

Abstract
Systemic administration of anti-amyloid-β (Aβ) antibodies results in reduced parenchymal amyloid but increased vascular amyloid and microhemorrhage in amyloid precursor protein (APP) transgenic mice. Here, we evaluate the effects of reducing effector interactions of the antibody via deglycosylation. Mice aged 20 months were treated weekly for 4 months and tested behaviorally before they were killed. APP transgenic mice receiving either anti-Aβ (2H6) or deglycosylated anti-Aβ (de-2H6) showed significant improvement in radial arm water maze performance compared with mice receiving a control antibody. Both groups receiving anti-Aβ antibodies showed significant reductions in total Aβ immunochemistry and Congo red. Significantly fewer vascular amyloid deposits and microhemorrhages were observed in mice administered the de-2H6 antibody compared with those receiving unmodified 2H6 antibody. Deglycosylated anti-Aβ antibodies may be preferable to unmodified IgG because they retain the cognition-enhancing and amyloid-reducing properties of anti-Aβ immunotherapy, while greatly attenuating the increased vascular amyloid deposition and microhemorrhage observed with unmodified IgG.
Introduction
For the past 7 years, immunotherapy has been investigated as a potential treatment for Alzheimer’s disease (AD) since it was initially described in 1999 (Schenk et al., 1999). Immunotherapy targets the amyloid-β (Aβ) protein, which forms amyloid plaques in the brains of AD patients and, according to the amyloid hypothesis, is thought to instigate a sequence of molecular and cellular events resulting in the pathology of AD (Hardy and Selkoe, 2002). Early reports of the effectiveness of immunotherapy used an active vaccination protocol in amyloid precursor protein (APP) (Janus et al., 2000) and APP/presenilin 1 (Morgan et al., 2000) transgenic mouse models, in which the Aβ peptide was injected along with an adjuvant, resulting in the production of circulating anti-Aβ antibodies. This active immunization later advanced to human clinical trials in which it was withdrawn in phase 2A because of a small but significant subset of patients developing meningoencephalitis (Orgogozo et al., 2003). Reports of pathology from several autopsies of patients receiving the immunization suggested that amyloid pathology may have been improved in these patients, despite an apparent autoimmune reaction resulting in CNS T-cell infiltration (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). More recently, passive immunization, in which anti-Aβ antibodies are administered, has been shown to be equally, if not more, effective at both reducing amyloid pathology and improving cognition than the active immunization (Bard et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002; Wilcock et al., 2004b). Given the lack of T-cell activation by this method, it is less likely to provoke an autoimmune reaction against Aβ than active immunization. However, several groups, including ours, have shown that systemic administration of anti-Aβ antibodies in three different APP transgenic mouse models increased the incidence of brain microhemorrhage in association with amyloid-laden vessels (Pfeifer et al., 2001; Wilcock et al., 2004c; Racke et al., 2005).
In the current study, we aimed to understand the basis for the increased vascular leakage, an effect likely undesirable in a human population. One question regarded the role of antibody interaction with its molecular effectors such as Fcγ receptors and the complement cascade. IgG molecules have a single carbohydrate moiety attached to the Fc region of the molecule, which is important in antibody binding to the Fcγ receptors. Deglycosylation results in defective binding of the Fcγ receptor to the antigen–antibody complex (Radaev and Sun, 2001) and impaired binding to complement C1q (Winkelhakeet al., 1980). In previous work (Carty et al., 2006), we demonstrated that deglycosylating anti-Aβ antibodies decreased their affinities for several effectors and diminished their activation of microglial cells in vivo, yet still retained amyloid clearing properties when injected intracranially. Here we show that long-term systemic administration of deglycosylated anti-Aβ effectively removes amyloid deposits in old APP transgenic mice despite impaired effector cell interaction. We also show that this deglycosylated antibody significantly reduces microhemorrhage incidence compared with the unmodified antibody and is associated with lower levels of cerebral amyloid angiopathy (CAA).
Materials and Methods
Experiment design.
Mice derived from APP Tg2576 mice (Hsiao et al., 1996) were obtained from our breeding program at University of South Florida started in 1996. Twenty APP transgenic mice aged 20 months were assigned to one of three groups. Two groups received weekly intraperitoneal injections of 2H6 (anti-Aβ33–40; IgG2b isotype; 9 nm affinity; n = 4; Rinat Neurosciences, San Francisco, CA) or de-2H6 (deglycosylated Aβ33–40; n = 5; Rinat Neurosciences). A third group received weekly intraperitoneal injections of 2908 [mouse monoclonal anti-Drosophila amnesiac protein (AMN); IgG2b isotype; n = 7; Rinat Neurosciences] for a period of 4 months. Six nontransgenic mice also aged 20 months were injected weekly with 2H6 or 2908 for a period of 4 months. The dose for all antibodies was 10 mg/kg.
Deglycosylation of 2H6.
