

Abstract
Mutations in MTMR2, the myotubularin-related 2 gene, cause autosomal recessive Charcot-Marie-Tooth type 4B1 (CMT4B1). This disorder is characterized by childhood onset of weakness and sensory loss, severely decreased nerve conduction velocity, demyelination in the nerve with myelin outfoldings, and severe functional impairment of affected patients, mainly resulting from loss of myelinated fibers in the nerve. We recently generated Mtmr2-nullneo mice, which show a dysmyelinating neuropathy with myelin outfoldings, thus reproducing human CMT4B1. Mtmr2 is detected in both Schwann cells and neurons, in which it interacts with discs large 1/synapse-associated protein 97 and neurofilament light chain, respectively. Here, we specifically ablated Mtmr2 in either Schwann cells or motor neurons. Disruption of Mtmr2 in Schwann cells produced a dysmyelinating phenotype very similar to that of the Mtmr2-nullneo mouse. Disruption of Mtmr2 in motor neurons does not provoke myelin outfoldings nor axonal defects. We propose that loss of Mtmr2 in Schwann cells, but not in motor neurons, is both sufficient and necessary to cause CMT4B1 neuropathy. Thus, therapeutical approaches might be designed in the future to specifically deliver the Mtmr2 phospholipid phosphatase to Schwann cells in affected nerves.
Introduction
Myelin is a highly specialized structure of vertebrates formed by the plasma membrane of Schwann cells in the peripheral nervous system, which enwraps larger axons in a spiral manner. This structure allows faster propagation of action potentials because of saltatory conduction. In the “axon-Schwann cell unit,” myelin-forming Schwann cells affect axonal health and regulate clustering of channels at the node of Ranvier, axonal diameter, cytoskeleton, and transport (Jessen and Mirsky, 2004). Axons provide signals for Schwann cell survival, proliferation, differentiation, and myelination (Poliak and Peles, 2003; Salzer, 2003; Jessen and Mirsky, 2004; Trapp and Kidd, 2004). Only a few genes that mediate this reciprocal relationship have been identified (Lappe-Siefke et al., 2003; Michailov et al., 2004).
Charcot-Marie-Tooth (CMT) disease represents a heterogeneous group of inherited disorders affecting myelinated axons in the peripheral nervous system (Suter and Scherer, 2003; Wrabetz et al., 2004). CMTs are characterized by progressive distal muscular atrophy and weakness with an age of onset between the first and second decades of life. Two main phenotypes have been classically recognized: primary demyelinating CMT type 1 (CMT1) with nerve conduction velocities (NCVs) <38 m/s and primary axonal CMT2 associated with normal values of NCV (>38 m/s) and decreased compound motor action potential (CMAP). Because of the intimate relationship between Schwann cells and axons, axonal loss and/or atrophy might also occur as a consequence of primary demyelination and vice versa. Moreover, CMT forms with “intermediate” slowing of NCV between 25 and 45 m/s have also been reported (Davis et al., 1978; Rouger et al., 1997). At least 22 responsible genes have been isolated thus far (http://www.molgen.ua.ac.be/CMTMutations/default.cfm). Although the pattern of expression of several CMT genes predicts phenotype (Schwann cell-specific with demyelinating, neuron-specific with axonal), for many CMT genes, it does not. For instance, mutations in MPZ, the myelin protein zero gene, which is specifically expressed in Schwann cells, are associated with primary demyelinating, axonal, and intermediate forms of CMT (Shy, 2004). The GDAP1 gene, the expression of which is highly enriched in neurons, is responsible for demyelinating, axonal, and intermediate CMTs (Pedrola et al., 2005). Other genes, although ubiquitously expressed in nerve, are specifically associated with primary axonal CMTs (e.g., MFN2, RAB7, HSP22/27, and GARS) (Antonellis et al., 2003; Verhoeven et al., 2003; Evgrafov et al., 2004; Irobi et al., 2004; Zuchner et al., 2004) or primary demyelinating CMTs [e.g., myotubularin-related 2 (MTMR2) and MTMR13] (Bolino et al., 2000; Azzedine et al., 2003; Senderek et al., 2003).
The MTMR2 and MTMR13 genes are responsible for the demyelinating autosomal recessive CMT type 4B1 (CMT4B1) and CMT4B2, respectively. CMT4B is a severe neuropathy characterized by childhood onset of weakness and sensory loss, severely decreased NCV, and, in the nerve, demyelination, loss of myelinated fibers, hypomyelination, and myelin outfoldings (Quattrone et al., 1996). MTMR2 and MTMR13 represent catalytically active and inactive phosphatase proteins, respectively, belonging to the myotubularin-related protein family, which comprises 14 members in human, named MTM1 and MTMR1–MTMR13 (Laporte et al., 2003). Although MTMRs share extensive homology with phosphotyrosine/dual-specificity phosphatases, they dephosphorylate phosphoinositides (Schaletzky et al., 2003).
We recently generated Mtmr2-nullneo mice, which recapitulate the CMT4B1 pathology (Bolino et al., 2004). These mutants develop a progressive neuropathy with myelin outfoldings and redundant loops of myelin originating mainly at paranodal regions and impaired spermatogenesis. However, this mouse model does not demonstrate whether loss of Mtmr2 in Schwann cell and/or neurons provokes the dysmyelination observed in the nerve. Moreover, a cell autonomous role for Mtmr2 in both Schwann cells and neurons might be predicted on the basis of expression as well as physical interactions. We recently showed that Mtmr2 is expressed in vivo in myelin-forming, non-myelin-forming Schwann cells and axons. We also provided evidence that Mtmr2 might interact in neurons with neurofilament light chain (NF-L), a protein mutated in other forms of CMT (Previtali et al., 2003). In Schwann cells, Mtmr2 interacts with discs large 1 (Dlg1)/synapse-associated protein 97 (SAP97), which is enriched in the paranodal cytoplasm. In Mtmr2-nullneo nerves, the Mtmr2/Dlg1 complex is disrupted (Bolino et al., 2004). To gain insight into the function of Mtmr2 in Schwann cells and neurons, we specifically ablated Mtmr2 in either Schwann cells or motor neurons using the Cre/loxP technology. Here, we report that inactivation of Mtmr2 in Schwann cells and not in motor neurons is sufficient and necessary to produce CMT4B1 neuropathy with myelin outfoldings.
Materials and Methods
Generation of Mtmr2 conditional mutants. The generation of an Mtmr2 Floxed allele has been reported previously (Bolino et al., 2004). To produce mice with specific ablation of Mtmr2 in either Schwann cells or motor neurons, Mtmr2Flneo (floxed with neo) mice were crossed with transgenics carrying the Flpe recombinase for excision of the neo cassette (see Fig. 1 A). Homozygous Mtmr2Fl mice were then crossed with heterozygous Mtmr2-nullneo mice (lacking exon 4 but still containing the neo cassette) carrying the P0-Cre or the HB9-Cre transgene. Mtmr2Fl/nullneo//P0-Cre or HB9-Cre mice are the conditional null in Schwann cells or in motor neurons, respectively. Homozygous Mtmr2Fl mice are normal, and no morphological alterations were observed in their nerves (data not shown). Congenic C57BL/6 mouse strains carrying the P0-Cre or HB9-Cre transgenes were used. Mice with conditional ablation of Mtmr2 were generated on a mixed C57BL/6/129SvPas genetic background (n = 3 or 4). All experiments involving animals were performed in accordance with Italian national regulations and covered by experimental protocols reviewed by local Institutional Animal Care and Use Committees.
