

Abstract
Neuronal communication requires the coordinated assembly of polarized structures including axons, dendrites, and synapses. Here, we report the identification of a ubiquitin ligase mind bomb 1 (Mib1) in the postsynaptic density and the characterization of its role in neuronal morphogenesis. Expression of Mib1 inhibits neurite outgrowth in cell culture and its gene deletion enhances synaptic growth at the neuromuscular junction in Drosophila. The analysis of Mib1 interactome by mass spectrometry revealed that Mib1 primarily interacts with membrane trafficking proteins [e.g., EEA1 (early endosomal antigen 1), Rab11-interacting proteins, and SNAP25 (synaptosomal-associated protein of 25 kDa)-like protein] and cell adhesion components (e.g., catenin, coronin, dystrobrevin, and syndecan), consistent with its previously reported function in protein sorting. More interestingly, Mib1 is associated with deubiquitinating enzymes, BRCC36 and the mammalian ortholog of fat facets, and a number of kinases, such as casein kinase II, MARK (microtubule affinity regulating kinase)/PAR1, and cyclin-dependent kinase 5 (CDK5). Further characterization of the Mib1-CDK5 interaction indicated that the N-terminal domain of Mib1 directly binds to the regulatory subunit p35 of the CDK5 complex. In cell culture, Mib1 induces the relocalization of p35/CDK5 without affecting its degradation. Surprisingly, p35/CDK5 downregulates the protein level of Mib1 by its kinase activity, and completely rescues the Mib1-induced inhibitory effect on neurite morphology. p35/CDK5 also genetically interacts with Mib1 in the fly according to the rough-eye phenotype. The data strongly support that the negative interplay between Mib1 and p35/CDK5 may integrate the activities of multiple pathways during neuronal development.
Introduction
During neuronal morphogenesis, genetic and environmental signals (Jan and Jan, 2003) are amplified in different transduction pathways, and may be eventually merged into common mechanisms to regulate cytoskeletonal organization and protein/membrane sorting (Luo, 2002; Horton and Ehlers, 2004). Although rapid progress has been made in identifying the signaling cascades, large gaps still remain in the connection of individual signaling components as well as in the coordination of multiple pathways. In addition to classic genetic approaches, recent advances in proteomics provide a complementary approach to search for important regulators. In this study, we used mass spectrometry to identify mind bomb 1 (Mib1), a ubiquitin ligase, in the sample enriched in postsynaptic density (PSD), a membrane compartment aligned with the presynaptic active zone at synaptic junction (Sheng and Hoogenraad, 2007).
Mib1 is an essential positive regulator in Notch pathway (Itoh et al., 2003) that is a conserved cell interaction mechanism for cell fate determinations during development (Weinmaster and Kintner, 2003). Notch signaling also negatively regulates neuronal morphogenesis, supported by numerous works in genetic models (Giniger et al., 1993; Fambrough et al., 1996; Wittenburg et al., 2000; Huang et al., 2005) and in cell cultures (Berezovska et al., 1999; Franklin et al., 1999; Sestan et al., 1999; Redmond et al., 2000). Mib1 was first cloned in genetic mutagenesis of zebrafish, with its loss-of-function mutant displaying developmental defects that resemble the loss of lateral inhibition in Notch signaling (Itoh et al., 2003). Instead of controlling protein degradation, Mib1 enhances the ubiquitination and endocytosis of the ligand Delta and, paradoxically, facilitates Notch activation. More studies showed that Mib1 may interact with all known Notch ligands, and knock-out of Mib1 in mice results in embryonic mortality (Koo et al., 2005b). Similar results were recapitulated in the fly model as well (Lai et al., 2005; Le Borgne et al., 2005; Wang and Struhl, 2005). Although the function of Mib1 in neurogenesis is well delineated, it is not clear whether Mib1 is involved in the morphogenesis of postmitotic neurons.
In addition, the establishment of proper neuronal morphology is dependent, at least partially, on the activity of cyclin-dependent kinase 5 (CDK5). CDK5 is a conserved multifunctional kinase activated by an atypical cyclin-like subunit p35 or p39 (Dhavan and Tsai, 2001; Cheung et al., 2006). Unlike other CDKs, CDK5 has the highest expression level and kinase activity in neurons and has crucial functions in a wide range of cellular events, including protein trafficking, cell death, neurite and synapse development, dopamine response, learning, and memory. Defects in axon guidance were observed in p35 knock-out mice (Kwon et al., 1999) and in Drosophila with abnormal CDK5 activity (Connell-Crowley et al., 2000). In cultured cortical neurons, suppression of CDK5 activity inhibited neurite outgrowth, whereas the introduction of exogenous p35 and CDK5 led to the elaboration of longer neurites (Nikolic et al., 1996). Here, we identified many components in Mib1 interactome and characterized the novel interaction between p35/CDK5 with Mib1 during neuronal morphogenesis.
