip Morris Incorporated (Y.F.V.). We thank Drs. J. Boulter and S. Rogers for generously providing constructs and antibodies used in this study. We also thank Dr. S. Bertrand and members of the Green and Bertrand laboratories for discussion and comments about this manuscript. We thank Dr. Gregory D. Jefford for his assistance with the confocal microscopy presented in this manuscript.
Correspondence should be addressed to William N. Green, Department of Neurobiology, Pharmacology, and Physiology, University of Chicago, 947 East 58th Street, Chicago, IL 60637. E-mail: wgreen{at}midway.uchicago.edu.
B. Buisson's present address: Department of Pharmacology and Toxicology, Trophos SA, Parc Scientifique de Luminy, Case 931, 13288 Marseille Cedex 9, France.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255563-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}