

Abstract
Changes in synaptic strength during ongoing activity are often mediated by neuromodulators. At the synapse between cerebellar granule cell parallel fibers (PFs) and Purkinje cells (PCs), brief bursts of stimuli can evoke endocannabinoid release from PCs and GABA release from interneurons that both inhibit transmission by activating presynaptic G-protein-coupled receptors. Studies in several brain regions suggest that synaptic activity can also evoke calcium signals in astrocytes, thereby causing them to release a transmitter, which acts presynaptically to regulate neurotransmitter release. In the cerebellum, Bergmann glia cells (BGs) are intimately associated with PF synapses. However, the mechanisms leading to calcium signals in BGs under physiological conditions and the role of BGs in regulating ongoing synaptic transmission are poorly understood. We found that brief bursts of PF activity evoke calcium signals in BGs that are triggered by the activation of metabotropic glutamate receptor 1 and purinergic receptors and mediated by calcium release from IP3-sensitive internal stores. We found no evidence for modulation of release from PFs mediated by BGs, even when endocannabinoid- and GABA-mediated presynaptic modulation was prominent. Thus, despite the fact that PF activation can reliably evoke calcium transients within BGs, it appears that BGs do not regulate synaptic transmission on the time scale of seconds to tens of seconds. Instead, endocannabinoid release from PCs and GABA release from molecular layer interneurons provide the primary means of feedback that dynamically regulate release from PF synapses.
Introduction
At many synapses throughout the brain, astrocytes are closely associated with neuronal membranes (Kettenmann and Ransom, 2005). Astrocytes express multiple types of ionotropic and metabotropic receptors (Porter and McCarthy, 1997; Verkhratsky and Steinhauser, 2000; Haydon, 2001; Fellin and Carmignoto, 2004; Schipke and Kettenmann, 2004), whose activation can lead to increases in intracellular calcium levels (Pasti et al., 1997; Haydon, 2001; Perea and Araque, 2005). In turn, calcium increases or G-protein activation can trigger the release of a transmitter from astrocytes (Nedergaard et al., 2002; Auld and Robitaille, 2003; Evanko et al., 2004; Newman and Volterra, 2004; Volterra and Steinhauser, 2004; Volterra and Meldolesi, 2005), which can control transmitter release at nearby synapses (Kang et al., 1998; Newman and Volterra, 2004; Volterra and Steinhauser, 2004; Pascual et al., 2005), activate postsynaptic receptors (Parri et al., 2001; Angulo et al., 2004; Fellin et al., 2004), or modulate local blood flow via vasodilation (Zonta et al., 2003; Takano et al., 2005).
In the cerebellum, Bergmann glia cells (BGs) extend processes throughout the molecular layer (Palay and Chan-Palay, 1974) and ensheath parallel fiber (PF) synapses formed by granule cells onto Purkinje cells (PCs) (Grosche et al., 1999; Xu-Friedman et al., 2001). BGs express high levels of glutamate transporters (Chaudhry et al., 1995; Ottersen et al., 1997) and are therefore particularly effective in confining glutamate to activated synapses (Bergles et al., 1997; Clark and Barbour, 1997; Huang and Bordey, 2004). In addition, exogenous agonist application (Muller et al., 1992; Kirischuk et al., 1995; Kirischuk et al., 1996) and synaptic activation (Grosche et al., 1999) via the release of nitric oxide (NO) (Matyash et al., 2001) or noradrenaline (Kulik et al., 1999) can lead to calcium increases in BGs. Although activation of metabotropic glutamate receptors (mGluRs) and purinergic receptors (P2Rs) via synaptically released glutamate and ATP are mediators of calcium signaling in other types of glia (Porter and McCarthy, 1996; Pasti et al., 1997; Newman, 2005), no such role has been has been described in BGs. Thus, the mechanisms triggering and mediating calcium signaling in BGs under physiological conditions and its functional consequences remain poorly understood.
Here we examined the effects of synaptic activation on calcium signals within BGs of young animals and tested whether these calcium signals play a role in modulating synaptic transmission on short time scales. Activation of PF synapses with brief bursts evoked calcium signals in BGs that lasted for tens of seconds. These transients were mediated by P2Rs and mGluR1s and were not prevented by disrupting NO signaling, or by blocking either calcium-permeable AMPA receptors (AMPARs) or α1 adrenoceptors receptors. We found that although glial calcium transients were reliably evoked by synaptic activation, they did not contribute to presynaptic inhibition of PF synapses. Instead, feedback inhibition was dominated by endocannabinoids liberated from PCs (Brown et al., 2003) and GABA released from interneurons (Dittman and Regehr, 1997). We conclude that ATP and glutamate acting through P2R and mGluR1 are necessary in triggering calcium signals in BGs but that these calcium signals do not lead to feedback inhibition of PF synapses.
Materials and Methods
Slice preparation.