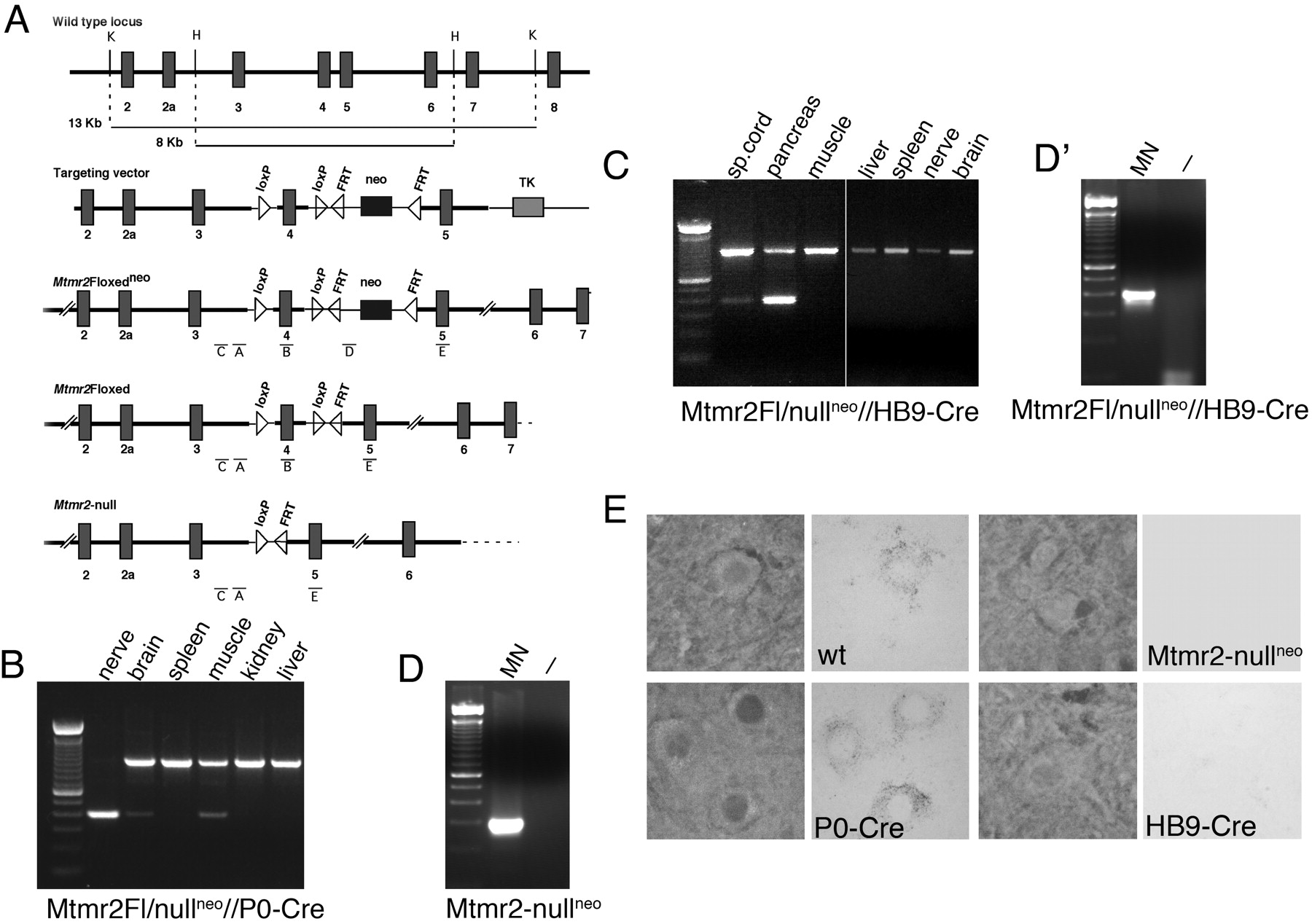
PCR analysis. Genotype analysis of tail genomic DNA used primer pairs C and B or A and D to distinguish between the (+/+) wild-type, Mtmr2Flneo, and Mtmr2-nullneo mice as described previously (Bolino et al., 2004). Excision of the neo cassette from the floxed allele was assessed using primers D (designed as Forward) and E (see Fig. 1 A). Finally, the conditional Mtmr2Fl/nullneo//P0-Cre or HB9-Cre mice were genotyped using primers C and B and specific primers to P0-Cre or HB9-Cre transgenes. Genomic DNA extracted from several tissues was amplified using primers A and E, which were able to amplify only the allele in which the neo cassette has been removed (Fl) (see Fig. 1 B, C).
Dissection of motor neurons from the spinal cord was performed by laser capture microdissection according to the instructions of the manufacturer (LS LMD; Leica, Northvale, NJ). Genomic DNA was extracted using the QIAamp DNA Microkit (Qiagen, Germantown, MD), and PCR was performed using standard conditions with 45 cycles of amplification. The following pairs of primers were used: primers A and D for Mtmr2-nullneo mice (see Fig. 1 D); primers A and E for Mtmr2Fl/nullneo//HB9-Cre mice (see Fig. 1 D'). In this way, a product of amplification was detected in both cases confirming that genomic DNA was always obtained from laser-dissected cells.
cDNA synthesis on total RNA extracted from sciatic nerve, muscle, and brain was performed as described previously (Bolino et al., 2002). At least three animals per genotype were analyzed for brain and muscle. For sciatic nerve, three independent RNA pools of four nerves (two animals) were used in a single reaction to increase the amount of the RNA analyzed.
The expression of selected mRNAs was determined by real-time PCR (RT-PCR). Samples were processed in triplicate, and reactions without target cDNA were used as negative control for each reaction. PCRs were performed on 96-well plates using the TaqMan Universal Master Mix (Applied Biosystems, Foster City, CA) and following the conditions of the manufacturer. TaqMan probes (Applied Biosystems) were used to amplify mouse Mtmr2, Mtmr13, Mtm1, Mtmr5, and Mtmr12 (3-phosphatase adapter protein) and the endogenous reference transcripts glyceraldehyde-3-phosphate dehydrogenase and PGK (phosphoglycerate kinase). RT-PCR efficiencies (E) of each transcript were first determined using at least eight different amounts of reverse-transcribed mRNA. To this aim, the comparative CT method (ABI PRISM Sequence Detection System User Bulletin 2; Applied Biosystems) and the ABI PRISM 7700 Sequence Detection System (version 1.6 software) were used. When log input total RNA amounts were plotted versus ΔCT (selected transcript CT minus endogenous reference CT), only slopes <0.1 were taken into account. As a calibrator, a control sample ΔCT was chosen for each selected transcript in each tissue. The ΔΔCT (ΔCT of each normalized selected transcript minus ΔCT of the calibrator) was calculated. Expression levels of each mRNA (the 2–ΔΔCT values) are indicated as “n-folds” relative to 1, which correspond to the 2–ΔΔCT value of the calibrator.
For statistical analysis, the StatView program (version 5.0) with the paired t test method was used. The SD was calculated for triplicate samples of each reaction, and SEM is indicated on the media of the determinations from different animals (see Fig. 9).
In situ hybridization analysis. A cDNA fragment of 500 bp containing exons 11–14 of Mtmr2 cDNA was amplified from mouse nerve total RNA and cloned into TA vector (Invitrogen, Carlsbad, CA). This product was then subcloned into pBluescript II KS- and used as a template to generate probes. The resulting plasmid was linearized by digestion with HindIII and transcribed with T7 RNA polymerase to obtain antisense probe and linearized with XhoI and transcribed with T3 RNA polymerase to obtain the sense probe. In situ hybridization experiments were performed as reported previously (Bulfone et al., 1993)
Behavioral analysis and electrophysiology. Rotarod test groups of mice 6 months of age were placed on a rotating cylinder that accelerated from 4 to 40 rotations per minute (rpm) over 5 min (Ugo Basile, Comerio, Italy). The animals were allowed to stay on the cylinder for a maximum of 700 s, and the time to fail was measured in different trials (two trials of three replicates per day for 3 d, nonconsecutively). For statistical analysis, the StatView program (version 5.0) with the paired t test method was used. Electrophysiology was performed as described previously (Bolino et al., 2004).
Morphological analysis. Three animals per genotype for each time point were analyzed. Semithin section analysis of sciatic, saphenous, quadriceps, and digital nerves was performed as reported previously (Wrabetz et al., 2000).
The proportion of fibers having outfoldings was counted as previously reported on at least 800–1000 fibers observed in three random microscopic fields from nerves of three animals. The proportion of fibers with more than three loops of redundant myelin was calculated as the percentage of fibers showing outfoldings. To perform morphometric analysis, digitalized images of fiber cross sections were obtained from corresponding levels of the sciatic or quadriceps nerves with a 100× objective and digital camera Leica DFC300F. At least three images from three different animals per genotype at 8 months of age were acquired (120 × 103 μm2 of sciatic nerve per each animal) and analyzed with the Leica QWin software (Leica Microsystem). The g-ratio was determined by dividing the mean diameter of an axon (without myelin) by the mean diameter of the same axon including myelin. Approximately 150 randomly chosen fibers per animal were analyzed. Myelinated axons in the first toe were counted in a cross section and the two plantar nerves (derived from sciatic nerve) were analyzed (Frei et al., 1999). Statistical analysis was performed using Statview 5.0 software (SAS Institute, Cary, NC). Ultrathin morphological analysis was performed as described previously (Wrabetz et al., 2000). Teased fiber preparation was performed as reported previously (Previtali et al., 2000).
Hematoxylin, eosin, and ATPase isozyme staining of posterior compartment muscles of the leg were performed as described previously (Wrabetz et al., 2000).
Antibodies. The following antibodies were used for immunohistochemistry: rabbit polyclonal anti-SAP97 (provided by Dr. T. Nakagawa, Massachusetts Institute of Technology, Boston, MA), rabbit polyclonal anti-Caspr-GOGI (provided by Dr. E. Peles, The Weizmann Institute of Science, Rehovot, Israel), mouse monoclonal anti-NaCh PAN (Sigma, St. Louis, MO), rabbit polyclonal anti-Kv1.1/1.2 (Alomone Labs, Jerusalem, Israel), mouse monoclonal anti-E-Cadh (Transduction-BD, Franklin Lakes, NJ), and mouse monoclonal anti-MAG (Chemicon, Temecula, CA). For Western blot analysis on sciatic nerve homogenates, performed as described previously, a mouse monoclonal anti-E-Cadh antibody (Transduction-BD) was used.
Secondary antibodies included fluorescein-conjugated (FITC) or rhodamine-conjugated (tetramethylrhodamine isothiocyanate) donkey anti-mouse or rabbit IgG-conjugated (Jackson ImmunoResearch, West Grove, PA) or peroxidase-conjugated goat anti-rabbit or rabbit anti-mouse IgG (Dako, Glostrup, Denmark).