Materials and Methods
Isolation of the PSD and affinity purification of Mib1-interacting proteins.
The PSD isolation and proteomic analysis were performed essentially as described previously (Cheng et al., 2006). The affinity purification using glutathione S-transferase (GST)-Mib1 fusion proteins was performed in the following steps. GST fusion proteins were expressed in Escherichia coli and purified by glutathione-sepharose resins (GE Healthcare, Piscataway, NJ). The resins with the bound proteins were directly used to pack affinity columns (1 bed volume = 1 vol, at least 1 mg fusion protein/ml resin). The columns were loaded with precleared rat-brain extract (20 vol, ∼10 mg/ml protein) in buffer A (20 mm HEPES, pH 7.2, 0.1 m NaCl, 0.1% Triton X-100, 1 mm DTT, 1 mm EDTA, 1 mm EGTA, 15% glycerol and protease inhibitors), washed extensively with 40 vol of buffer A, eluted with 4 vol of buffer B (the same as buffer A except with 0.6 m NaCl and 0.5% Triton), and cleaned with 4 vol of 2% SDS. The eluates were analyzed by an SDS gel followed by silver staining.
Identification of Mib1 interactome by mass spectrometry.
The eluted proteins by buffer B above were identified with an SDS gel coupled with liquid chromatography (LC)-tandem mass spectrometry (MS/MS) (Peng and Gygi, 2001). The proteins were first separated on a 12% SDS polyacrylamide gel, and the gel lane was cut into six pieces and subjected to in-gel trypsin digestion. The peptides recovered from gel pieces were analyzed on a 75 μm (internal diameter) fused-silica C18 capillary column, coupled with an LCQ-DECA XP ion trap mass spectrometer (Thermo Finnigan, San Jose, CA). Sequest algorithm (Eng et al., 1994) was used to search MS/MS spectra against the rat reference database (ftp.ncbi.nih.gov/refseq, released on May 11, 2005; 23,983 proteins). The database was concatenated with another randomized rat database to evaluate the frequency of false-positive matches (Peng et al., 2003). The searching parameters were set to allow precursor ion mass tolerance (3.0 Da), no enzymatic restriction, and dynamic mass change of methionine (+16 Da, oxidation). To remove false positives during the database search, we filtered the result using more stringent criteria than described previously (Washburn et al., 2001; Peng et al., 2003). (1) Nontryptic peptides or singly charged peptide matches were discarded. (2) The minimum length of peptides was seven. (3) The ΔCn score was at least 0.08, and the XCorr cutoff was adjusted to reduce the estimated false positive rate to 0%, namely, no peptides matched from the randomized database survived in the filtering process. (4) Proteins that were identified by less than three peptides were removed.
The relative abundance of an identified protein is roughly evaluated by its spectral count, which is the total numbers of MS/MS spectra identifying the protein. Although the spectral counts increase linearly with protein abundance (Liu et al., 2004), larger proteins have higher probability of being detected because more peptides are generated during the digestion. This bias is normalized by the formula (spectral counts/molecular weight) × 50,000, assuming the average size of proteins is 50 kDa (Peng et al., 2004).
Cell culture and transfection.
Human embryonic kidney 293 (HEK293) and COS7 cells were cultured in DMEM containing 10% fetal bovine serum. Hippocampal or cortical neurons were dissected from embryonic day 18 (E18) Sprague Dawley rat embryos, plated onto clean glass coverslips coated with 30 μg/ml poly-d-lysine, and cultured in Neurobasal medium (Invitrogen, Eugene, OR) with B27 (Invitrogen) and 0.5 mm glutamine with or without the addition of 2.5% fetal bovine serum during seeding. After 2 d, the cells were fed with the same serum-free medium. Cultured cells were transfected with appropriate amount of DNA with the method of calcium phosphate precipitation (Xia et al., 1996).
Western blotting and immunofluorescence.