Rats (postnatal days 13–19) were anesthetized with halothane and decapitated, and sagittal or transverse cerebellar slices (250 μm) were obtained as described previously (Atluri and Regehr, 1996). Slices were cut in a sucrose solution containing the following (in mm): 81.2 NaCl, 23.4 NaHCO3, 69.9 sucrose, 23.3 glucose, 2.4 KCl, 1.4 NaH2PO4, 6.7 MgCl2, and 0.5 CaCl2. Slices were incubated at 32°C for 25 min and then transferred to a saline solution for 25 min at 32°C containing the following (in mm): 125 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 25 glucose, 2 CaCl2, and 1 MgCl2. Experiments were performed at 30–33°C using an inline heater (Warner Instruments, Hamden, CT) while perfusing the bath with saline solution at 3–4 ml/min using a Minipulse 3 pump (Gilson, Middleton, WI).
Electrophysiology.
Patch pipettes (2.5–4 MΩ) were pulled from borosilicate glass and were filled with an internal solution containing the following (in mm): 130 K-gluconate, 10 NaCl, 10 HEPES, 0.5 EGTA, 2 MgCl2, 0.16 CaCl2, 4 Na-ATP, 0.4 Na3-GTP, and 14 Tris-creatine phosphate. To measure calcium signals, the intracellular solution was supplemented with 50 or 100 μm fura-2 (Invitrogen, Eugene, OR). Whole-cell recordings were obtained from visually identified BGs with an Olympus BX50WI microscope (Olympus Optical, Tokyo, Japan), equipped with a 60× 0.9 numerical aperture water-immersion objective. Cells were held in voltage clamp at −80 mV using an Axopatch 200B or a Multiclamp 700A amplifier (Molecular Devices, Union City, CA).
Stellate cells were recorded with a voltage-clamp internal solution containing the following (in mm): 127 CsMeSO3, 10 CsCl, 10 HEPES, 0.5 EGTA, 2 MgCl2, 0.16 CaCl2, 2 Mg-ATP, 0.4 NaGTP, and 14 Tris-creatine phosphate (310 mOsm). Cells were held at −70 mV, and recordings were performed in artificial CSF (ACSF) containing 0 mm MgCl2. Purkinje cells were recorded with an internal solution containing the following (in mm): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, and 0.2 D600 (methoxyverapamil). Cells were held at −40 mV.
Parallel fiber tracts were stimulated with glass pipettes (2–3 MΩ) filled with ACSF. Single stimuli had durations of 200 μs and were delivered with a stimulus isolation unit (World Precision Instruments, Sarasota, FL). In some experiments, receptor agonists were locally applied via a puffer pipette (1–2 s) controlled by a Picrospritzer II (General Valve Corporation, Fairfield, NJ) and placed 50 μm above the slice.
Fluorescence measurements.
Fura-2 was excited at 380 nm with a monochromator (Polychrome IV; TILL Photonics, Gräfelfing, Germany). The filter set used was 415 nm dichroic (TILL Photonics) and 515LP for emission (Omega Optical, Brattleboro, VT). Images were acquired with 25 ms exposures at 5 or 20 Hz using a SensiCam CCD camera (PCO Computer Optics, Kelheim, Germany). Photobleaching was estimated by comparing images collected at 359 nm before and after acquisition and was usually <1%.
Presynaptic calcium measurements.
Magnesium Green AM (Invitrogen) was loaded into parallel fibers as described previously (Regehr, 2000), with loading times of 4–5 min. Slices were then incubated for 1–2 h at room temperature. Fluorescence transients in activated parallel fiber bands were measured using a photodiode. The excitation wavelength was 470 nm, and the filter set used was 510DRLP dichroic and 535DF35 for emission.
Data acquisition and analysis.
All signals were digitized with a 16-bit analog-to-digital converter (ITC-16; InstruTech, Port Washington, NY). Whole-cell currents were filtered at 5 kHz and digitized at 20 kHz. Photodiode signals were filtered at 800 Hz and digitized at 5 kHz. All analysis was performed using custom macros written in Igor Pro (Wavemetrics, Lake Oswego, OR) or Vision software (TILL Photonics). Values are given as mean ± SEM.
Results
Brief bursts of PF activity trigger calcium signals in Bergmann glia
We tested whether realistic patterns of PF synaptic activity trigger calcium signals in BGs. BGs were identified by the location of their cell bodies in the PC layer. When recorded with a K-gluconate internal solution, BGs had a low resting membrane potential (less than −80 mV), a low input resistance (<30 MΩ), and showed no spontaneous synaptic currents. Dye-filled BGs displayed several processes that extended toward the pia, where they terminated with characteristic end feet (Fig. 1A).

Brief bursts of PF stimulation evoke calcium responses in BGs. Data shown here and in Figure 2 were obtained in sagittal slices. A, 2PLSM image of BGs filled with Alexa-594. The area outlined by the box indicates the region selected for calcium imaging. B, Close-up of BG process, as visualized with either 2PLSM (top) or CCD optics (bottom). The white box outlines the area used for the fluorescence measurement in C. The dot indicates site of stimulus (stim) electrode. C, Calcium response (top) and current (bottom) evoked by train of stimuli (10 pulses, 50 Hz). The inset shows the current recorded during stimulation on an expanded time scale.