A deglycosylated version of 2H6 (de-2H6) was generated by enzymatic removal of N-linked glycans. 2H6 was incubated at 37°C for 1 week with peptide-N-glycosidase F (0.05 U/mg antibody; QA-Bio, San Mateo, CA) in 20 mm Tris-HCl, pH 8.0, and 0.01% Tween. The deglycosylated antibody was purified by protein A chromatography, and endotoxin was removed by Q-Sepharose. Completeness of deglycosylation was verified by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and protein gel electrophoresis (Carty et al., 2006).
Behavioral analysis.
After the 4 months of treatment, the mice were subjected to a 2 d radial arm water maze paradigm, followed by 1 d of an open-pool visible platform task. The apparatus was a six-arm maze as described previously (Gordon et al., 2001). The radial arm water maze task was run as described previously (Wilcock et al., 2004c). Briefly, on day 1, 15 trials were run in three blocks of five. Mice were further run in cohorts of four mice, permitting a short rest between each trial (as the other three mice were run) and a longer break between the blocks, when another cohort of four mice were run. This permitted rapid testing of even aged mice without development of fatigue. Moreover, the spaced practice of the trials appears to enhance the rate of acquisition compared with daily massed trials over 10–14 d. The start arm was varied for each trial, with the goal arm remaining constant for both days. For the first 11 trials, the platform was alternately visible and then hidden and remained hidden for the last four trials. On day 2, the mice were run in exactly the same manner as day 1, except that the platform was hidden for all trials. The numbers of errors (incorrect arm entries) were measured in each trial over a 1 min time frame. As in previous studies, to avoid confounds caused by inactive mice, we assigned one error for every 20 s a mouse failed make an arm selection. However, no mice in this study had to be assigned an error in this manner. Each mouse’s errors for three consecutive trials were averaged, producing five blocks of trials for each day. Trial blocks were analyzed statistically by ANOVA using StatView (SAS Institute, Cary, NC). After the 2 d of radial arm water maze, the mice received 15 trials in an open-pool task with a visible platform to identify whether poor scores in the radial arm maze could be attributed to sensory or performance deficits rather than memory impairment. In the current study, all mice performed well on the visible platform version of the maze, and none were excluded because of sensory or performance deficits.
ELISA analysis of serum Aβ.
Serum was diluted and incubated in 96-well microtiter plates (MaxiSorp; Nunc, Rosklide, Denmark), which were precoated with antibody 6E10 (Signet, Dedham, MA) at 5 μg/ml in PBS buffer, pH 7.4. The secondary antibody was biotinylated 4G8 (Signet) at a 1:5000 dilution. Detection was done using a streptavidin–horseradish peroxidase conjugate (Amersham Biosciences, Arlington Heights, IL), followed by tetramethylbenzidine substrate (Sigma-Aldrich, St. Louis, MO). Standard curves of Aβ1–40 (American Peptide, Sunnyvale, CA) scaling from 6 to 400 pm were used.
Tissue preparation and histology.
On the day the mice were killed, they were weighed, overdosed with 100 mg/kg Nembutal sodium solution (Abbott Laboratories, North Chicago, IL) and intracardially perfused with 25 ml of 0.9% sodium chloride. Brains were rapidly removed, and the left half of the brain was immersion fixed for 24 h in freshly prepared 4% paraformaldehyde in 100 mm KPO4, pH 7.2, for histopathology. The hemibrains were then incubated for 24 h in 10, 20, and 30% sucrose sequentially to cyroprotect them. Horizontal sections of 25 μm thickness were collected using a sliding microtome and stored at 4°C in Dulbecco’s PBS with sodium azide, pH 7.2, to prevent microbial growth. A series of eight equally spaced tissue sections 600 μm apart were randomly selected spanning the entire brain and stained using free-floating immunohistochemistry for total Aβ [rabbit polyclonal anti-Aβ, raised at University of South Florida, 1:10,000 dilution (described by Gordon et al., 2001)] and CD45 (rat monoclonal anti-CD45, 1:5000 dilution; Serotec, Raleigh, NC) as described previously (Gordon et al., 2001; Wilcock et al., 2004b). A second series of tissue sections 600 μm apart were stained using 0.2% Congo red solution in NaCl-saturated 80% ethanol. Another set of sections were also mounted and stained for hemosiderin using 2% potassium ferrocyanide in 2% hydrochloric acid for 15 min, followed by a counterstain in a 1% neutral red solution for 10 min.