Polyclonal antibodies were raised in rabbits using a mixture of two Mib1 peptides (CSLQDSEGDTPLHDAISKKRD, named M20, and CRKAIERRILLY, named C12). A cysteine residue was tagged to the N terminus of the M20 peptide to enable conjugation to keyhole limpet hemocyanin as a carrier protein. The resulting antisera from two rabbits (a and b) were purified sequentially by two affinity columns that were made of the M20 and C12 peptides coupled to SulfoLink resins (Pierce Biotechnology, Rockford, IL), respectively. Four purified antibodies were collected (M20a, M20b, C12a, and C12b) in which M20b had the highest specificity and recognized a single band with the size of Mib1 from total rat-brain lysate in Western blotting. The other three antibodies also recognized the Mib1 protein in the lysate. Moreover, all four antibodies had very similar patterns of immunofluorescence in the cells examined, such as HeLa, COS7, HEK293, and primary neuronal culture. M20 antibodies also recognize dMib1 in the fly. Other primary antibodies used for the analyses are as follows: monoclonal anti-HA (Abgent, San Diego, CA); monoclonal anti-FLAG (Sigma, St. Louis, MO), monoclonal anti-early endosomal antigen 1 (EEA1; BD Biosciences, San Jose, CA), anti-α-tubulin (Sigma), anti-β-tubulin (clone E7; Hybridoma Bank at the University of Iowa, Iowa City, Iowa), anti-synaptophysin (Millipore, Temecula, CA), anti-p35 (Nikolic et al., 1996), anti-CDK5 (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-PSD-95 and anti-glutamate receptor 2 (GluR2; gifts from Dr. Morgan Sheng, Massachusetts Institute of Technology, Cambridge, MA).
Kinase assay.
Purified GST fusion proteins (∼0.5 μg) were incubated with recombinant p35/CDK5 kinase purified with the Baculovirus expression system (Lee et al., 2000) in a 20 μl reaction including 10 mm HEPES, pH 7.0, 5 mm MgCl2, 1 mm DTT, 0.1 μg/μl BSA, 5 μm unlabeled ATP, and 2 μCi of [γ-32P]ATP (3000 Ci/mmol; ICN Biochemicals, Costa Mesa, CA) at room temperature for 20 min. Reactions were terminated by SDS loading buffer with 10 mm EDTA, and then analyzed by SDS-PAGE followed by autoradiography.
Analysis of neurite morphology in primary neuronal culture.
Neurite morphological analysis was performed as described previously (Yu and Malenka, 2003). Green fluorescent protein (GFP)-expressing neurons or immunofluorescence-labeled neurons were imaged using a Zeiss (Oberkochen, Germany) LSM510 confocal microscope system through a 40× [1.3 numerical aperture (NA)] or 63× (1.4 NA) objective. Axons and dendrites were differentiated by their distinct morphology. Data analysis was performed using MetaMorph software (Universal Imaging, West Chester, PA) by tracing the neurites and branches with the MetaMorph built-in drawing tool. The entire length for dendrites was measured by accumulation during the tracing process. All filopodia longer than 3 μm were considered dendritic branch tips and counted manually. The p values were calculated by Student's unpaired t test with a two-tailed distribution.
Genetics analysis in Drosophila melanogaster.
A P-element insertion (EY09760, y[1] w[67c23]; P(w[+mC] y[+mDint2] = EPgy2)CG5841[EY09780]/TM3, Sb[1] Ser[1]) was used and quantification of neuromuscular synapses was performed as described previously (Jin et al., 2004). After staining with cysteine-string protein antibody, the boutons at type 1b terminals from muscle 6/7 were counted and analyzed. For scanning electron microscopy images of the fly eye, whole flies were dehydrated in ethanol, dried with hexamethyldisilazane (Sigma), and analyzed with an LaB6 microscope. GMR-Gal4 driver was used and females were selected for phenotypic analysis. Other fly lines, UAS-CDK5-FLAG and UAS-Dp35-Myc (Connell-Crowley et al., 2000) (inserted in chromosome 3) were gifts from Dr. Edward Giniger (National Institutes of Health).
Results
Mib1 is localized in the PSD and its overexpression inhibits neurite outgrowth in cultured neurons
To identify regulatory proteins in neuronal morphogenesis, we used MS to analyze the PSD fraction from rat brains, revealing several hundreds of components in multiple signaling pathways (Peng et al., 2004; Cheng et al., 2006). Recently, protein ubiquitination has emerged as an important regulatory mechanism in the formation and remodeling of synaptic morphology, but only a few ubiquitin-related proteins were characterized (DiAntonio and Hicke, 2004). We performed additional proteomic analyses and found a ubiquitin ligase Mib1 in the PSD (Fig. 1 A). To validate the result, we analyzed subcellular localization of Mib1 by biochemical fractionation and immunostaining. During the PSD fractionation, Mib1 was enriched to the level similar to GluR2, a well known receptor in the PSD, whereas a synaptic vesicle marker synaptophysin was totally removed (Fig. 1 B). Consistently, Mib1 immunostaining showed partial (∼50%) but statistically significant overlap (p < 10−4) with PSD-95 in hippocampal neurons (Fig. 1 C–I). In contrast, the random overlap between Mib1 and tubulin staining was much lower, although tubulin was distributed in a larger area than PSD-95. Moreover, immunoelectron microscopy (Fig. 1 J) indicated that Mib1 antibodies recognized some vesicle-like structures adjacent to the electron-dense region of the PSD. Additional experiments of Northern blotting and Western blotting indicated that Mib1 is present in all tested mouse tissues, but is most abundant in the brain and the testis (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In Western blot analysis, the affinity purified Mib1 antibodies recognized a single protein band in total brain lysate, and the band showed the predicted size of native Mib1 (115 kDa) and migrated identically to recombinant Mib1 on an SDS gel, indicating the high specificity of Mib1 antibodies.