To examine how BGs respond to realistic patterns of PF activity, a stimulating electrode was placed 10–20 μm from one of the main processes in the outer half of the molecular layer (Fig. 1B). Brief trains (50 Hz, 3–10 stimuli) reliably evoked calcium signals in BGs, as shown in a representative experiment (Fig. 1C, top). At 32°C, relative to the onset of stimulation, their latency to onset and peak were 0.9 ± 0.1 and 1.7 ± 0.1 s, respectively, and their half-decay time was 7.1 ± 0.8 s (n = 12). Responses were blocked by TTX (500 nm, n = 7; data not shown), confirming that they were evoked by synaptic activity. In contrast, stimulus-evoked currents commenced with short latency after activation, peaked immediately after stimulation, and decayed rapidly, as shown in a representative experiment (Fig. 1C, bottom). Under our experimental conditions, no spontaneous calcium increases were observed.
Spatial spread and stimulus dependence of glial calcium signal
Previous work in glial cultures and acute slices has shown that calcium increases can propagate slowly within and between astrocytes (Cornell-Bell et al., 1990; Charles et al., 1991; Newman, 2001). By monitoring synaptically evoked changes in calcium within glial processes, we found that bursts of PF stimulation evoked an initial calcium transient in a localized dendritic region before spreading through glial branches, at speeds of 50 ± 4 μm/s (n = 12). This is shown for a representative experiment (Fig. 2A–C). A glial process was stimulated by activating PFs with a nearby stimulus electrode (Fig. 2A), which evoked calcium increases in the glia (Fig. 2B). Calcium increases were initially limited to a localized dendritic region (Δt = 1 s) but 500 ms later spread through the entire glial process in the field of view (Fig. 2C). Calcium levels were then uniform throughout the glial process and gradually returned to resting levels. Thus, synaptically evoked calcium signals in BGs propagate slowly from a local initiation site.

Characteristics of BG calcium signals. A–C, A representative experiment is shown that characterizes the spatiotemporal spread of calcium. A, 2PLSM (left) and CCD image (right) of BG process. B, Calcium response evoked by PF stimulus burst (10 pulses, 50 Hz), in region indicated by red line in A. The arrow indicates onset of stimulus train. C, Spatial spread of calcium wave. The sequence of fluorescence CCD images (50 ms frame duration) shows initiation and spread of calcium response (top row), followed by a more uniform decay (bottom row). Times indicated are relative to the onset of the stimulus train. The color bar indicates fluorescence change (% ΔF/F). D–G, In another experiment, the effect of different numbers of stimuli on BG calcium transients was assessed. D, 2PLSM (left) and CCD images (right) of a BG process. E, CCD images show peak fluorescence changes in glial process evoked by 50 Hz stimulus trains of either 1, 3, or 10 stimuli. Times indicated are relative to stimulus onset. F, Calcium transients evoked by trains of 3 (left) or 10 stimuli (right), in region proximal (black) and distal (gray) to initiation site. Regions of measurements are outlined in D. G, Stimulus dependence of peak fluorescence change at proximal (black) and distal (gray) site outlined in D, evoked by 50 Hz trains. stim, Stimulus location.
We also examined the dependence of the calcium signals on the stimulus parameters. In these experiments, we varied the number of stimuli of a 50 Hz train and monitored the magnitude and spatial extent of the resulting BG calcium signals. This is shown for a representative experiment (Fig. 2D–G). Single stimuli did not evoke calcium signals (Fig. 2E), whereas bursts of three to seven stimuli evoked calcium signals that remained confined to a small region (Fig. 2E–G). In contrast, trains with more than eight stimuli led to calcium increases that spread throughout the BG process (Fig. 2E–G). In general, trains with a range of stimuli led to calcium signals that always initiated in the same local region (data not shown), indicating that the changes in spatial spread were not attributable to fiber recruitment. A qualitatively similar behavior was observed in an additional four of four cells examined. These experiments illustrate that the initial localized calcium response may not propagate throughout the BG process unless synaptic activation is of sufficient duration. Therefore, to obtain reliable calcium signals, we evoked calcium transients with 10 stimuli at 50 Hz for all subsequent experiments.
Mechanisms underlying neuron-to-glia signaling
BGs express a variety of ionotropic and metabotropic receptors that could play a role in detecting synaptic activity. To examine the mechanisms underlying the calcium responses in BGs, we recorded from cells in transverse slices and stimulated PF tracts in the outer half of the molecular layer, at lateral distances of 50–100 μm from the BG process.
We first examined the role of α1 adrenoceptors and NO, which have been implicated by previous studies. One study reported that BG calcium increases evoked by granule layer stimulation were blocked by the α1 adrenoceptor antagonist prazosin (Kulik et al., 1999), suggesting that activity in afferent fibers from the locus ceruleus trigger BG calcium signals. In contrast to these studies, calcium signals evoked by PF stimulation were not affected by prazosin (Fig. 3A) (n = 5), indicating that BG calcium transients evoked by molecular layer stimulation do not require α1 adrenoreceptor activation. Another study reported that PF activation evoked calcium signals in BGs that were blocked by the NO-synthase inhibitor N-ω-nitro-l-arginine methyl ester (l-NAME) (Matyash et al., 2001). This suggested that release of NO from activated parallel fibers triggers calcium influx. However, we found that l-NAME had no effect on BG calcium signals (Fig. 3B) (n = 5).