Quantification of Congo red staining, CD45 and Aβ immunohistochemistry was performed using the Image-Pro Plus (Media Cybernetics, Silver Spring, MD) software to analyze the percentage area occupied by positive stain. Images with a field size of 600,000 μm2 were collected using the 10× objective on an Olympus Optical (Thornwood, NY) microscope. One region of the frontal cortex and three regions of the hippocampus were analyzed (to ensure that there was no regional bias in the hippocampal values). The initial analysis of Congo red was performed to give a total value. A second analysis was performed after manually editing out all of the parenchymal amyloid deposits to yield a percentage area restricted to vascular Congo red staining. To estimate the parenchymal area of Congo red, we subtracted the vascular amyloid values from the total percentage. For the hemosiderin stain, eight equally spaced sections were examined, the numbers of Prussian blue-positive deposits were counted on all sections, and the average number of hemosiderin deposits per section were calculated. To assess possible treatment-related differences, the values for each treatment group were analyzed by one-way ANOVA, followed by Fisher’s least significance difference means comparisons using StatView (SAS Institute).
Results
Both intact and deglycosylated antibodies eliminate spatial learning deficits
The radial arm water maze task is a behavioral test that reliably detects spatial learning and memory deficits in aged transgenic mice (Gordon et al., 2001). It has also been shown to detect elimination of these cognitive deficits after treatment with anti-Aβ immunotherapy (Morgan et al., 2000; Wilcock et al., 2004c). In the current study, APP transgenic mice were tested after 4 months of treatment with 2H6, de-2H6, or 2908. Included in the task were age-matched nontransgenic mice treated with either 2H6 or 2908. We found that the APP transgenic mice receiving 2908, the control antibody, were significantly impaired when compared with the nontransgenic mice (Fig. 1). However, APP transgenic mice treated with either the intact 2H6 or the de-2H6 performed significantly better than the control APP transgenic mice treated with 2908. Both groups treated with anti-Aβ antibodies were indistinguishable from the nontransgenic mice at the end of the second day of testing, averaging less than one error, our criterion for stable acquisition of this task (Fig. 1).