Identification of Mib1 in the isolated PSD by mass spectrometry and subsequent validation by immunostaining. A , Rat PSD proteins were purified and analyzed by an SDS gel followed by LC coupled with MS/MS, leading to the identification of one fully tryptic peptide (WTFNPAVLTK) assigned to Mib1. B , Western blotting analysis of multiple fractions (∼10 μg loaded) during the PSD purification. Rat forebrains were homogenized and the lysate was differentially centrifuged to make a crude synaptosomal fraction (Syn). The synaptosomal fraction was further separated by sucrose gradient and double Triton extraction, resulting in a Triton-soluble fraction containing presynaptic and vesicle proteins (Pre), and a Triton-resistant fraction enriched in PSD. GluR2, a subunit of AMPA receptor in PSD, was analyzed as a positive control, whereas synaptophysin is an immunocytochemical marker of the synaptic vesicle and was, thus, used as a negative control. C–E , Rat hippocampal neurons (∼DIV21) were double immunostained with antibodies of Mib1 (red) and PSD-95 (green). F–H , The neurons were double immunostained with antibodies of Mib1 (red) and α-tubulin (negative control; green). I , The degree of colocalization was measured from >30 neurites using MetaMorph software: α-tubulin over Mib1, 25 ± 14%; PSD over Mib1, 60 ± 19%; Mib1 over α-tubulin, 39 ± 14%; Mib1 over PSD-95, 54 ± 15%. Data are shown as SEM and statistical significance (p value) was evaluated by Student's t test. J , The immunoelectron microscopy of Mib1, using a section of adult mouse cortex and diaminobenzidine as a chromagen, displaying mitochondria (mt), PSDs (arrows), and Mib1-stained regions (circled with dashed lines).
Because Mib1 is required for efficient Notch signaling during embryogenesis (Koo et al., 2005b) and activation of the Notch pathway inhibits neurite outgrowth in cultured neurons (Sestan et al., 1999; Redmond et al., 2000), we tested the role of Mib1 in neurite development in primary neuronal culture. Cortical neurons of 3 d in vitro (DIV3) were transiently cotransfected with enhanced GFP and Mib1 constructs followed by analysis at DIV5 (Fig. 2). When compared with the control GFP cells, GFP/Mib1-coexpressing cells had much less complex neurite morphology, as indicated by significant reduction in total neurite length (more than twofold; p < 10−14) and branch tip numbers (more than twofold; p < 10−13). The similar result was obtained in five repeated experiments and in other similar experiments using GFP-tagged Mib1 and cultured hippocampal neurons (data not shown). In contrast, a loss-of-function Mib1 mutant (C985S) that was documented in zebrafish (Itoh et al., 2003) did not affect neurite morphology, suggesting that the E3 ligase activity of Mib1 is essential for its inhibitory effect on neurite outgrowth.

Overexpression of Mib1 in neurons causes the reduction of neurite length and branching. A–C , Cultured cortical neurons were transfected with enhanced GFP alone (0.5 μg), or cotransfected with another plasmid (1.5 μg) expressing HA-tagged Mib1 or its mutant form (C985S) that lacks ligase activity. The GFP signals were visualized and captured for the analysis of neurite morphology. D , The histogram of total neurite length in the control GFP cells (n = 68) was compared with that in Mib1-transfected neurons (n = 66). The neurite lengths measured by MetaMorph software were grouped in 14 bins and then fitted to a Gaussian distribution. E , F , The dendritic morphology was evaluated and compared by the total length of all neurites and total neurite tip number without considering axons. Data are shown as SEM. The p values were calculated compared with the GFP-transfected cells using Student's t test.