Immunohistochemistry and confocal microscopy. Immunofluorescence on cryosections was performed as described previously (Previtali et al., 2003) and examined with confocal (Bio-Rad MRC 1024; Bio-Rad, Hercules, CA) or fluorescent (Olympus BX; Olympus Optical, Tokyo, Japan) microscopy. For immunohistochemistry, sciatic nerves were removed and rapidly snap-frozen in liquid nitrogen, either unfixed or previously fixed in buffered 4% paraformaldehyde. For teased fiber preparation, sciatic nerves were removed, fixed on ice in freshly prepared buffered 4% formaldehyde, as described by Bolino et al. (2004). The slides were washed in PBS and incubated with appropriate secondary antibodies (Jackson ImmunoResearch) and mounted with Vectashield (Vector Laboratories, Burlingame, CA). Quantification of the average of signal intensity was performed on fibers from at least two animals per genotype using the ImageJ program (version 1.32j; http://rsb.info.nih.gov/ij/). For MAG and Dlg1 staining, the average intensity of nonsaturated signals on images acquired with the same parameters between wild-type and mutant fibers was measured. The area corresponding to the Caspr staining in double-labeling experiments Dlg1/Caspr and MAG/Caspr was chosen. The average of signal intensity of the background (other regions without specific signals; e.g., internodes) was subtracted from the average of signal intensity of MAG and Dlg1. Measurements of NaCh cluster size were determined on NaCh/Caspr double immunostaining and normalized using NaCh cluster diameter for each node. Caspr clusters distance and Caspr clusters length were measured on E-cadh/Caspr double immunostaining and normalized for Caspr diameter. Kv1.1/Kv1.2 cluster length and distance were measured on NaCh and Kv1.1/Kv1.2 double immunostaining and normalized with the NaCh cluster diameter. For Kv1.1/Kv1.2 cluster staining, fixed thresholds across all samples (upper and lower limits) of saturated signal intensity were chosen. Statistical analysis was performed using the StatView program (version 5.0) and the paired t test method.
Image analysis. Micrographs were acquired using a digital camera (Leica F300), and figures were prepared using Adobe Photoshop version 8.0 (Adobe Systems, San Jose, CA).
Results
Generation of a conditional targeted allele for tissue-specific ablation of Mtmr2
A Floxedneo Mtmr2 allele was produced in which exon 4 was flanked by lox-P sites in a pFlrt1 vector carrying a neo cassette between FRT sites (Bolino et al., 2004). Excision of exon 4 introduces a frameshift from both ATG start sites of translation (exon 1 and 3) and is predicted to code for a short truncated protein, lacking all functional domains of the protein (Fig. 1A). To produce mice with Mtmr2-specific ablation in either Schwann cells or motor neurons, we crossed homozygous Mtmr2Fl mice with heterozygous Mtmr2-nullneo mice carrying the P0-Cre or the HB9-Cre transgenes.
P0-Cre produces somatic recombination of floxed genes specifically in Schwann cells and not in dorsal root or spinal motor neurons that contribute axons to peripheral nerves (Feltri et al., 1999, 2002). To confirm Schwann cell-specific ablation in Mtmr2Fl/nullneo//P0-Cre mice, we performed a PCR assay on genomic DNA from nerve and other tissues, which demonstrated that exon 4 was nearly completely excised in the sciatic nerve (remaining non-recombined signal probably originates from perineurium), whereas there was no recombination signal in the spleen and liver. The faint recombination signal in muscle is attributable to the presence of nerves in this tissue (Fig. 1B). Recombination in a few cells in the brain mediated by P0-Cre has been reported for some loci (M. L. Feltri, unpublished results).
HB9-Cre produces somatic recombination at embryonic day 9.5 (E9.5) in motor neurons and the pancreas (Arber et al., 1999; Li et al., 1999; Yang et al., 2001). To show Mtmr2 motor neuron-specific ablation in Mtmr2Fl/nullneo//HB9-Cre mice, PCR analysis on genomic DNA was used to demonstrate no recombination signal in sciatic nerve (containing nuclei of Schwann cells but not of neurons), brain, liver, spleen, and muscle. Exon 4 was nearly completely excised in the pancreas and partially in the spinal cord, where other neuronal cell types and glial cells are located (Fig. 1C). To confirm motor neuron-specific ablation in the spinal cord, we selectively dissected motor neurons from spinal cord sections of Mtmr2Fl/nullneo//HB9-Cre mice using laser-capture microdissection. A PCR assay was then performed on genomic DNA from laser-dissected cells and Mtmr2 ablation in motor neurons from both Mtmr2-nullneo mice (Fig. 1D) and Mtmr2Fl/nullneo//HB9-Cre mice (Fig. 1D') was confirmed. Finally, by in situ hybridization analysis, we detected Mtmr2 mRNA in the spinal cord and specifically in ventral horn motor neurons of Mtmr2Fl/nullneo//P0-Cre mice, at levels similar to wild-type and not in Mtmr2Fl/nullneo//HB9-Cre or Mtmr2-nullneo motor neurons (Fig. 1E). Thus, Mtmr2 was completely and specifically ablated in either Schwann cells or motor neurons in the two conditional mutants.
Mtmr2 disruption in Schwann cells is sufficient to provoke dysmyelination with myelin outfoldings
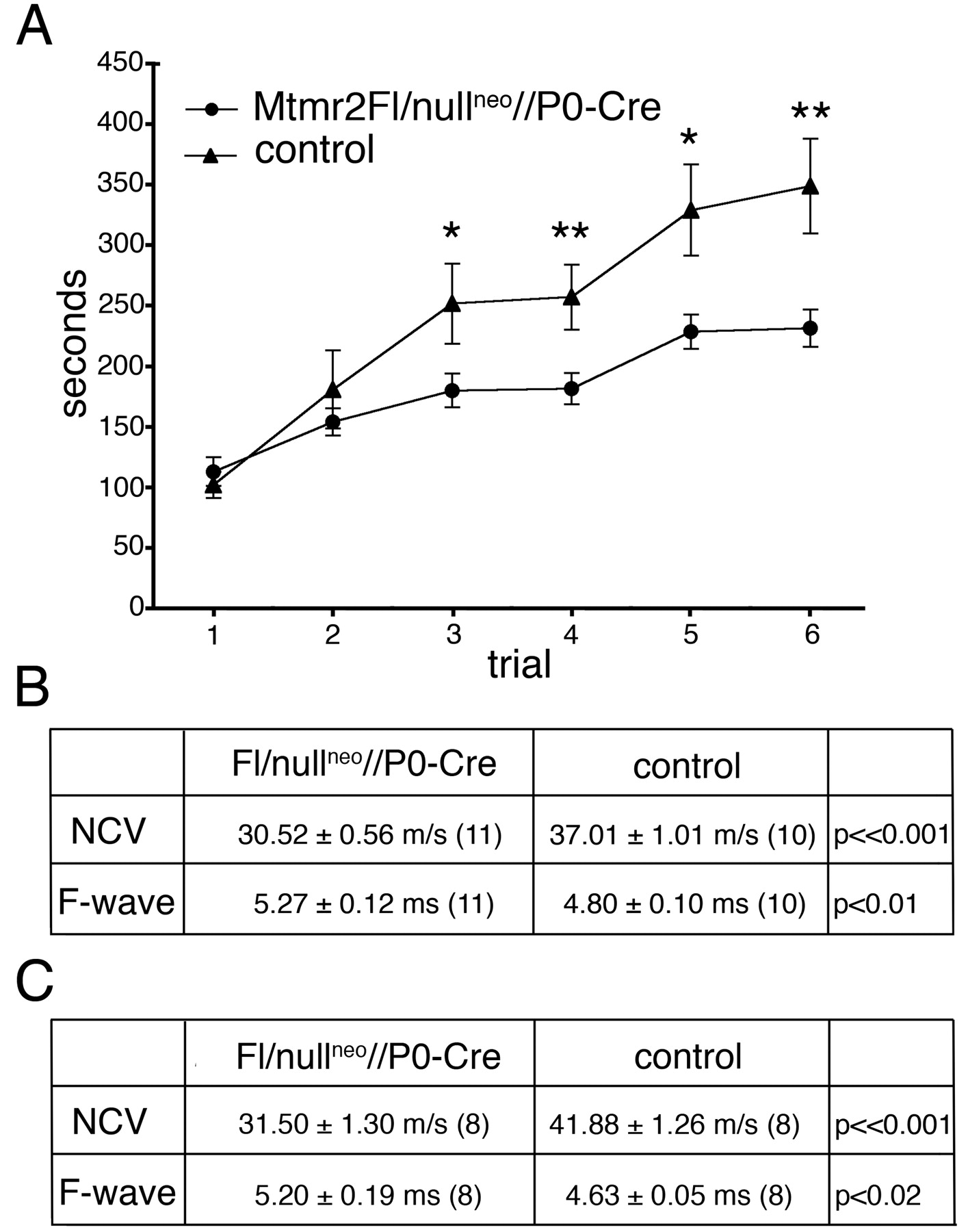
Mtmr2Fl/nullneo//P0-Cre mice are viable and born according to the expected ratio. At external examination, no signs of tremor or functional disability were observed. Mutant and normal littermates showed comparable weights at the different time points, different to what was observed in Mtmr2-nullneo mice (Bolino et al., 2004). Rotarod tests were therefore performed by placing 6-month-old mice on a horizontal cylinder rotating at increasing speed, and the time that they remain on the cylinder was measured in six different tests. Mutant animals remained on the cylinder significantly less than wild-type littermates, suggesting a neuromuscular defect in Mtmr2Fl/nullneo//P0-Cre mice (Fig. 2A). Electrophysiological examination of these mutants at 6 months of age revealed a significant reduction of nerve conduction velocities and prolonged F-wave latencies (Fig. 2B). Similar results have been obtained on Mtmr2-nullneo mice, as reported previously (Bolino et al., 2004). The amplitudes of proximal and distal compound motor action potentials were normal. At 12 months, no additional changes in nerve conduction velocities were observed in Mtmr2Fl/nullneo//P0-Cre mutants (Fig. 2C).