Deglycosylated and intact 2H6 anti-Aβ antibody eliminate spatial learning deficits in APP transgenic mice. Blocks are the average of three trials. The graph shows number of the average number of errors for APP transgenic mice treated with intact anti-Aβ antibody (○, dashed line), deglycosylated 2H6 anti-Aβ antibody (□, dashed line), or control 2908 antibody (▪, solid line) and nontransgenic mice receiving either control antibody or intact 2H6 anti-Aβ antibody (•, solid line). *p < 0.05, **p < 0.01 compared with APP transgenic mice receiving control antibody anti-AMN (2908). The level of significance is indicated for both the 2H6 and deglycosylated 2H6 anti-Aβ antibody treatment groups. Error bars indicate SEM.
Intact and deglycosylated antibodies increase levels of circulating Aβ
One day after the final antibody injection, circulating Aβ levels were dramatically increased in both anti-Aβ antibody treatment groups compared with the group receiving control antibody in which Aβ was barely detectable in the serum (Fig. 2). Mice treated with the intact 2H6 antibody showed a significantly greater increase (almost twofold) in circulating Aβ than the mice treated with the deglycosylated 2H6 (Fig. 2).
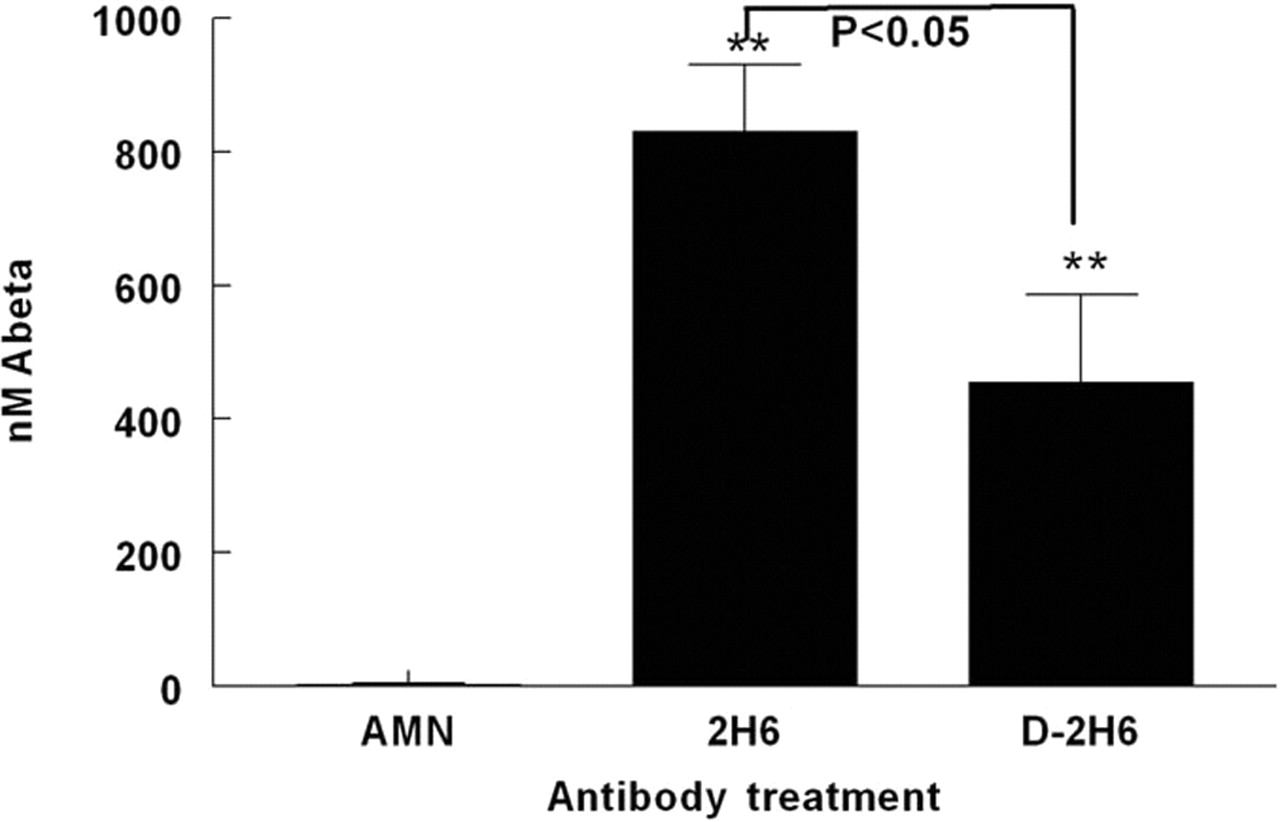
Deglycosylated (D-2H6) and intact 2H6 anti-Aβ antibody treatments result in increased circulating Aβ levels. Graph shows concentration of Aβ in serum 24 h after the final antibody injection. **p < 0.01 compared with mice receiving control antibody anti-AMN (2908). Also shown is the p value for the comparison between intact and deglycosylated anti-Aβ antibody treatments. Error bars indicate SEM.
Intact and deglycosylated antibodies reduce total Aβ in the brain
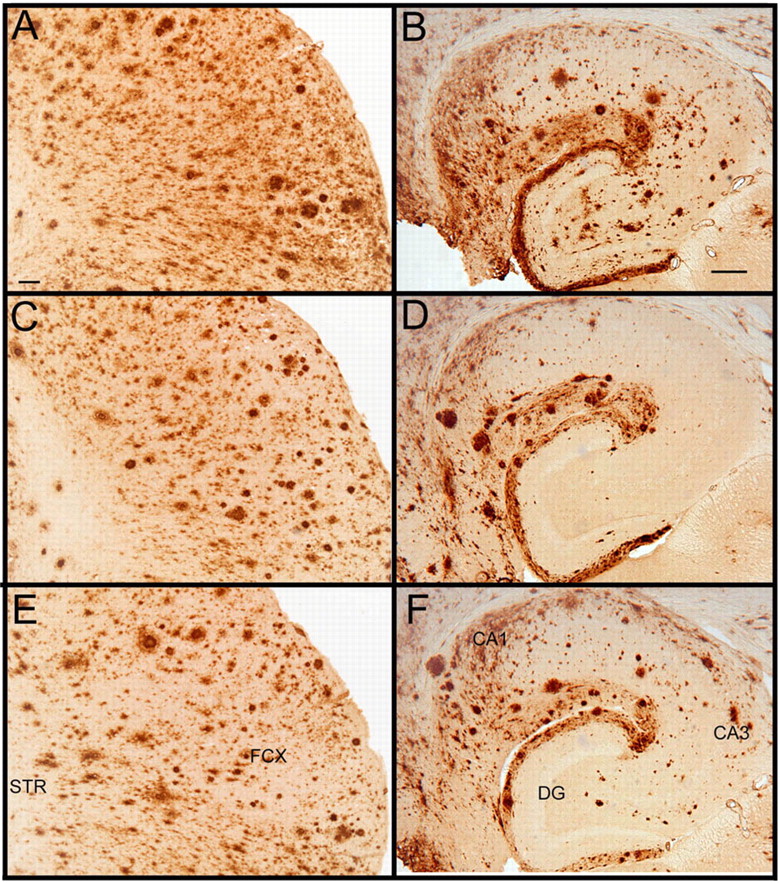
Immunohistochemistry detecting total Aβ, both compact and diffuse deposits, in APP transgenic mice treated with control antibody showed a typical staining pattern for mice of this age. In the frontal cortex, we observed high levels of diffuse Aβ staining as well as less frequent, more intensely stained deposits that are typically found to be positive when stained with Congo red or thioflavine-S (Fig. 3A). In the hippocampus, Aβ deposits are concentrated around the hippocampal fissure and the CA1 region with fewer deposits throughout the dentate gyrus and remainder of hippocampal tissue (Fig. 3B). APP transgenic mice treated with anti-Aβ antibodies 2H6 (Fig. 3, frontal cortex, C; hippocampus, D) or deglycosylated 2H6 (Fig. 3, frontal cortex, E; hippocampus, F) showed significant reductions in total Aβ immunohistochemistry of similar magnitude in both the frontal cortex and hippocampus with both antibodies. Quantification of the percentage area occupied by positive staining showed a 50% reduction in total Aβ in the frontal cortex (Fig. 4A) and a 55% reduction in the hippocampus (Fig. 4B).