Identification and characterization of the interaction between Mib1 and p35/CDK5
To investigate how Mib1 regulates neurite outgrowth, we isolated Mib1-interacting proteins by GST affinity purification (Fig. 3). Mib1 has many characteristic motifs including five kelch repeats (Adams et al., 2000) and one ZZ zinc finger (Ponting et al., 1996) in the N-terminal domain (ND), 12 ankyrin repeats (Sedgwick and Smerdon, 1999) in the middle domain (MD), and three RING finger motifs (Joazeiro and Weissman, 2000) in the C-terminal domain (CD) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). All domains are predicted to fold into different structures that mediate protein-protein interactions. The kelch repeats predict a β-propeller structure, which may be involved in substrate recognition, reminiscent of the WD40-repeat proteins of the cullin-RING ligase family (Angers et al., 2006). The RING fingers are proposed to interact with ubiquitin conjugating enzymes. Recombinant GST fusion proteins with individual domains were generated as an affinity matrix to pull down interacting components from rat brain lysate (Fig. 3 C). Whereas GST alone, GST-MD, and GST-CD captured only a few proteins, GST-ND was associated with numerous proteins, consistent with its role in substrate recruitment. Mass spectrometry of the ND interactome revealed 24 major proteins, most of which act in membrane sorting and cell adhesion (Fig. 3 D). Intriguingly, deubiquitinating enzymes and kinases were also identified in the list. CDK5 was selected for subsequent characterization, because CDK5 has been shown to promote neurite outgrowth (Nikolic et al., 1996).

Identification of Mib1-interacting proteins by affinity purification and mass spectrometry. A , Domain structures of Mib1 recombinant proteins. The diagrams depict the full-length Mib1 of 1006 aa, its ND (1–401 aa), MD (384–801 aa), and CD (802–1006 aa). The Mib1 protein sequence contains five kelch repeats (in gray boxes), one ZZ motif (in hashed box), 12 ankyrin repeats (in dotted boxes), and three RING finger motifs (in black boxes). See details in supplemental Figure 2, available at www.jneurosci.org as supplemental material. B , Purified recombinant GST fusion proteins of Mib1, silver stained on an SDS gel. Molecular marker (M) is shown in kDa. The precise position of recombinant proteins on the gel is indicated by arrows. C , Purification of Mib1-associated proteins by affinity chromatography made of different GST fusion proteins. The columns were loaded with rat-brain lysates, washed extensively, and eluted with a buffer with 0.6 m NaCl and 0.5% Triton. The eluates were analyzed on a 6–16% gradient gel followed by silver staining. The proteins were eluted by high salt with mild detergent instead of SDS to avoid contamination with huge amounts of GST fusion proteins. D , The list of major Mib1-interacting proteins identified by tandem mass spectrometry. Proteins associated with the Mib1 N-terminal domain were analyzed by one-dimensional gel LC-MS/MS to determine their protein sequences. In the list, the proteins were first sorted by functional categories and then by relative abundance. CDK5 was sequenced by three tryptic peptides (LLGTPTEEQWPAMTK, LPDYKPYPMYPATTSLVNVVPK, and VRLDDDDEGVPSSALR).
CDK5 is present in cells as an inactive monomer, or in an active form by the association of regulatory subunits p35 or p39 (Dhavan and Tsai, 2001). Under some physiological and pathological conditions, the p35 subunit is cleaved by the calpain protease into two fragments (p10 and p25). The p25 fragment remains associated with CDK5 and has much longer half-life than p35. Thus, this cleavage event extends the duration of CDK5 activation. To examine whether Mib1 directly interacts with CDK5 or other regulatory subunits, affinity columns made of GST-Mib1 and the truncated domains were used to isolate proteins from a mixture of recombinant p35/CDK5 and p25/CDK5 complexes. Only Mib1 full-length and the ND were capable of enriching p35/CDK5, but not to p25 (Fig. 4 A). Because p25/CDK5 can form a stable complex, the result indicated that Mib1 interacts with neither p25 nor CDK5, instead, it is directly associated with p35 presumably through the p10 region. In addition, GST-p35 or p35 antibodies could pull down the Mib1 protein from total brain lysate, but GST-p25 did not (Fig. 4 B,C). Finally, during a glycerol gradient centrifugation of the brain lysate, a small portion of CDK5 was cofractionated with p35 and Mib1, whereas the majority of CDK5 protein existed in a monomer form in low-molecular-weight fractions (Fig. 4 D).

Analysis of the interacting domains between Mib1 and the p35/CDK5 complex. A , Glutathione beads were first coated with proteins including GST, GST-Mib1, or the truncated proteins (GST-ND, -MD, or -CD), incubated with a mixture of two recombinant protein complexes, p35/CDK5 and p25/CDK5, and then washed and eluted by 2% SDS. The recombinant CDK5 complexes were generated by coexpressing two subunits in the Baculovirus system as described previously (Lee et al., 2000). The p35 subunit includes the N-terminal p10 region and the C-terminal p25 region. The p25 region has an atypical cyclin domain that interacts with the CDK5 subunit. The load (10% loaded on the gel) and the eluted proteins (20% loaded) were analyzed by Western blotting using antibodies against CDK5 and p35. The p35 antibodies recognize both p35 and its proteolytic form p25. B , Protein affinity purification using GST-25 and GST-p35 as bait and rat-brain lysate as the load. The eluents were analyzed by immunoblotting using Mib1 antibodies. C , Immunoprecipitation (IP) from rat-brain lysate using p35 antibodies or control HA antibodies. The IP complexes were probed with antibodies to detect Mib1 and CDK5, whereas the IgG signal was used as the loading control. D , Cofractionation of Mib1 and the p35/CDK5 complex during glycerol gradient centrifugation. Rat-brain lysate was resolved in 24 fractions in a velocity centrifugation (1, the bottom fraction with protein complexes of large size and high velocity during centrifugation; 24, the top fraction with small proteins). The fractions were analyzed by Western blotting to detect the distribution of Mib1, p35, and CDK5 proteins.