Generation of mice with Mtmr2 conditional ablation. A, First row, Genomic structure of Mtmr2 encompassing exon 4 (the wild-type Mtmr2 locus). Second row, Targeting construct in which Mtmr2 genomic fragments are represented by thick lines and vector sequences are represented by thin lines (the targeting vector). Third row, Mtmr2 locus after homologous recombination in ES cells as reported by Bolino et al. (2004) (the Mtmr2Flneo allele). Fourth row, Targeted allele without the neo cassette, removed in vivo using the Flpe transgene (the Mtmr2Fl allele). Fifth row, Targeted allele without neo and exon 4 (the Mtmr2-null allele). FRT, Flp recognition target; H, HindIII; K, KpnI; TK, thymidine kinase gene. B, PCR on genomic DNA from different tissues of Mtmr2Fl/nullneo//P0-Cre mice performed using primers A+E. Exon 4 was nearly completely excised in the sciatic nerve. C, PCR on genomic DNA from different tissues of Mtmr2Fl/nullneo//HB9-Cre mice performed using primers A+E. Exon 4 was nearly completely excised in the pancreas (as shown in previous studies) and only partially in the spinal cord (sp. cord), in which other neuronal cell types, endothelial and glial cells, are located. No recombination was noted in other tissues. D, PCR on genomic DNA from laser capture-microdissected motor neurons (MN) of Mtmr2-nullneo mice. The primer pair A+D was used. In these mice, exon 4 was removed from both Mtmr2-targeted alleles in which the neo cassette is still present. D', PCR on genomic DNA from laser capture-microdissected motor neurons (MN) of Mtmr2Fl/nullneo//HB9-Cre mice. The primer pair A+E was used. In these mice, exon 4 was absent in motor neurons from the spinal cord (MN), and a band of ∼300 bp was detected. If exon 4 was present, a band of ∼800 bp (containing exon 4 and one loxP site) would have been amplified, as in the case of the non-recombined tissues in B and C. Empty lanes (–) in D and D' correspond to the negative control of the PCRs. E, In situ mRNA hybridization analysis using, as a probe, a portion of the Mtmr2 cDNA-containing exons 11–14. Mtmr2 mRNA is detected in motor neurons from wild-type (wt) and Mtmr2Fl/nullneo//P0-Cre mice but not in Mtmr2Fl/nullneo//HB9-Cre or Mtmr2-nullneo motor neurons.
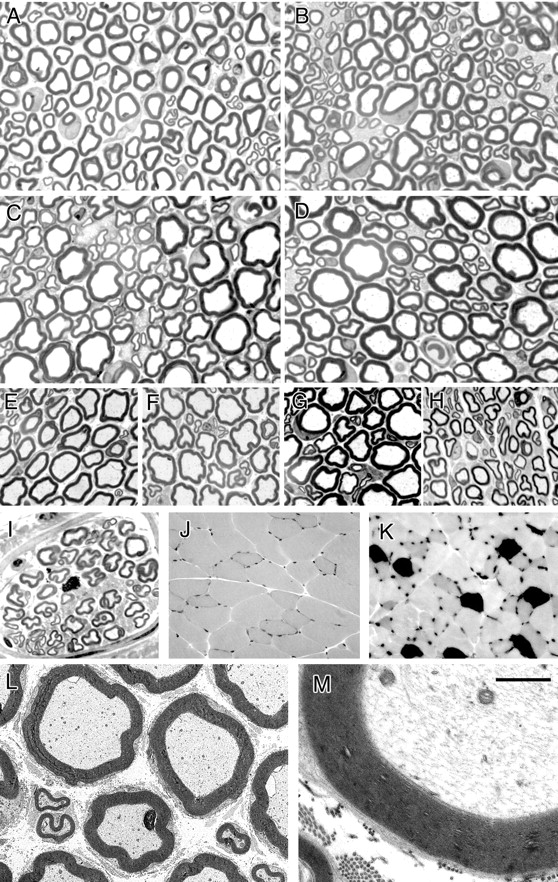
Myelin outfoldings were noted in nerves of Mtmr2Fl/nullneo//P0-Cre mice with similar shape and number as seen in Mtmr2-nullneo mouse nerves at 3–4 weeks after birth (Fig. 3A–F). The number of fibers showing myelin outfoldings and redundant loops increases with age (in transverse semithin sections, 5% of myelinated fibers at 2 months, 10% at 4 months, 16% at 6 months, and 26% at 12 months). The complexity of the dysmyelinating morphology also increased with age as the number of fibers showing three or more satellite loops increased from 7.5% at 2 months, 11.8% at 6 months, and up to 21.7% at 12 months of age. Remyelinated axons were observed at 12 and 15 months of age (Fig. 3C,D). Occasional axonal degeneration was also noted in sciatic nerves of these mutants. A reduction of 33% of myelinated axons was noted in plantar nerves of the first toe examined at 12 months of age (Fig. 3E,F).
Longitudinal semithin sections of sciatic nerves showed that redundant loops of myelin arise not only at paranodal/juxtaparanodal myelin but also at Schmidt–Lanterman incisures (Fig. 3G,H). By electron microscopy, the node of Ranvier, paranodal loops, and axo-glial junctions between paranodal loops of Schwann cell membrane and axolemma appeared normal (Fig. 3I,J). At Schmidt–Lanterman incisures evaginations contain both Schwann cell cytoplasm and axoplasm, contiguous with the main myelinated fiber (Fig. 3K,L).

Behavioral and electrophysiological analysis of Mtmr2Fl/nullneo//P0-Cre mice. A, Rotarod test analysis performed on mutant and control animals at 6 months of age. For statistical analysis, the StatView program (version 5.0) with the paired t test method was used. *p < 0.05; **p < 0.01. B, Electrophysiological analysis of Mtmr2Fl/nullneo//P0-Cre mice and controls at 6 months of age. Reduction of NCV and prolonged F-wave latencies in mutant mice are comparable with those obtained for Mtmr2-nullneo mice at the same age, as reported by Bolino et al. (2004). C, Electrophysiological analysis of Mtmr2Fl/nullneo//P0-Cre mice and controls at 12 months of age.
Dysmyelinating neuropathies in Mtmr2Fl/nullneo//P0-Cre and Mtmr2-nullneo mice are similar
Electrophysiological and morphological analyses were performed on Mtmr2-nullneo mice until 6 months of age. This mutant showed a dysmyelinating phenotype with myelin outfoldings similar to those observed in Mtmr2Fl/nullneo//P0-Cre mice (Bolino et al., 2004). To compare further the neuropathy in the two mutants, Mtmr2-nullneo mice were also examined at 12 and 15 months of age. At 12 months, Mtmr2-nullneo mice showed reduced nerve conduction velocities [30.90 ± 1.59 m/s (n = 5) vs 38 ± 2.52 m/s (n = 5); p < 0.05] and prolonged F-wave latencies [5.06 ± 0.25 ms (n = 5) vs 4.50 ± 0.11 ms (n = 5); p < 0.05]. These values are very similar to those obtained with Mtmr2Fl/nullneo//P0-Cre mice at the same age (compare with Fig. 2B). The number of fibers with myelin outfoldings increases up to 20% at 15 months of age in complete null mice. The nerve morphology and shape of myelin outfoldings became more complex with age as shown in Figure 4A–D at 12–15 months of age. Signs of axonal degeneration or de-remyelination were occasionally noted (Fig. 4E). Reduction of the number of myelinated axons was also observed in these mutants at 12 months of age in distal nerves such as plantar nerves of the first toe (data not shown). Teased fiber analysis as well as longitudinal semithin section of sciatic nerves at 6 and 12 months of age, respectively, demonstrated that redundant loops of myelin arise at Schmidt–Lanterman incisures in addition to paranodal/juxtaparanodal regions, as observed in Mtmr2Fl/nullneo//P0-Cre mouse nerves (Fig. 4F–J). Thus, the dysmyelinating neuropathy of Mtmr2-nullneo mice is very similar to that of Mtmr2Fl/nullneo//P0-Cre mice, with Mtmr2-specific ablation in Schwann cells.