Total Aβ immunohistochemistry is reduced by systemic administration of anti-Aβ antibodies. A–F show Aβ immunohistochemistry from APP transgenic mice in the frontal cortex (A, C, E) and hippocampus (B, D, F). Mice were treated with control antibody anti-AMN (2908) (A, B), intact anti-Aβ antibody 2H6 (C, D), or deglycosylated anti-Aβ antibody 2H6 (E, F). Scale bars: A (for A, C, E), B (for B, D, F), 120 μm. FCX, Frontal cortex; STR, striatum; CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus.
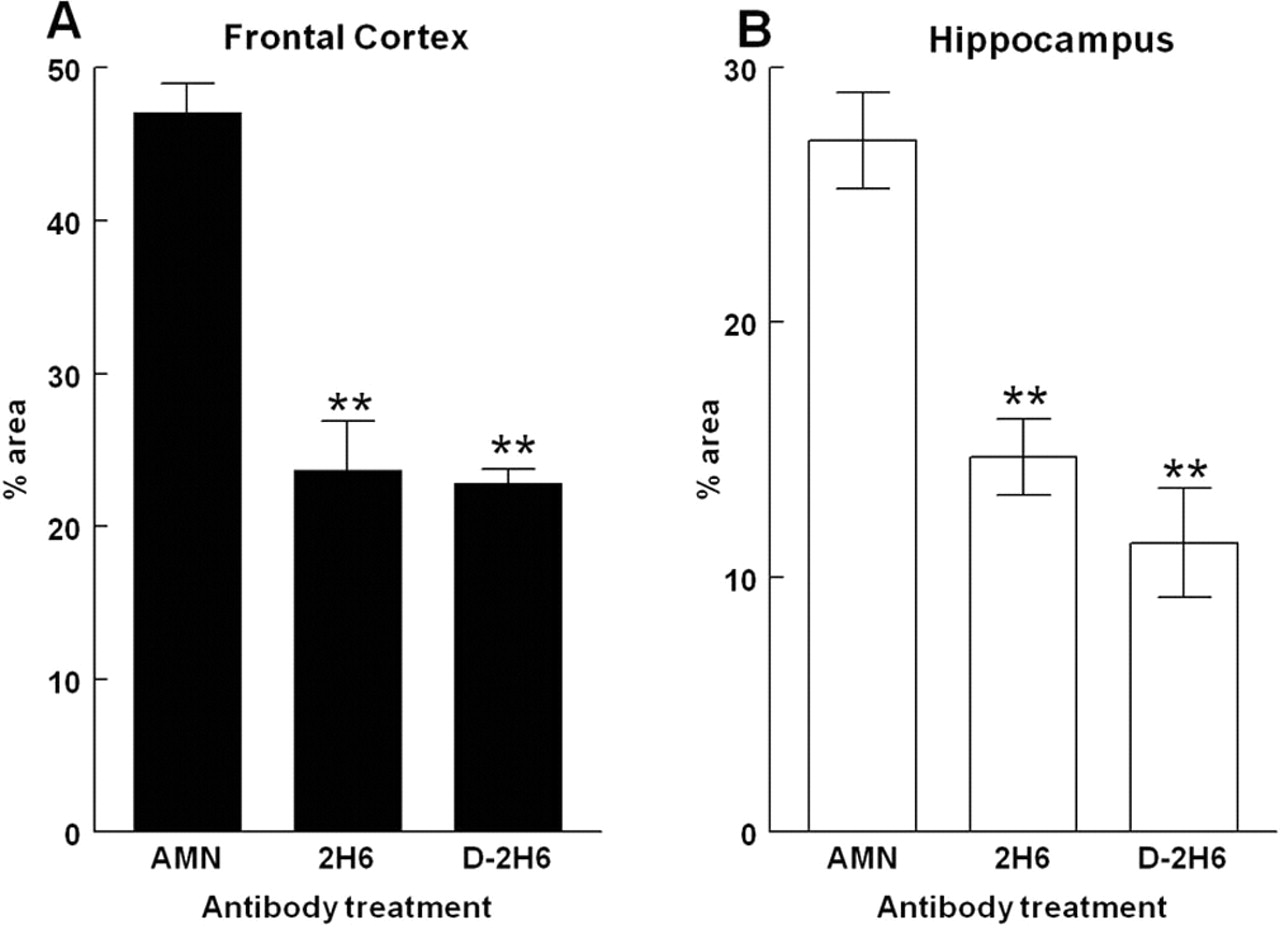
Total Aβ immunohistochemistry is significantly reduced in both the frontal cortex and hippocampus by systemic treatment with either intact or deglycosylated anti-Aβ (D-2H6) antibodies. A shows quantification of total Aβ immunohistochemistry in the frontal cortex. B shows quantification in the hippocampus. **p < 0.01 when compared with mice treated with control antibody anti-AMN (2908). Error bars indicate SEM.