Direct interaction between Mib1 and p35 was further confirmed by colocalization analysis in COS7 cells. HA-tagged Mib1 (HA-Mib1) exhibited an obvious punctate staining pattern analogous to native Mib1 (Fig. 5 A,B). However, the expression of p35 tagged with GFP at its C terminus (p35-GFP) showed membrane-associated staining that may be caused by its N-terminal myristoylation (Dhavan and Tsai, 2001) (Fig. 5 C). When HA-Mib1 and p35-GFP were coexpressed, p35-GFP was redistributed into vesicle-like structures and almost completely colocalized with Mib1 (Fig. 5 D–F). Moreover, we found that p35-GFP was partially colocalized and internalized with HA-tagged Mib1 ND (Fig. 5 G–I), but not with the MD or CD in transfected cells (Fig. 5 J–O). Together, the data were highly consistent with the results of the biochemical binding assays. Efficient redistribution of p35 requires the interaction with Mib1 and the function of full-length Mib1.

Mib1 is colocalized with p35 and induces p35 relocalization in transfected cells. Cultured COS7 cells were transfected with plasmids expressing various proteins for 1–2 d and immunostained with antibodies and DAPI (4,6,diamidino-2-phenylindole; DNA staining, blue). A , endogenous Mib1 without transfection. B , HA-Mib1 alone. C , p35-GFP alone. D–F , HA-Mib1 and p35-GFP. G–I , HA-Mib1-ND with p35-GFP. J–L , HA-Mib1-MD with p35-GFP. M–O , HA-Mib1-CD with p35-GFP. P–R , HA-Mib1 and native EEA1.
The nature of the Mib1-staining vesicles was tested by a number of organelle markers: EEA1 for early endosomes, Rab9 for late endosomes, and Lamp1 for lysosomes. Mib1 showed the best colocalization with EEA1 (Fig. 5 P–R), and some overlapping with the other two markers (data not shown). In some cases, the size of punctate staining of overexpressed Mib1 was larger than the native size, possibly because of vesicle fusion or clustering (Fig. 5 P). These results are in good agreement with the previous finding that Mib1 is possibly associated with endosomal vesicles according to immunostaining of Rab9 and RhoB, other GTPases involved in membrane trafficking (Itoh et al., 2003). Indeed, EEA1 was identified by mass spectrometry to be one of the major interacting proteins of Mib1 (Fig. 3 D), and more endosomal proteins were found in the Mib1 interactome, including several Rab11-interacting proteins that are proposed to regulate endocytic sorting and recycling (Tarbutton et al., 2005), and NipSnap1, which is a SNAP25 (synaptosomal-associated protein of 25 kDa)-like protein detected in the PSD sample (Satoh et al., 2002).
We then examined whether Mib1 colocalizes with the p35/CDK5 complex in cells. As negative controls, overexpressed p25 or CDK5 alone had a very distinct staining pattern from Mib1 (Fig. 6 A–F). When an adequate amount of p35 was coexpressed, CDK5 was recruited to the Mib1 loci, as shown in the triple staining of cultured COS7 cells and neurons (Fig. 6 G–N), implicating that p35 interacts with CDK5 by the C-terminal p25 region, as well as Mib1 through the N-terminal p10 region.

Colocalization of Mib1 and the p35/CDK5 complex in transfected COS7 cells and neurons. Cultured cells were cotransfected with plasmids expressing various proteins for 1–2 d and immunostained with antibodies. In some cases, the nuclei were stained with DAPI (4,6,diamidino-2-phenylindole; blue). A–C , Recombinant HA-Mib1 and p25 proteins in COS7 cells. D–F , HA-Mib1 and CDK5 in COS7 cells. G–J , Triple immunofluorescence of recombinant HA-Mib1, p35-GFP and DNK5 (a dominant negative CDK5 mutant, in which Asp144 was replaced by Asn) in COS7 cells. The loss-of-function mutant of CDK5 was used because active CDK5 led to Mib1 degradation. K–N , Triple staining of HA-Mib1, p35-GFP and DNK5 in cortical neurons. The neurons at DIV3 were transfected and stained at DIV5.