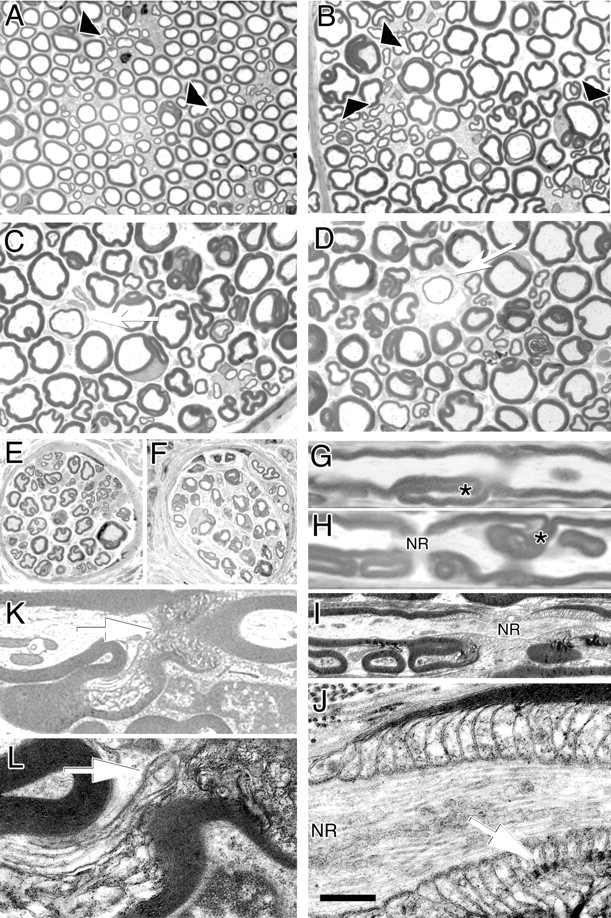
Morphological analysis of Mtmr2Fl/nullneo//P0-Cre mice. A–D, Semithin analysis of sciatic nerves from mutant mice in a transverse section at 7 weeks and at 6, 12, and 15 months. Arrowheads indicate myelin outfoldings and redundant loops. Arrows indicate remyelinated axons at older ages. E, F, Digital nerves (plantar) from normal (E) and mutant (F) mice at 12 months of age. G, H, Semithin analysis of sciatic nerves in a longitudinal section at 12 months of age. Myelin infoldings (H) and outfoldings (G), which arise at Schmidt–Lanterman incisures, are indicated by an asterisk. H, Myelin outfoldings also arise at paranodal regions. NR, Node of Ranvier. I, J, Electron microscopy of a sciatic nerve analyzed at 12 months of age showing myelin outfoldings at paranodal regions (I), which appeared normal with axo-glial junctions and adherens junctions in register (arrow) (J, enlarged). K, L, Electron microscopy showing myelin outfoldings at Schmidt–Lanterman incisures (arrows), enlarged in L. In the outfold, beneath the compact myelin sheath, Schwann cell cytoplasm containing vesicles and axoplasm contiguous with the main myelinated axon might be observed. Scale bar: A–F, 25 μm; G, H, 6 μm; I, 2 μm; J, 400 nm; K, 3.5 μm; L, 1.5 μm.
Disruption of Mtmr2 in motor neurons
To assess whether axons contribute to the CMT4B1 pathology and whether Mtmr2 has a cell-autonomous role in neurons, we also generated mice with Mtmr2-specific inactivation in motor neurons.
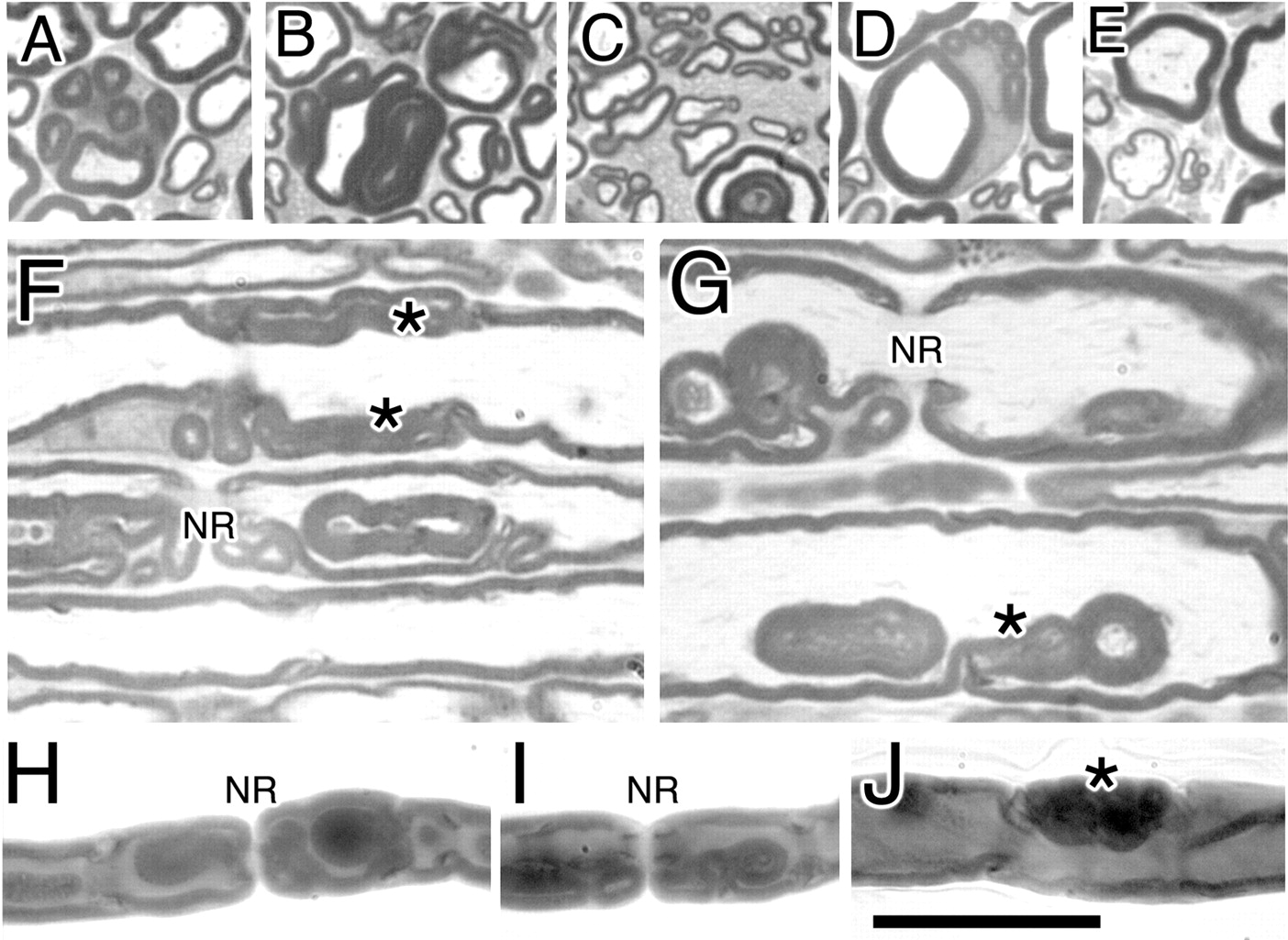
Morphological analysis of Mtmr2-nullneo mice at 12 and 15 months of age. A–E, Different shape of myelin outfoldings in Mtmr2-nullneo mouse sciatic nerves at older ages when the complexity of the dysmyelinating phenotype progresses. In A and D, upto five satellite loops are noted around a main myelinated fiber, which also appear around the nucleus (D). C, Myelin outfoldings in small caliber-myelinated fibers are shown. E, A thin myelinated fiber is depicted. F, G, Semithin analysis in a longitudinal section of mutant sciatic nerves at 12 months of age. Asterisks indicate myelin loops arising at Schmidt–Lanterman incisures. H–J, Teased fiber analysis of Mtmr2-nullneo sciatic nerves at 6 months of age. Myelin outfoldings originate at paranodal regions but also at Schmidt–Lanterman incisures (asterisk). NR, Node of Ranvier. Scale bar: (in J) A–E, 30 μm; F–J, 8 μm.
Mtmr2Fl/nullneo//HB9-Cre mice are viable and born according to the expected ratio. Mutant and normal littermates showed comparable weights at the different time points. No signs of tremor or functional disability were observed on these mutants examined at different ages. Rotarod tests performed at 6 months of age did not reveal any significant difference between mutant and control mice (Fig. 5A). Accordingly, no alterations in the motor nerve conduction velocity and F-wave latency, which represent electrophysiological properties of motor axons, were demonstrated in Mtmr2Fl/nullneo//HB9-Cre mice at 6 months of age (Fig. 5B). Transverse semithin section analysis of sciatic, quadriceps, saphenous, and digital nerves were performed at different time points (1, 2, 4, 6, 8, and 12 months). Myelin outfoldings and infoldings were not demonstrated in nerves of these mutants (Fig. 6A–I). No signs of axonal degeneration were observed. The number of myelinated axons in plantar nerves of the first toe was similar to that of controls (∼80 fibers in two plantar nerves). A morphometric analysis was performed on sciatic and quadriceps nerves of mutant mice and controls at 8 months of age. No differences in the axonal diameter and g-ratio were noted in either sciatic or quadriceps nerves (data not shown). Myelin structure also appeared normal when observed by electron microscopy (Fig. 6L,M). Moreover, no axonal swelling or any evident alteration in the axoplasm was noted. In peripheral neuropathies, loss of peripheral nerve fibers causes a rearrangement of the target muscle, including atrophic angulated fibers and, eventually, type grouping, resulting from degenerative-regenerative processes and collateral sprouting. No atrophic fibers and fiber-type grouping were noted in quadriceps muscle samples from mutant mice (Fig. 6J,K). In conclusion, Mtmr2Fl/nullneo//HB9-Cre mice do not show either dysmyelination or axonopathy.