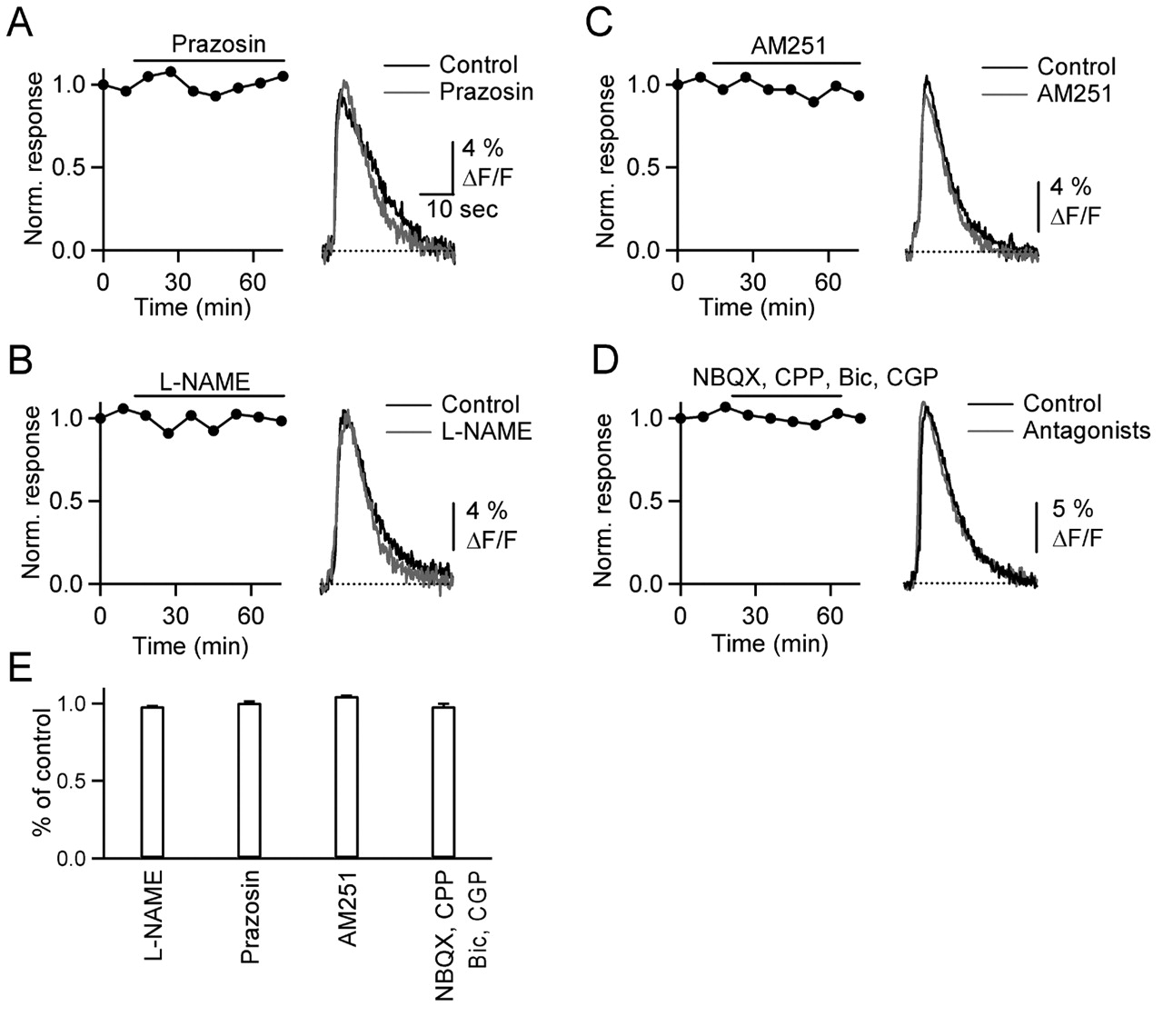
PF-evoked calcium transients are not triggered by activation of α1-adrenergic receptors, nitric oxide release, CB1 receptors, or AMPA, NMDA, or GABA receptors. Data shown here and in the following figures, except for Figure 6, were obtained in transverse slices. A–D, Graphs on the left plot normalized peak fluorescence transients in BG processes evoked by PF stimulation (10 stimuli, 50 Hz) every 9 min, during bath application of the α1-adrenergic receptor antagonist prazosin (A) (100 μm), the nitric oxide synthase blocker L-NAME (B) (100 μm), the CB1 receptor antagonist AM251 (C) (2 μm), and NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline) (D) (5 μm), CPP 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (5 μm), bicuculline (20 μm), and CGP 55845A (2 μm). Traces (right) show responses from same experiments under control conditions (black) and after bath application of antagonists (gray). E, Summary of effects of antagonists on the amplitude of fluorescence transients (n = 5 cells for each antagonist). Bic, Bicuculline; CGP, CGP 55845A.