p35/CDK5 downregulates Mib1 and rescues its inhibitory effect on neurite outgrowth
Consistent with the notion that Mib1 induces the internalization of ligands rather than degradation during Notch signaling (Itoh et al., 2003), we observed that the ubiquitin ligase controlled the localization of p35 (Figs. 5, 6), but did not affect the abundance of p35 in cells (data not shown). Surprisingly, when Mib1 was cotransfected with p35 in cells, Mib1-staining vesicles appeared to become sparse, compared with the transfection of Mib1 alone (Fig. 5, compare B, D). Western blotting indicated that the Mib1 level was reduced in a dose-dependent manner until virtually undetectable with increasing amounts of p35 (Fig. 7 A). The degradation of Mib1 by p35 expression was observed in six repeated experiments and in transfection using other plasmids including untagged Mib1, FLAG-Mib1, and p35-GFP (data not shown). In contrast, p35 expression did not significantly change the protein level of several other cotransfected Mib1 truncations or C985S, an Mib1 mutant without ligase activity (Fig. 7 B,C). The C985S mutant was expressed at a higher level than the wild-type Mib1 when the same amount of plasmids was introduced in cells (data not shown). Moreover, when transfected cells were treated with a selective, potent proteasome inhibitor PS-341, a dipeptidyl boronic acid analog approved for clinical treatment of cancer (Lee and Goldberg, 1998) (Fig. 7 D), Mib1 was accumulated during the titration of the inhibitor, but was not restored to the original level in the absence of p35. This may be explained by the fact that p35, a short-lived protein that is degraded by proteasome (Dhavan and Tsai, 2001), was markedly increased during the treatment (Fig. 7 D), which compromised the effect of PS-341 on the accumulation of Mib1. Overall, the results suggest that the p35-stimulated degradation of Mib1 involves its own ligase activity and the ubiquitin-proteasome system.

The downregulation of Mib1 by p35 is dependent on their physical interaction, the ligase activity of Mib1 and may be mediated by proteasome. HEK293 cells were transfected with the combination of plasmids (0.1 μg unless specified; low, 0.05 μg; high, 0.2 μg), harvested after ∼2 d and analyzed by Western blotting with antibodies. Enhanced GFP construct (0.1 μg) was used as a control for transfection efficiency. The transfection efficiency was ∼30% without large variation by visualizing cotransfected GFP and Western blotting. A , Cotransfection of plasmids expressing HA-Mib1 and p35-FLAG. B , Cotransfection of plasmids expressing p35-FLAG and HA-Mib1-truncations. C , Cotransfection of p35-FLAG with a HA-Mib1 mutant C985S lacking ligase activity. D , Cells were transfected with HA-Mib1 and p35-FLAG, incubated for 1 d and then treated with a potent proteasome inhibitor PS-431 (low, 3 μm; high, 10 μm) for another day before harvest.
It should be emphasized that the level of regulatory subunits (p35 and p39) is the limiting factor for endogenous CDK5 kinase activity. The activity is extremely low in HEK293 cells because the regulatory subunits were below the level of detection, although CDK5 catalytic subunit is present in the cells (Fig. 7 D). It is assumed that p35 downregulates Mib1 through the endogenous CDK5. Unlike p35, p25 did not have such an effect (Fig. 8 A), recapitulating the significance of direct interaction with Mib1. To examine the requirement of CDK5 kinase activity for Mib1 downregulation, we coexpressed Mib1, p35, and CDK5, or a dominant-negative mutant of CDK5 (DNK5), in cells. As expected, Mib1 level was decreased with the expression of CDK5, but not the mutant (Fig. 8 B). Furthermore, in vitro kinase assay indicated that the Mib N-terminal and C-terminal domains were weakly phosphorylated, and the middle domain was clearly modified by recombinant p35/CDK5 in the presence of radiolabeled ATP, whereas GST alone was not labeled as a negative control, (Fig. 8 C).

The downregulation of Mib1 is regulated by the kinase activity of p35/CDK5. A , HEK293 cells were transfected with Mib1 plasmids (0.1 μg) and p25 (low, 0.05 μg; high, 0.2 μg). The cells were harvested after ∼2 d and the expressed proteins were probed by Western blotting. B , Cotransfection of HA-Mib1 (0.4 μg), p35-FLAG (0.05 μg), and CDK5, or a dominant-negative CDK5 mutant [DNK5, D144N in its catalytic domain (Nikolic et al., 1996); low, 0.05 μg; high, 0.2 μg]. C , In vitro kinase assay by incubating recombinant p35/CDK5 with GST fusion proteins, including GST, GST-ND, GST-MD, and GST-CD. The reaction was initiated with the addition of ratio-labeled ATP and terminated by SDS loading buffer. The samples were separated on an SDS gel followed by autoradiography. CDK5 and p35 were also self-phosphorylated and the signals were similar in all samples, indicative of addition of equal amount of p35/CDK5 protein.