Compact Congophilic amyloid deposits are reduced by intact or deglycosylated anti-Aβ 2H6 antibody administration, whereas vascular amyloid is increased
In APP transgenic mice, the histological dye Congo red labels only compact amyloid deposits and stains ∼5% of the material stained by immunohistochemistry for total Aβ. As displayed in Figure 5, the distribution of Congophilic deposits resembled that observed for total Aβ. In the frontal cortex, compact amyloid deposits could be seen throughout the cortical tissue with several Congo red-positive blood vessels also showing Congo red staining (Fig. 5A). In the hippocampus, there was a concentration of deposits around the hippocampal fissure and also in the CA1 region (Fig. 5B). Mice treated with the 2H6 antibody showed significant reductions in compact amyloid deposits in both the frontal cortex and hippocampus, but there was an obvious increase in amyloid deposits in the vasculature of both regions (Fig. 5, frontal cortex, C; hippocampus, D). Mice treated with de-2H6 antibody also showed significant reductions in compact amyloid deposits in the frontal cortex and hippocampus, but more staining in the vasculature could also be observed in both regions (Fig. 5, frontal cortex, E; hippocampus, F).

Compact Congophilic amyloid deposits are reduced by intact or deglycosylated anti-Aβ 2H6 antibody administration, whereas vascular amyloid is increased. A, C, and E show Congo red staining in the frontal cortex. B, D, and F show Congo red staining in the hippocampus. Staining is shown from APP transgenic mice treated with control antibody anti-AMN (2908) (A, B), intact anti-Aβ antibody 2H6 (C, D), and deglycosylated anti-Aβ antibody (E, F). Scale bars: A (for A, C, E), B (for B, D, F), 120 μm. FCX, Frontal cortex; STR, striatum; CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus.
Quantification of total Congo red area stained shows significant reductions after 14 weeks of treatment of APP transgenic mice with either intact 2H6 [80% in frontal cortex and hippocampus (Fig. 6A)] or deglycosylated 2H6 [70% in frontal cortex and 55% in hippocampus (Fig. 6A)]. Parenchymal Congo red levels were calculated by subtracting vascular Congo red values from total Congo red values for each section. These show that dramatic reductions in Congophilic compact plaque loads in the group treated with 2H6 was dramatically reduced [95% in both frontal cortex and in hippocampus (Fig. 6B)]. The reduction of parenchymal deposits after de-2H6 treatment was less robust than that observed with the intact 2H6 but was still highly significant [75% in frontal cortex and 65% in hippocampus Fig. 6B)]. When we quantified vascular Congophilic staining by manually deselecting the parenchymal deposits, we observed significant increases in APP transgenic mice treated with the intact 2H6 antibody. These increases were 3.5-fold in the frontal cortex and threefold in the hippocampus (Fig. 6C). In comparison, mice treated with the de-2H6 antibody showed significantly less vascular amyloid than the mice treated with the unmodified 2H6 antibody (Fig. 6C). Nonetheless, the vascular deposits in the mice treated with de-2H6 were increased twofold in the frontal cortex and 1.5-fold in the hippocampus (Fig. 6C) when compared with the mice administered the control antibodies.

Total and parenchymal Congo red staining of APP transgenic mice is reduced, whereas vascular Congo red is increased after treatment with intact or deglycosylated anti-Aβ 2H6 (D-2H6). A shows quantification of total Congo red in the frontal cortex (filled bars) and hippocampus (open bars). B shows quantification of parenchymal Congo red in the frontal cortex (filled bars) and hippocampus (open bars). C shows quantification of vascular Congo red in the frontal cortex (filled bars) and hippocampus (open bars). *p < 0.05, **p < 0.01 when compared with mice receiving control antibody anti-AMN (2908). Error bars indicate SEM.
Intact anti-Aβ antibody 2H6 increases microhemorrhage levels in APP transgenic mice, whereas deglycosylation attenuates this effect
Associated with the increased vascular amyloid after intact 2H6 antibody treatment was a dramatic, 5.5-fold increase in the incidence of vascular microhemorrhage, as detected by Prussian blue staining, with approximately three microhemorrhages per hemibrain section examined (Fig. 7). These microhemorrhages were primarily located in the outer laminas of the cerebral cortex with occasional profiles observed in the hippocampal tissue. Because of the nature of the stain, we are unable to show the staining distribution because the profiles are small and undetectable at a low magnification. The incidence of microhemorrhage in the de-2H6-treated mice was 67% less than that found in the 2H6-treated mice. Nonetheless, this was still an increase in microhemorrhage compared with transgenic mice treated with the control antibody (Fig. 7).