The stimulus bursts that trigger BG calcium transients also evoke endocannabinoid release from PCs (Brown et al., 2003). Most effects of cannabinoids in the CNS are mediated by the CB1 cannabinoid receptor (CB1R). However, we found that the CB1R antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) had no effect on glial calcium transients (Fig. 3C) (n = 5).
BGs express ionotropic receptors, including calcium-permeable AMPA receptors lacking the GluR2 subunit, which could play a role in BG calcium transients (Muller et al., 1992). Furthermore, hippocampal astrocytes express GABAB receptors whose activation by synaptically released GABA can trigger increases in calcium levels (Kang et al., 1998). We therefore tested whether ionotropic glutamate or GABA receptors expressed on glia mediate calcium signals. Bath application of antagonists for AMPA, NMDA, GABAA, and GABAB receptors did not eliminate the calcium transients (Fig. 3D) (n = 5). These studies indicate that the BG calcium transients do not require activation of ionotropic glutamate receptors or GABA receptors (Fig. 3E).
Several lines of evidence suggest that mGluRs and P2Rs could play a role in BG calcium signaling. Synaptically released glutamate and ATP can trigger calcium transients in several types of glia (Porter and McCarthy, 1996; Pasti et al., 1997; Newman, 2005), although no such role has been established in BGs. High-frequency activation of PFs leads to glutamate release, pooling, and activation of extrasynaptic metabotropic glutamate receptors on PCs (Brasnjo and Otis, 2001) and local interneurons (Karakossian and Otis, 2004). The release of ATP after PF stimulation is less well established, but several possible sources of ATP exist, including PF vesicles, interneurons, and glia. We therefore tested whether mGluR1s or P2Rs mediate BG calcium signaling. Bath application of the mGluR1 antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), completely and reversibly blocked calcium transients in all cells tested (Fig. 4A,C) (n = 10). In contrast, MPEP [2-methyl-6-(phenylethynyl)-pyridine], an mGluR5 antagonist, had no effect on the calcium signals (Fig. 4C) (n = 5). Bath application of the P2R antagonist pyridoxal phosphate-6-azophenyl-2,4-disulfonic acid (PPADS) also reliably and reversibly blocked BG calcium transients (Fig. 4B) (n = 10). Together, our results suggest that activation of both mGluR1 and P2R is necessary for synaptically evoked BG calcium transients.

Calcium signals in BGs require mGluR1 and P2R activation. A, B, Graphs on the left plot normalized peak fluorescence transients in BG process, evoked by PF stimulation (10 stimuli, 50 Hz), during bath application of the mGluR1 antagonist CPCCOEt (A) (100 μm) and the P2R antagonist PPADS (B) (50 μm). Traces (right) show responses under control conditions (black) and after bath application of antagonists (gray). C, Summary of effects of antagonists on the amplitude of fluorescence transients [n = 10 cells for CPCCOEt and PPADS, n = 5 for 2-methyl-6-(phenylethynyl)-pyridine (MPEP)].
Calcium transients are mediated by calcium release from internal stores
Activation of metabotropic receptors can lead to the activation of phospholipase C (PLC), production of IP3, and ultimately calcium release from IP3-sensitive internal stores, in both neurons and glia (Pozzan et al., 1994). Here we tested whether calcium release from internal stores underlies synaptically evoked calcium transients. We found that bath application of the calcium ATPase blockers thapsigargin (Fig. 5A) (n = 5) and CPA (Fig. 5B) (n = 5) blocked glial calcium signals. In agreement with the known properties of these drugs, CPA was reversible, whereas thapsigargin was not.

Calcium signals are mediated by release from IP3-sensitive internal stores. A–D, Graphs plot normalized peak fluorescence transients in BG process, evoked by PF stimulus trains (10 stimuli, 50 Hz), during bath application of thapsigargin (A) (10 μm), CPA (B) (20 μm), ryanodine (C) (50 μm), and 2-APB (D) (50 μm). Traces show responses under control conditions (black) and after bath application of antagonists (gray). E, Summary of effects of antagonists on the amplitude of fluorescence transients (n = 5 cells for each antagonist).
Calcium can be liberated from internal stores either by calcium-induced calcium release (CICR) or by IP3. Bath application of ryanodine, which blocks CICR, did not significantly alter calcium transients (Fig. 5C,E) (n = 5), suggesting that CICR does not contribute to BG calcium signals. In contrast, bath application of the IP3 receptor antagonist 2-APB eliminated calcium signals in all cells tested (Fig. 5D,E) (n = 5), indicating that calcium release from IP3-sensitive internal stores underlies these events. Furthermore, preincubating the slices with U73122 [1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione], a broad-spectrum PLC inhibitor, eliminated calcium signals (Fig. 5E) (n = 5). Together, these findings show that after mGluR1 and P2Rs activation, PLC activation leads to IP3 synthesis, which leads to calcium release from internal stores.