Behavioral and electrophysiological analysis of Mtmr2Fl/nullneo//HB9-Cre mice. A, Rotarod test analysis performed on mutant and control animals at 6 months of age. No significant differences were observed in motor performances using the accelerating rod method. B, Electrophysiological analysis of Mtmr2Fl/nullneo//HB9-Cre mice and controls at 6 months of age. NCV, F-wave latencies, and CMAP registered in mutant animals were similar to controls.
Loss of Mtmr2 is associated with defects in non-compact myelin
Inactivation of Mtmr2 in Schwann cells and not in motor neurons causes myelin outfoldings and redundant loops of myelin, which arise at regions of non-compact myelin such as paranodal/juxtaparanodal regions and Schmidt–Lanterman incisures in both Mtmr2-null and Mtmr2Fl/nullneo//P0-Cre mouse nerves. Western blot analysis on sciatic nerve homogenates from Mtmr2-nullneo mice demonstrated decreased levels of both MAG and Dlg1, an interactor of Mtmr2 in Schwann cells located at regions of non-compact myelin (Bolino et al., 2004). To evaluate the distribution of these molecules on normal and Mtmr2-nullneo nerves, we performed immunohistochemistry on mouse teased fibers. Dlg1 stains cytoplasm at paranodal regions of Schwann cells as well as flanking incisures (Fig. 7A–A”). In Mtmr2-nullneo nerves, a faint Dlg1 signal was still detected at paranodes, whereas it was completely absent from flanking incisures (Fig. 7B–B”). Consistent with this, the Dlg1 average staining intensity at paranodes of control fibers was 58.1 ± 3.7 pixel/μm2, whereas in mutant fibers, it was 10.2 ± 1.7 pixel/μm2 (p < 0.0001). MAG was properly distributed at incisures, ad-axonal Schwann cell membrane, and paranodal loops of Mtmr2-nullneo fibers (Fig. 7C–C”, D–D”). However, staining intensity at paranodal loops appeared to be reduced in mutant nerves compared with controls (Fig. 7D–D”). Quantification of MAG immunostaining demonstrated that the average of the staining intensity at paranodes was 45.3 ± 2.7 pixel/μm2 in control fibers and 19.7 ± 1.6 pixel/μm2 in mutant nerves (p < 0.0001).
Myelin outfoldings arise at paranodal regions and at Schmidt–Lanterman incisures in Mtmr2-nullneo nerves. Electron microscopy performed on Mtmr2-nullneo fibers demonstrated that both electron-dense adherens junctions between loops of Schwann cell membrane as well as axo-glial junctions between Schwann cell and axolemma in paranodal regions were in register and normally formed. Architecture of Schmidt–Lanterman incisures was preserved in longitudinal sections of Mtmr2-nullneo mouse nerves. To better evaluate the molecular organization of these regions in mutant nerves, we observed the distribution and staining intensity of E-cadherin, a known component of adherens junctions in Schwann cells (Fannon et al., 1995) and of Caspr (Paranodin)-demarcating axo-glial junctions on the axolemma. Normal E-cadherin expression in Mtmr2-nullneo nerves was detected at Schmidt–Lanterman incisures, mesaxons, and as a more punctuate staining at paranodal loops of Schwann cells (Fig. 7E–E”, F–F”). A normal level of E-cadherin expression was further confirmed by Western blot analysis on sciatic nerve homogenates (data not shown). No difference in number and staining intensity of Caspr clusters was detected between mutant and normal teased fibers. No dispersion of Caspr clusters into the nodal region was noted (Fig. 7, compare C' and D', E' and F', G' and H').

Morphological analysis of Mtmr2Fl/nullneo//HB9-Cre mice. A–D, Semithin analysis of sciatic nerves from mutants at 1 month (A), 2 months (B), 6 months (C), and 12 months (D) of age. No myelin outfoldings were noted. E–G, Quadriceps nerves from mutant mice at 5 months (E), 8 months (F), and 12 months (G) of age and a saphenous nerve at 5 months of age (H) in which no myelin outfoldings were observed. I, Digital nerve at 12 months without signs of dysmyelination (plantar nerve with ∼40 myelinated fibers). J, K, ATPase isozyme staining of posterior compartment muscles of the leg shows that no type grouping occurred. L, M, Electron microscopy of quadriceps nerves at 12 months of age in which compact-myelin structure appears normal, and no axonal swelling was noted in the axoplasm. Scale bar: (in M) A–I, 25 μm; J, K, 80 μm; L, 8 μm; M, 3.5 μm.
Although the architecture of nodal and paranodal regions appeared normal in Mtmr2-nullneo nerves, a significant decrease in nerve conduction velocity (∼19%) was demonstrated in both Mtmr2-nullneo and Mtmr2Fl/nullneo//P0-Cre mutants. To assess whether this reduction might be related to the alteration of the number and/or distribution of NaCh and/or Kv1.1/Kv1.2 channel clusters, an expression analysis of NaCh and Kv1.1/Kv1.2 clusters was also performed. No difference in the number and staining intensity of NaCh and Kv1.1/Kv1.2 clusters was noted between mutant and normal fibers. NaCh clusters appeared normally distributed in the nodal region (Fig. 7G–G”, H–H”). Kv1.1/Kv1.2 cluster staining was also properly detected at juxtaparanodal regions, and no dispersion throughout the internode was observed (Fig. 7I–I”, J–J”). Consistently, NaCh cluster size, Caspr cluster distance, Caspr cluster length, Kv1.1/Kv1.2 cluster length, and Kv1.1/Kv1.2 cluster distance showed similar values between normal and mutant fibers (Fig. 8).
Expression analysis of members of the MTMR family
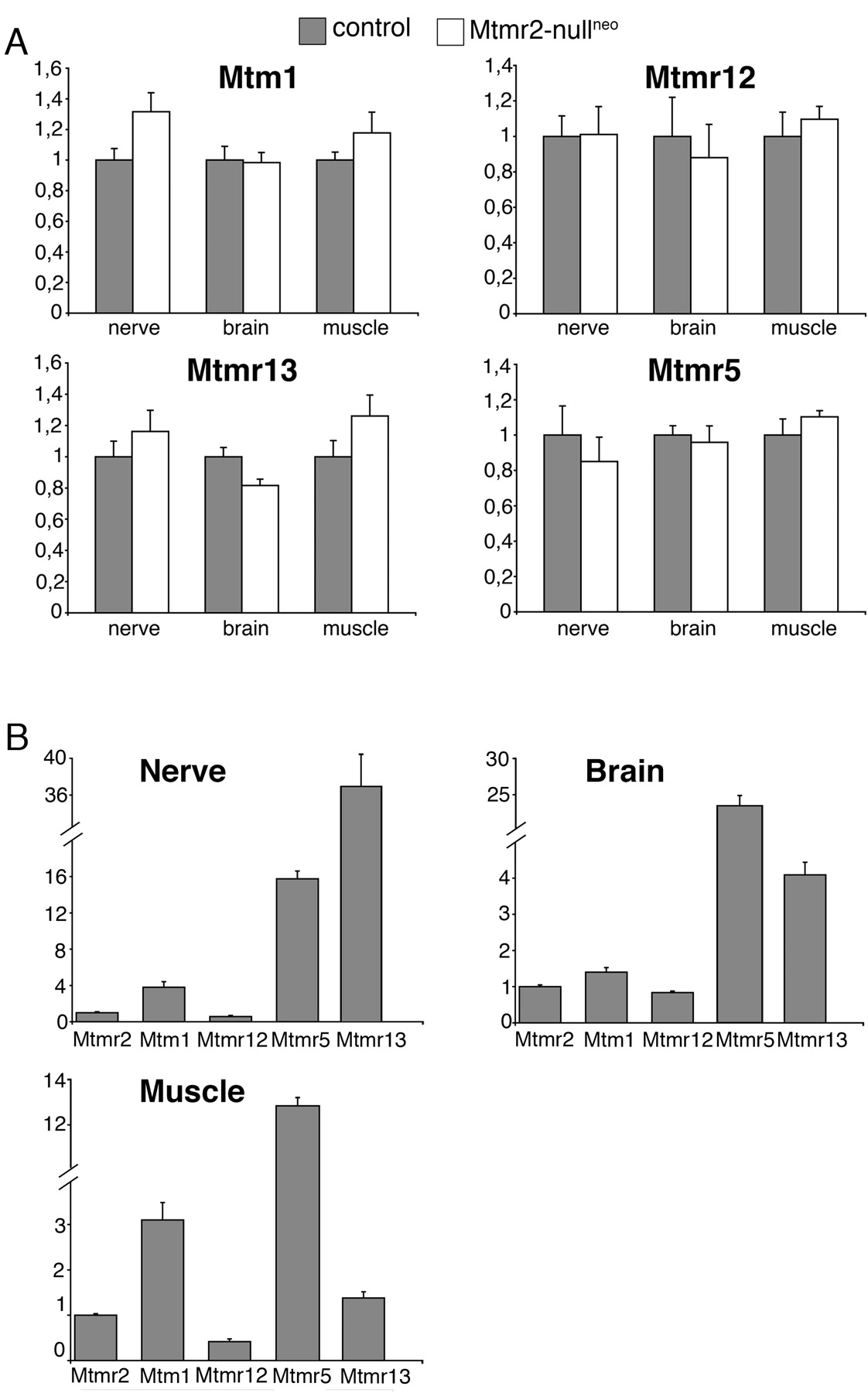
Overall, the neuropathy observed in both Mtmr2-nullneo and Mtmr2Fl/nullneo//P0-Cre mice is less severe than human CMT4B1. To evaluate the possibility of a partial compensation by Mtm1, highly homologous to Mtmr2 and also expressed in myelin-forming Schwann cells (Bolino et al., 2004), we performed a quantitative expression analysis of Mtm1 mRNA in sciatic nerve by RT-PCR. Mtm1 expression level was significantly increased in mutant nerves compared with controls (1.32-fold increase; p < 0.05) (Fig. 9A). It has been reported that Mtm1 might heterodimerize in vitro with its catalytically inactive partner Mtmr12 (Nandurkar et al., 2003). To test the hypothesis that Mtm1 in the nerve might compensate loss of Mtmr2 only if Mtmr12 is also expressed, we first confirmed Mtmr12 expression in Schwann cells by qualitative RT-PCR analysis. Mtmr12 expression levels were then quantified, and no difference was observed between normal and mutant nerves (Fig. 9A). Levels of both Mtm1 and Mtmr12 mRNAs were also measured in other tissues, such as brain and muscle, which are not affected in CMT4B1. The expression levels of these two genes were similar in mutant and normal tissues, suggesting that the lack of phenotype in other tissues in which Mtmr2 is absent might not be attributable to functional compensation by Mtm1 (Fig. 9A).