Intact anti-Aβ antibody 2H6 increases microhemorrhage levels in APP transgenic mice, whereas deglycosylation (D-2H6) attenuates this effect, although still increased. Graph shows quantification of Prussian blue staining by measuring the total number of positive profiles per section analyzed. *p < 0.05, **p < 0.01 when compared with mice receiving control antibody anti-AMN (2908). Error bars indicate SEM.
Microglial activation is increased around remaining deposits in mice treated with intact 2H6 anti-Aβ antibodies
CD45 immunohistochemistry stains activated microglia and was shown to be increased 3 d after intracranial anti-Aβ antibody injection (Wilcock et al., 2003) and also after 2 months of systemic anti-Aβ antibody administration (Wilcock et al., 2004b). In the current study, we found no differences in the total levels of microglial activation (detected by percentage area of positive CD45 immunohistochemistry) after treatment with either the intact or the deglycosylated 2H6 antibodies. However, because of the dramatic reductions in Congophilic and total amyloid levels, one would predict that microglial activation caused by the amyloid deposits would decrease. When examining the sections stained by CD45, we observed an increase in the amount of staining as well as the intensity of CD45 staining around the few remaining amyloid deposits in mice receiving the intact 2H6 antibody (both parenchymal and vascular) in both the frontal cortex (Fig. 8A,C) and hippocampus (Fig. 8B,D). In calculating the ratio of CD45 to total Congo red to essentially give a value of microglial activation per deposit, we found that this ratio is significantly increased in those mice receiving the intact anti-Aβ antibody compared with those mice receiving control antibody in both the frontal cortex and hippocampus (Fig. 8E). The mice treated with deglycosylated antibody show a slight increase in this CD45 to Congo red ratio, although not statistically significant.

Microglial activation is increased around remaining deposits in mice treated with intact 2H6 anti-Aβ antibodies. A–D show CD45 immunohistochemistry in APP transgenic mice from the frontal cortex (A, C) and hippocampus (B, D). Staining is shown from mice receiving control antibody anti-AMN (2908) (A, B) and intact 2H6 anti-Aβ antibody (C, D). Scale bar, 50 μm. F, Hippocampal fissure; DG, dentate gyrus. E shows ratio of CD45/total Congo red in the frontal cortex and hippocampus. *p < 0.05, **p < 0.01 when compared with mice receiving control antibody anti-AMN (2908). D2H6, Deglycosylated anti-Aβ 2H6. Error bars indicate SEM.
Discussion
We have shown previously that passive amyloid immunotherapy reduces diffuse and compact amyloid deposits, transiently activates microglia (Wilcock et al., 2004b,c), and increases CAA and the incidence of microhemorrhage (Wilcock et al., 2004c). We also shown elimination of cognitive deficits using the radial arm water maze paradigm (Wilcock et al., 2004c). In the current study, we have shown that deglycosylation of an anti-Aβ antibody retains the ability of the antibody to reduce amyloid deposition and eliminate cognitive deficits with considerable reduction in the potentially adverse vascular changes, such as CAA and microhemorrhage.
IgG antibody molecules are glycoproteins with a conserved glycosylation site in the CH2 domain. This carbohydrate chain enhances the interaction of IgG with Fcγ receptors on effector cells such as macrophages and microglia (Radaev and Sun, 2001). It has been shown previously that deglycosylation of human IgG1 and IgG3 monoclonal antibodies inhibited interaction with Fcγ receptors I and II using human monocytic cells (Walker et al., 1989). Deglycosylation of IgG has also been shown to impair the ability of IgG to bind complement component C1q (Winkelhakeet al., 1980). The advantage of deglycosylation over the generation of F(ab′)2 fragments for evaluating the importance of antibody interactions with effector molecules is that the deglycosylated IgG has a comparable plasma half-life to the intact IgG molecule, whereas F(ab′)2 fragment half-lives are greatly abbreviated (this results from increased renal clearance because of the lower molecular weight of the fragments). Thus, distinct from F(ab′)2 fragments, deglycosylated antibody can be administered at the same dose and frequency as the unmodified antibody.
Our group has demonstrated recently that intracranial injection of the same deglycosylated anti-Aβ antibody used in this study results in clearance of compact and diffuse amyloid deposits comparable with that observed with the intact IgG. Interestingly, however, there was no significant activation of microglia as detected by CD45 immunohistochemistry. Furthermore, there was no upregulation of Fcγ receptor immunostaining as was observed with the unmodified antibody (Carty et al., 2006). Direct binding studies of the de-2H6 and 2H6 to murine Fcγ receptors and human C1q in vitro demonstrated reduced (but not zero) affinities for these interactions after deglycosylation (Carty et al., 2006). These data suggest that the deglycosylated antibody used here is less able to interact with Fcγ receptors or complement components. In the current study, we have shown that systemic administration of de-2H6 reduces diffuse amyloid deposits as effectively as its intact counterpart and also produces significant reductions in compact amyloid deposits. It also elevates the circulating Aβ levels in the same manner as the intact antibody, although to a lesser magnitude. We have shown previously that the microglial activation after adoptive transfer of Aβ antibodies is transient. It is elevated up to 2 months after the start of treatment but returns to control levels after 3 months of treatment (Wilcock et al., 2004b). It is plausible that, in the current study, microglia were transiently activated as observed in the previous study examining the time course of pathology after systemic administration of anti-Aβ antibodies. However, the data from Carty et al. (2006) showed that this deglycosylated antibody did not activate microglia 3 d after intracranial administration of de-2H6. This is a time point at which we typically see extensive microglial activation with intact anti-Aβ antibodies, including 2H6. Thus, it would appear that engagement of microglial effector proteins with high-affinity antibodies is not essential for the clearance of amyloid deposits in the brains of APP transgenic mice.