Functional consequences of calcium increases in BGs
Recent studies in the hippocampus have shown that synaptic activity or application of a metabotropic glutamate receptor agonist causes astrocytes to release glutamate that can be detected in pyramidal cells as slow inward currents (SICs), mediated by extrasynaptic NMDA receptors (Angulo et al., 2004; Fellin et al., 2004). Here we tested whether glutamate release from BGs can generate responses in nearby neurons, via activation of extrasynaptic receptors. We recorded from cerebellar stellate cells in voltage clamp and activated PFs with 10–50 stimuli at 50 Hz. Although these stimulus trains reliably evoked calcium signals in BGs, SICs were never observed (0 of 30 cells, 3–5 trials per cell; data not shown).
We tested whether local application of a receptor agonist would provide a more robust means of activating BGs and eliciting SICs. We first established our ability to reliably evoke calcium transients under those conditions. Slices were bathed in TTX to block synaptic transmission, and BGs were activated with the group 1 metabotropic glutamate receptor agonist 3,4-dihydroxyphenylglycine (DHPG) or the P2 receptor agonist ATP, applied via a puffer pipette placed near the recorded cell above the slice (Fig. 6A). DHPG reliably evoked calcium transients in BGs (Fig. 6B) (n = 5), which were similar in time course to synaptically evoked calcium transients. Calcium signals evoked by DHPG were blocked by bath application of CPCCOEt (Fig. 6C,F) (n = 5), whereas PPADS application had no effect (Fig. 6F) (n = 5). Similarly, brief applications of ATP triggered calcium transients (Fig. 6D) (n = 5) that were blocked by PPADS (Fig. 6E,F) (n = 5) but were unaffected by CPCCOEt (Fig. 6F) (n = 5). ATP-evoked calcium transients were blocked by the IP3 receptor antagonist 2-APB (Fig. 6F) (n = 5), confirming that ATP triggers calcium release from IP3-sensitive internal stores (Kirischuk et al., 1995), mediated by the activation of P2Y receptors.

Agonist-induced calcium transients in BGs. A, Experimental setup. DHPG (100 μm) or ATP (100 μm) were puffed onto slice for 1–2 s. Bath contained 500 nm TTX. B, DHPG-evoked calcium transient in BG processes (black) is blocked by bath application of CPCCOEt (100 μm; gray). The time of DHPG application is indicated by horizontal bar. C, Time course of experiment shown in B. D, ATP-evoked calcium transient (black) is blocked by PPADS (gray). E, Time course of experiment shown in D. F, Summary of effects of antagonists (n = 5 for each condition) on calcium signals evoked by DHPG (open bars) and ATP (filled bars). G, DHPG evoked slow EPSC recorded in a voltage-clamped stellate cell. Bath contained 0 mm MgCl2 and 500 nm TTX.
We then recorded from stellate cells within the region of DHPG application, in the presence of TTX and with 0 mm external Mg2+ in the bath. In 7 of 64 neurons, DHPG evoked one or two SICs (Fig. 6G). Their decay time constant was 121 ± 25 ms (n = 9), and their amplitude was 182 ± 56 pA. In contrast, the decay time constant of mEPSCs recorded in stellate cells was 0.7 ± 0.04 ms (n = 33). These data suggest that in a small number of neurons, DHPG evokes synaptic events that could arise from glia. However, the mechanisms underlying SICs could not be determined, because SICs in individual cells could never be evoked repeatedly.
BGs do not control presynaptic release on short time scales
Release of a transmitter from BGs in response to brief bursts of parallel fiber activity could lead to activation of presynaptic receptors, thereby altering evoked calcium influx. To test this possibility, we loaded parallel fibers with the fluorescent calcium indicator Magnesium Green AM. We then monitored evoked calcium transients before and after a conditioning burst (10 stimuli, 50 Hz) known to elicit calcium responses in BGs. Under control conditions, bursts led to a strong reduction in calcium influx, which was largely blocked by the application of AM251 (Fig. 7A) (n = 7 experiments). This is consistent with previous studies demonstrating the release of endocannabinoids and the activation of presynaptic CB1 receptors after brief PF bursts (Brown et al., 2003). The remaining short-lived reduction was blocked by the GABAB antagonist (2S)-3[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP 55845A) (Fig. 7B), indicating that it is mediated by GABA released from molecular layer interneurons acting on presynaptic GABAB receptors (Dittman and Regehr, 1997). These data show that modulation of transmitter release via two distinct mechanisms can be reliably detected by monitoring presynaptic calcium influx. With both GABAB and CB1 receptors blocked, no modulation of presynaptic calcium influx after bursts could be detected (Fig. 7B).