Catalytically inactive MTMR5 interacts with MTMR2 in vitro (Kim et al., 2003). However, in vivo, the most likely interactor of MTMR2 in the nerve is the catalytically inactive protein MTMR13, because mutations in MTMR13 also cause CMT4B2 with myelin outfoldings. One might hypothesize that loss of Mtmr2 in mouse specifically affects Schwann cells and not other cell types because of the different tissue distribution of the two catalytically inactive partners Mtmr5 and Mtmr13. However, both genes were detected in muscle, brain, and nerve, and no significant changes in expression levels were appreciated between wild-type and mutant samples (Fig. 9A). The normal expression levels of Mtm1, Mtmr2, Mtmr5, Mtmr12, and Mtmr13 were also compared in the different tissues analyzed (Fig. 9B). In conclusion, we provide evidence that a significant increase of Mtm1 expression in Mtmr2-nullneo nerves might partially compensate loss of Mtmr2 in mouse Schwann cells.
Discussion
Mtmr2 conditional disruption in Schwann cells but not in motor neurons provokes dysmyelination
We previously reported that Mtmr2 inactivation in mice reproduces CMT4B1 disease. Mtmr2-nullneo mice develop a dysmyelinating neuropathy with myelin outfoldings and infoldings, which arise (starting from 3–4 weeks after birth) at paranodal/juxtaparanodal regions and at Schmidt–Lanterman incisures. Whether Schwann cells and/or neurons contribute to the dysmyelination remained to be assessed. A Schwann cell-autonomous role for Mtmr2 was suggested by the interaction between Mtmr2 and Dlg1/SAP97, a MAGUK (membrane associated guanilate kinase-like) protein specifically detected in the cytoplasm of both myelin-forming and nonmyelin-forming Schwann cells and not localized in axons (Bolino et al., 2004). In Mtmr2-null mouse nerves, Dlg1 staining at paranodal cytoplasm as well as at flanking incisures is almost lost. However, other evidence supports a cell autonomous role of Mtmr2 in neurons. Expression of Mtmr2 has been detected in vivo in axons, motor neurons, and sensory neurons. Also, we provided evidence that Mtmr2 might interact in neurons with NF-L, a protein mutated in either pure axonal forms of CMTs or in more severe forms with signs of primary demyelination (Jordanova et al., 2003; Previtali et al., 2003).

Expression analysis of nodal, paranodal, and juxtaparanodal markers in Mtmr2-nullneo sciatic nerves. Immunohistochemistry was performed on teased fibers from Mtmr2-nullneo and normal sciatic nerves at 2 months of age. The following stainings were performed: Dlg1, on normal (A–A“) and mutant (B–B”) fibers. Double labeling of Dlg1 (green) and NaCh channel clusters (red), which mark the node of Ranvier, was performed. Dlg1 is almost absent from paranodal loops and flanking incisures in Mtmr2-nullneo fibers (B–B“). MAG, on normal (C–C”) and mutant (D–D“) fibers. Double labeling of MAG (red) and Caspr (green), which mark paranodal regions on the axolemma, was performed. MAG staining is reduced in paranodes from null fibers (compare the merge in C” and D“). Reduced stainings of both Dlg1 and MAG in Mtmr2-nullneo fibers were further confirmed using quantification of the average signal intensity. E-cadherin (E-cadh) on normal (E–E”) and mutant (F–F“) fibers, double labeled with E-cadh (red) and Caspr (green), is shown. No difference in signal intensity and distribution was detected between normal and mutant fibers (compare the merge in E” with F“). NaCh clusters on normal (G–G”) and mutant (H–H“) fibers are shown. Double labeling of NaCh (red) and Caspr (green) was performed. Kv1.1/1.2 clusters (green) on normal (I–I”) and mutant (J–J“) fibers costained with NaCh (red) are shown. No difference in staining intensity or distribution of NaCh, Caspr, and Kv1.1/1.2 clusters was appreciated between normal and mutant fibers. Scale bar: (in J”) A–F, 10 μm; G–J”, 5 μm.
We now report the full characterization of two mouse models in which Mtmr2 was specifically disrupted in either Schwann cells or motor neurons. Mtmr2Fl/nullneo//P0-Cre mice develop a dysmyelinating neuropathy very similar to that observed in Mtmr2-nullneo nerves. Myelin outfoldings arise at paranodal regions and at Schmidt–Lanterman incisures. The number of outfoldings increases with age, and the complexity progresses, being more pronounced at distal nerves. Occasional small onion bulbs were also noted at 12 and 15 months. In this model, it is interesting that even when Mtmr2 is ablated specifically in Schwann cells, axoplasm is found in the outfolds and recurrent loops of myelin. Thus, a primary genetic defect only in Schwann cells produces compensatory growth of axonal membrane. This may have a functional consequence. Although NaCh and Kv1.1/Kv1.2 clusters at nodal and juxtaparanodal regions, respectively, are not altered, a significant decrease in nerve conduction velocity (∼19%) was demonstrated in Mtmr2-nullneo and Schwann cell-null mice. Axoplasm is found in most of the redundant loops observed in mutant nerves. It is possible that axonal branches will increase the capacitance, resulting in a decrease of the current spreading along the axon.

A, Analysis of protein clusters at nodal, paranodal, and juxtaparanodal regions of Mtmr2-nullneo sciatic nerves. B, Na cluster size (wild type, 1.25 ± 0.04μm; knock-out, 1.46 ± 0.04 μm average) was normalized using the Na cluster diameter for each node (wild type, 1.85 ± 0.05 μm; knock-out, 2.04 ± 0.06 μm average); n, Number of nodes analyzed. Caspr cluster length (wild type, 3.80 ± 0.09μm; knock-out, 4.06 ± 0.09μm average) was normalized using Caspr diameter (wild type, 2.59 ± 0.09μm; knock-out, 2.57 ± 0.07μm average). n refers to paranodes analyzed. Kv1.1/1.2 cluster length (wild type, 8.22 ± 0.47 μm; knock-out, 6.91 ± 0.40 μm average) was normalized with the Na cluster diameter (wild type, 2.75 ± 0.09 μm; knock-out, 2.48 ± 0.11 μm average). n refers to juxtaparanodes analyzed. Kv1.1/1.2 cluster distance (wild type, 6.47 ± 0.33 μm; knock-out, 6.54 ± 0.29 μm average) was normalized using Na cluster diameter as for Kv1.1/1.2 cluster length. n is the number of fibers analyzed. The difference in Kv1.1/1.2 cluster distance between mutant and normal fibers was not statistically significant.
In contrast, Mtmr2Fl/nullneo//HB9-Cre mouse nerves did not show any sign of the CMT4B1 dysmyelinating neuropathy. These findings suggest that loss of Mtmr2 in Schwann cells is sufficient and necessary to generate myelin outfoldings.
Axonal defects were not noted in Mtmr2Fl/nullneo//HB9-Cre mouse nerves. Therefore, what is the role of Mtmr2 in motor neurons? Two scenarios might be envisaged. First, Mtmr2 could have no cell-autonomous role in these cells. In this case, the marked axonal loss observed in CMT4B1 patient nerves might be the consequence of the extensive demyelination and not the effect of a cell-autonomous role of the phosphatase in motor neurons. It has to be noted that, contrary to human CMTs, demyelination in mice does not necessarily result in marked axonal degeneration (Martini, 1997). In Mtmr2-nullneo mouse nerves, occasional small onion bulbs were noted only at 12–15 months of age. In both Mtmr2-nullneo and Mtmr2Fl/nullneo//P0-Cre but not in Mtmr2Fl/nullneo//HB9-Cre, signs of axonal degeneration in sciatic nerves were noted at an older age when a reduction of the number of myelinated fibers in more distal nerves was also revealed. Second, Mtmr2 has a cell-autonomous role in motor neurons in both humans and mice. The lack of axonal defects in the Mtmr2Fl/nullneo//HB9-Cre mouse, which is generated by excision of exon 4 at E9.5, might suggest that other proteins are able to compensate Mtmr2 loss in motor neurons at this stage of development. Other possibilities might be that primary axonal neuropathies are not so easily reproducible in mice or that Mtmr2 function in motor neurons is related to particular physiological conditions of axons (for instance, after nerve injury), as reported previously for other proteins (Perlson et al., 2005). Additional experiments are necessary to test the latter hypothesis.