It has been suggested previously by our group and others that microglial cells are important for anti-Aβ immunotherapies to be effective (Schenk et al., 1999; Bard et al., 2003; Wilcock et al., 2003, 2004a). However, most data were based on intracranial studies and ex vivo assays. Other groups have suggested that this is not necessary, using F(ab′) fragments of anti-Aβ antibodies (Bacskai et al., 2002) or immunizing APP transgenic mice crossed with Fcγ receptor knock-out mice (Das et al., 2003). Other mechanisms may be occurring without the requirement of complete microglial responses. The peripheral sink hypothesis suggests that anti-Aβ antibodies can bind Aβ in the plasma, thereby creating a concentration gradient essentially drawing the Aβ from the brain (DeMattos et al., 2001). Also, a direct, catalytic disaggregation of the deposits via interaction between the antibody and the Aβ fibrils in the brain may occur without an inflammatory response (Solomon et al., 1997). Disaggregation of amyloid deposits may make the Aβ molecule more accessible to degrading enzymes such as neprilysin and insulin-degrading enzyme or enhance clearance by other mechanisms. In the current study, we show that a deglycosylated C-terminal anti-Aβ antibody improves cognition and removes amyloid almost as efficiently as the intact version of this antibody. It also elevates the level of circulating Aβ, consistent with a peripheral sink mechanism. The data presented here suggest that systemic administration of anti-Aβ immunotherapy does not require maximal involvement or activation of microglial cells to observe efficacy, both cognitively and for improvement in pathology. However, microglial mechanisms may facilitate this process, because clearance was slightly greater in animals administered unmodified antibodies. This is further supported by the observation of increased microglial activation around the remaining deposits in mice receiving the intact anti-Aβ antibody compared with mice receiving control antibody. It is further not clear whether the rate of clearance is modified by the deglycosylation. It will be necessary to perform detailed time course and concentration–response studies to better evaluate the role of effector molecule interactions in the amyloid-reducing effects of anti-Aβ immunotherapies.
To date, there have been three reports of anti-Aβ immunotherapy-induced microhemorrhage in APP transgenic mice (Pfeifer et al., 2001; Wilcock et al., 2004c; Racke et al., 2005). In addition, we further showed that associated with this increase in microhemorrhage is an increase in vascular amyloid levels (Wilcock et al., 2004c). This phenomenon appears to be dependent on the duration of treatment and is of concern to the advancement of human clinical trials using passive amyloid immunotherapy. It is conceivable that, after anti-Aβ immunotherapy, microglial activation plays a role in the increase in vascular amyloid, the generation of vascular leakage (by creating inflammation near these deposits), or both. In an attempt to minimize the microglial involvement in these processes, we compared intact and deglycosylated 2H6 antibodies on the development of vascular amyloid and microhemorrhage. The deglycosylated version of this antibody clearly diminished these outcomes but did not eliminate them entirely. These data suggest that some component of the antibody–effector molecule interactions is strongly contributing to these effects on the vasculature. Although the most parsimonious explanation is that there is less recruitment of microglia to perivascular deposits, it may equally well be the case that a slower rate of amyloid clearance (because of the reduction of microglial-mediated mechanisms) is less likely to be associated with these vascular events. Future studies will attempt to dissect whether it is the rate of clearance or mechanisms of clearance that is the critical feature leading to the unwanted vascular events associated with anti-Aβ immunotherapy.
In summary, we show here that deglycosylation of an anti-Aβ antibody shows an improved profile with regards to potential adverse effects while maintaining efficacy when compared with the intact antibody. We conclude that antibodies minimizing interactions with effector molecules, so called “inert” antibodies, may be a safer alternative for Aβ immunotherapy in the clinic.
Footnotes
-
This work was supported by National Institutes of Health Grants AG 15490 and AG 18478. D.M.W. is the Benjamin Franklin Scholar in Alzheimer’s Disease Research.
- Correspondence should be addressed to Dave Morgan, Department of Pharmacology and Molecular Therapeutics, University of South Florida, 12901 Bruce B. Downs Boulevard, MDC Box 9, Tampa, FL 33612. Email: dmorgan{at}hsc.usf.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}