BGs do not control neurotransmitter release from PFs. Parallel fibers were loaded with Magnesium Green AM, and stimulus-evoked calcium transients were recorded with a photodiode. Summary graphs (left column) show normalized calcium transients evoked by single stimuli (0.5 Hz) before and after a burst (10 stimuli, 50 Hz) delivered at t = 0 s. Representative traces (right columns) show calcium transients from single experiments preceding (Pre; black) and after (gray) burst stimulation, under control condition (left column) and after drug application (right column). A, Reduction in presynaptic calcium influx after burst is partially blocked by AM251. B, Remaining reduction is completely blocked by CGP 55845A. Data in A and B are from same set of experiments. C, In the presence of AM251 and CGP 55845A, the addition of CPCCOEt has no effect on presynaptic calcium transients. D, CPA has no effect on the transient reduction of transmitter release that is mediated by GABAB and CB1 receptors. E, Calcium transients in BGs do not lead to modulation of PF EPSC in PCs. PCs were recorded in voltage clamp, and EPSCs were evoked by stimulating two independent PF pathways (S1 and S2). Graph plots normalized responses (n = 5 PCs) evoked by low-frequency (0.5 Hz) stimulation of S1. Arrow indicates onset of burst stimulation (10 stimuli, 50 Hz) of S2. Traces (middle) show sample EPSCs evoked in S1 before (black) and 5 s after (gray) burst stimulation of S2. Right, Experimental setup.
It is possible that repeated activation at high frequencies led to a fatigue of transmitter release from BGs, which could account for the negative result in the above experiment. We therefore repeated the experiments, by eliciting stimulus bursts at longer interburst intervals (every 4 min) and including AM251 and CGP 55845A at the beginning of the experiment (Fig. 7C) (n = 8). Similar to the experiments above, stimulus bursts did not evoke a significant reduction in calcium influx. Moreover, bath application of the mGluR1 antagonist CPCCOEt to block glial calcium signals did not lead to a change in evoked calcium influx after bursts.
Glutamate release from BGs might not influence presynaptic receptors but could activate high-affinity metabotropic glutamate receptors on PCs. Previous results have shown that the activation of metabotropic glutamate receptors significantly enhances endocannabinoid release from PCs (Brown et al., 2003; Brenowitz and Regehr, 2005; Hashimotodani et al., 2005), and it is possible that this is at least partly mediated by glutamate released from BGs. To address this possibility, we monitored presynaptic calcium influx after a stimulus burst as described above and then added CPA to block calcium transients in BGs. This treatment had no effect on the endocannabinoid-mediated reduction in calcium influx (Fig. 7D) (n = 5). Thus, the activation of mGluRs on PCs is entirely caused by glutamate released from PFs, without a significant contribution by BGs.
BGs might control transmitter release at PF synapses by influencing intraterminal calcium levels or by acting downstream of calcium influx, mechanisms that would have remained undetected by monitoring evoked calcium influx as described above. To further test for a possible modulation of presynaptic transmitter release by BGs, we performed a second set of experiments. PCs were recorded in voltage clamp, and PF EPSCs were evoked in two independent pathways, S1 and S2 (Fig. 7E, right). EPSCs were recorded by activating S1 at low frequencies (0.5 Hz), before and after a conditioning burst (10 stimuli, 50 Hz) applied to S2 (Fig. 7E). As shown in Figure 2, these bursts will lead to propagating calcium transients in BG processes and could therefore trigger transmitter release from BGs and presynaptic modulation at synapses activated by S1. However, under our conditions, S1 EPSC amplitude remained unchanged after conditioning bursts applied to S2 (n = 6 PCs).
In summary, our data do not support a role of BGs in the modulation of presynaptic or postsynaptic receptors on the timescale of seconds to tens of seconds.
Discussion
Here we have shown that brief high-frequency bursts of parallel fiber activity can evoke calcium signals in BGs, which initiate in local domains before spreading through glial processes. We found that the activation of both mGluR1s and P2Rs is necessary for the initiation of calcium signals. We also tested whether the activation of BGs leads to the release of a transmitter that could influence release probability on PF synapses. We found that on the time course of seconds to tens of seconds, activation of BGs did not appreciably influence transmitter release from PFs.
Mechanisms of neuron-to-glia signaling
The observation that blocking either mGluR1 or P2R eliminates synaptically evoked BG calcium transients indicates that activation of both types of receptors is necessary for calcium signaling in BGs (cf. Piet and Jahr, 2005). mGluR1 is a Gq-coupled receptor whose activation elevates IP3 levels and liberates calcium from internal stores (Finch and Augustine, 1998; Takechi et al., 1998). Most types of P2YRs are also Gq coupled, suggesting that ATP and glutamate act in parallel in activating Gq to elevate IP3 levels and trigger calcium release from internal stores. This is consistent with the observation that depletion of internal stores eliminates BG calcium signals, as does the disruption of IP3 production. Although we found that for brief trains of PF stimuli both receptor types must be activated, the ability of an exogenous agonist to evoke calcium transients indicates that more powerful activation of mGluR1 or P2YR alone is sufficient to evoke calcium transients.
Both glutamate and ATP have been implicated previously in triggering glial calcium signaling (Porter and McCarthy, 1996; Pasti et al., 1997; Newman, 2005). In the cerebellum, there are several possible sources of synaptically evoked ATP, including vesicles at PF synapses, interneurons, and BGs. The dominant source of glutamate is more apparent. It is well established that parallel fiber activation evokes glutamate release from closely spaced parallel fibers that leads to glutamate pooling and activation of mGluR1 expressed on PC and stellate cell dendrites (Brasnjo and Otis, 2001; Karakossian and Otis, 2004). It is therefore likely that parallel fibers are the main source of glutamate that gives rise to BG calcium signals. The observation that mGluR1 mediates BG calcium signals raises the possibility that some forms of long-term plasticity that are blocked by bath application of mGluR1 antagonists could involve BG calcium signaling.