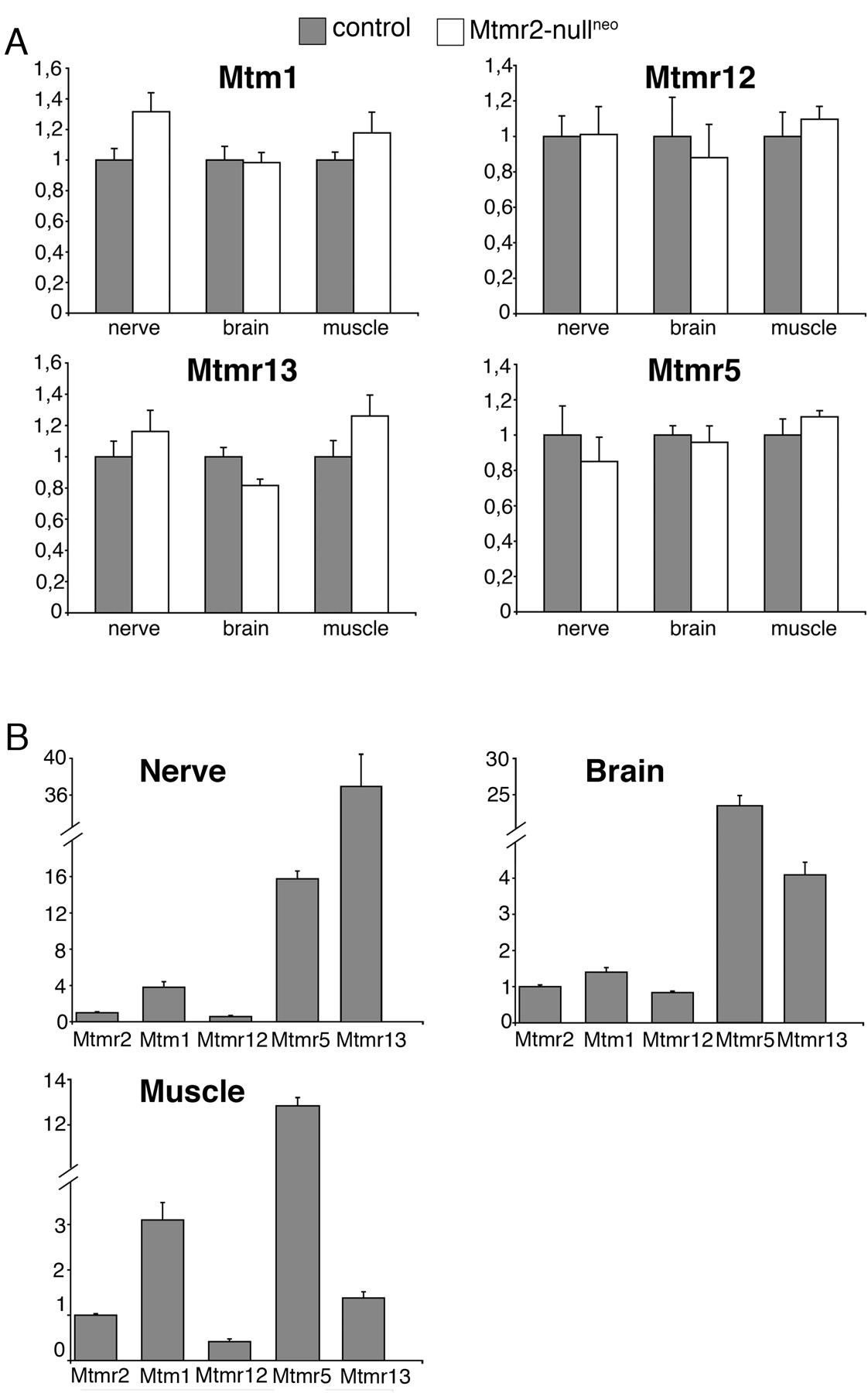
Expression analysis of MTMR family members. A, The expression of Mtm1, Mtmr12, Mtmr13, and Mtmr5 mRNAs was determined by RT-PCR in nerve, brain, and muscle from Mtmr2-nullneo and wild-type animals. TaqMan probes (Applied Biosystems) and the comparative CT method (ABI PRISM Sequence Detection System User Bulletin 2; Applied Biosystems) were used. mRNA expression levels (2–ΔΔCT values) in mutant tissues are indicated as n-folds with respect to the levels in normal tissues, taken as 1. Diagrams show the average of expression levels calculated on at least three animals per genotype in muscle and brain and, for the nerve, on at least three separate pools of two animals each. SD was calculated for triplicate samples of each reaction, and SEM is indicated on the media of the determinations from different animals. B, mRNA expression levels of Mtmr2, Mtm1, Mtmr12, Mtmr5, and Mtmr13 in normal nerve, brain, and muscle. Mtm1, Mtmr12, Mtmr5, and Mtmr13 levels are indicated relative to Mtmr2, taken as 1. The mRNA expression levels of different genes were compared at the same CT value, using the same endogenous references.
Alterations at sites of non-compact myelin
Decreased levels of MAG and Dlg1 were initially demonstrated by Western blot analysis on Mtmr2-nullneo nerve homogenates. Consistently, immunohistochemical localization of MAG and Dlg1 on teased fibers demonstrated a reduction of MAG staining at paranodal regions. Dlg1 was almost absent from paranodes and flanking Schmidt–Lanterman incisures of null nerves. These molecular alterations suggest a defect at the level of non-compact myelin in which myelin outfoldings arise (at paranodal regions and incisures) in both mutants, Mtmr2-nullneo, and Mtmr2Fl/nullneo//P0-Cre. Electron microscopy revealed that the general architecture of these regions is preserved in these mice. This observation was also confirmed by immunohistochemical analysis of E-cadherin and Caspr, NaCh, and Kv1.1/Kv1.2 clusters. Quantitative RT-PCR analysis of MAG and Dlg1 mRNA levels did not show any difference between wild-type and mutant nerves (data not shown). Therefore, reduced protein synthesis, increased degradation, or altered recruitment to incisures and paranodal loops might be responsible for reduction of MAG and Dlg1 levels. Dlg1 interacts with Mtmr2 in Schwann cells and, therefore, when Mtmr2 is lost, it is also possible that Dlg1 is degraded. Whether the decreased level of MAG in null nerves is a consequence of the dysmyelination or whether it might be functionally related to the Mtmr2/Dlg1 complex remains to be assessed. A generalized defect in transport to Schmidt–Lanterman incisures or paranodal loops is not likely to occur, because other molecules located at paranodal regions (e.g., Caspr) were normally expressed and localized in mutant nerves.
Mtmr2 is a phospholipid phosphatase showing preferential activity toward PtdIns3,5P2, a key regulator of vacuolar homeostasis and intracellular trafficking at the level of multivesicular bodies/late endosomes (Odorizzi et al., 1998). Dlg1/SAP97 is a component of adherens junctions in vertebrate epithelial cells, and it is involved in polarization, cell proliferation, and cancer (Humbert et al., 2003). In Schwann cells, Dlg1 is located at paranodal loops and Schmidt–Lanterman incisures in which several types of junctions are also located. Mtmr2/Dlg1 complex is disrupted in Schwann cells when Mtmr2 is absent. The pathogenesis of CMT4B1 is strictly related to the loss of Mtmr2/Dlg1 function in Schwann cells, perhaps involving membrane homeostasis/vesicular trafficking targeted to adherens junctions of paranodes and incisures where myelin outfoldings arise. The finding that CMT4B1 neuropathy is caused by loss of Mtmr2 specifically in Schwann cells might help to specifically direct the efforts aimed at clarifying the molecular mechanism at the basis of this pathology. Moreover, therapeutical approaches based on the delivery of the Mtmr2 enzyme in Schwann cells of affected nerves might be designed.
Footnotes
This study was supported by grants from Telethon, Italy (A. Bolino, S.C.P., M.L.F., and L.W.), the National Institutes of Health (M.L.F. and L.W.), Compagnia San Paolo (A. Bolino), and the Fondazione Mariani, Italy (L.W.). A. Bolino is a recipient of a Telethon Assistant Scientist Career Award. We acknowledge the kind collaboration of Silvia Arber (University of Basel, Basel, Switzerland) and Thomas Jessell (Columbia University, New York, NY), who provided the HB9-Cre transgenic mouse. We thank Marina Scarlato, Stefano Grassi, and Felipe Court (San Raffaele Scientific Institute, Milan, Italy) for their help. We also thank Elior Peles (The Weizmann Institute of Science, Rehovot, Israel) and Terunaga Nakagawa (Massachusetts Institute of Technology, Boston, MA) for antibodies.
Correspondence should be addressed to Dr. Alessandra Bolino, Dulbecco Telethon Institute, Department of Biological and Technological Research, San Raffaele Scientific Institute, Via Olgettina 58, 20132 Milano, Italy. E-mail: bolino.alessandra{at}hsr.it.
DOI:10.1523/JNEUROSCI.2493-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258567-11$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}