It is also notable that several signaling systems that have previously been implicated in BG calcium signaling do not contribute to BG calcium increases under our experimental conditions. We found that blocking α1 adrenoceptors does not alter BG calcium signals, suggesting that the release of noradrenaline by locus ceruleus fibers is not required for BG calcium signals (Kulik et al., 1999). Similarly, the insensitivity of BG calcium signals to the blockade of NO synthase indicates that BG calcium signals do not require NO release (Matyash et al., 2001). In addition, calcium-permeable AMPARs do not play a prominent role in the synaptically evoked calcium signals that we observed. This was surprising because BGs express calcium-permeable AMPARs that can be activated with glutamate to elevate calcium levels within BGs (Muller et al., 1992). Calcium signals mediated by AMPARs must be much smaller in amplitude and more spatially restricted compared with the signals mediated by release from internal calcium stores.
Neurotransmitter release from glia: postsynaptic effects
There is increasing evidence that glia can release a transmitter to influence nearby synapses. Studies in the hippocampus, retina, thalamus, and other areas suggest that the most likely candidates for a transmitter released from astrocytes are glutamate and ATP (Kang et al., 1998; Guthrie et al., 1999; Parri et al., 2001; Newman, 2003; Angulo et al., 2004; Fellin et al., 2004; Gordon et al., 2005). Release of these transmitters appears to be driven by calcium-dependent vesicle-mediated exocytosis, although other mechanisms have been postulated (Evanko et al., 2004).
Synaptic activation of astrocytes in the hippocampus can trigger the release of glutamate, which then acts on extrasynaptic NMDA receptors expressed in a subset of nearby neurons to generate slow inward currents (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005). SICs can be detected spontaneously or in response to tetanic stimulation or agonist application.
By recording from stellate cells, which express both AMPA and NMDA receptors, we tested whether SICs occur in response to calcium signals generated in BGs. We found that in a small number of neurons, agonist application led to SICs, with properties similar to the ones observed in hippocampus. However, synaptic stimulation never evoked SICs, and SIC could never be evoked repeatedly, suggesting that this type of signaling may not generalize to the cerebellum.
It is possible that glutamate released from BGs is rapidly taken up by both neurons and BGs before acting on NMDA receptors expressed by stellate cells. We therefore tested a second potential target of glutamate release from BGs, namely metabotropic glutamate receptors expressed in PCs. However, we found that synaptically evoked endocannabinoid release from PCs whose magnitude strongly depends on the extent of postsynaptic mGluR activation was not affected by blocking glial calcium signals. Thus, glutamate binding to the high-affinity mGluR on PCs likely originates entirely from PF synapses. In conclusion, our results provide little support for the activation of postsynaptic NMDA or mGluR receptors by glutamate released from BGs.
Glial control of transmitter release
Studies performed at the neuromuscular junction (Robitaille, 1998), the retina (Newman and Volterra, 2004), the hippocampus (Kang et al., 1998; Pascual et al., 2005), and hypothalamus (Gordon et al., 2005) suggest that transmitter release from glia can also modulate ongoing synaptic transmission by acting on presynaptic receptors. Here, we tested this idea for BGs, by monitoring presynaptic calcium influx at PFs before and after brief PF bursts that trigger calcium signals in BGs. Our results do not support a role of BG calcium signals in modulating transmitter release at PF synapses, at least on the time scale of seconds to tens of seconds. PF express kainate (Delaney and Jahr, 2002), group III metabotropic glutamate (Kreitzer and Regehr, 2001; Neale et al., 2001), and adenosine (Dittman and Regehr, 1996) receptors and should therefore be able to respond to glutamate or adenosine released by glia.
Previous studies have shown that brief bursts of PF activity can lead to endocannabinoid release from PCs and GABA release from local interneurons, modulating presynaptic calcium influx via activation of CB1 and GABAB receptors, respectively (Dittman and Regehr, 1997; Brown et al., 2003). Thus, realistic patterns of PF activity can lead to presynaptic modulation via two distinct mechanisms that can be reliably detected by monitoring presynaptic calcium influx. In contrast, our results show that calcium signaling in BGs does not contribute to the modulation of synaptic release on rapid time scales. Instead, calcium increases might trigger long-term structural processes such as changes in glial coverage of synapses, resulting in changes in synaptic efficacy (Iino et al., 2001; Oliet et al., 2004).
Footnotes
-
This work was supported by National Institutes of Health Grants RO1-NS044396 and RO1-NS432405 to W.G.R. We thank Claudio Acuna-Goycolea, Aaron Best, Stephan Brenowitz, Megan Carey, John Crowley, Michael Myoga, and Patrick Safo for comments on this manuscript.
- Correspondence should be addressed to Wade G. Regehr, Goldenson 307, Department of Neurobiology, Harvard Medical School, 220 Longwood Avenue, Boston, MA 02115. Email: wade_regehr{at}hms.harvard.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}