To determine the effect of p35/CDK5 on native Mib1 in neurons, we transfected cultured hippocampal neurons with p35-GFP. In p35-GFP-expressing neurons, Mib immunoreactivity was reduced slightly in the soma and more dramatically in the neurites, and this effect could be inhibited by the CDK5 inhibitor roscovitine (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In addition, during the development of cultured hippocampal neurons, CDK5 level stays stable, but the expression of p35 is enhanced and therefore increases CDK5 activity (Tsai et al., 1994). Interestingly, the protein level of native Mib1 was reduced during this process (Fig. 9 A), and quantification of the immunoblot revealed reverse correlation of the abundance of p35 and Mib1: from DIV1 to DIV7, Mib1 level was decreased approximately twofold and the amount of p35 was increased approximately twofold (Fig. 9 B).

p35/CDK5 suppresses the negative activity of Mib1 in neurons. A , Immunoblotting of native Mib1, p35, CDK5, and β-tubulin in lysates of primary hippocampal neurons ranged from DIV1 to DIV7. B , Quantification of immunoblotting signals of p35 and Mib1 using Scion Image program (National Institutes of Health, Bethesda, MD). Intensities of protein bands were first normalized to β-tubulin and then to the strongest (100%). C , D , Overexpression of p35 rescues Mib1-mediated inhibition of neurite outgrowth. Cultured cortical neurons were transfected with enhanced GFP alone (0.4 μg), HA-Mib1 (0.4 μg), p35-FLAG (1.2 μg), or the combination at DIV3 and stained at DIV5. The CDK5 inhibitor roscovitine was added at 10 μm after transfection. The dendritic morphology was compared by measuring total length of all neurites and counting the total neurite tip number without considering axons. The p values with no asterisks are transfected cells compared with the control GFP cells according to Student's t test; p values marked with one asterisk are derived from the comparison of Mib1-transfected cells with or without p35 overexpression; and the p values marked with two asterisks are computed from the double-transfected cells in the absence and presence of roscovitine treatment.
We then investigated how two opposite activities of Mib1 and p35/CDK5 contribute to neurite outgrowth. When DIV3 cortical neurons were transfected with p35 to affirm the stimulatory effect of p35/CDK5 on neurite outgrowth (Nikolic et al., 1996), no obvious effect was detected (data not shown). The discrepancy could be explained by the possibility that our culture condition contained some serum that stimulated the expression of endogenous p35 to the range of almost saturated level, leading to resistance to ectopically expressed p35, as p35 was found to be upregulated by numerous trophic factors (Cheung et al., 2006). To test this possibility, the culture condition was slightly modified by omitting serum during the plating, resulting in slower development of neurite morphology (compare Figs. 2 E, 9 C). After the modification, p35 expression could facilitate neurite outgrowth with respect to total neurite length (p < 0.005) and tip number (p < 10−4) (Fig. 9 C,D). Meanwhile, the introduction of Mib1 had less, but still significant, negative effects on neurite outgrowth. More importantly, the inhibition by Mib1 was totally rescued by the cotransfection of p35, and this effect was abolished by the treatment of the CDK5 inhibitor roscovitine, suggesting that the activity of p35/CDK5 can counteract the negative impact on neuronal morphogenesis resulted from Mib1 expression in the cell culture.
Genetic interaction of Mib1 and p35/CDK5 in the fly
We used a Drosophila model with Mib1 loss-of-function to further examine its physiological role and interaction with p35/CDK5. A P-element insertion was identified in the fly gene of CG5841, the Drosophila ortholog of Mib1 (dMib1). The homozygote was affirmed to be a dMib1-null allele, because it showed pupal lethality that was rescued by precise removal of the transposon in the 5′ untranslated region of the gene, or by the expression of transgenic dMib1 (Lai et al., 2005; Le Borgne et al., 2005). We also validated the lacking of full-length dMib expression in the allele by Western blotting (Fig. 10 A). In Drosophila, the synaptic structure at the larval neuromuscular junction (NMJ) is a well defined system with which to study synaptic structure and neurotransmission, and the number of synaptic boutons is an established index for synaptic growth. By counting synaptic boutons in the wild-type (n = 8; 48 ± 9) and the mutant larvae (dmib1−/−; n = 8; 89 ± 11), we found that the loss of dMib1 causes the synaptic overgrowth, and the number of synaptic boutons increased 85% (p < 10−5). This finding suggests that Mib1 plays a conserved negative role in the formation of synaptic structure.
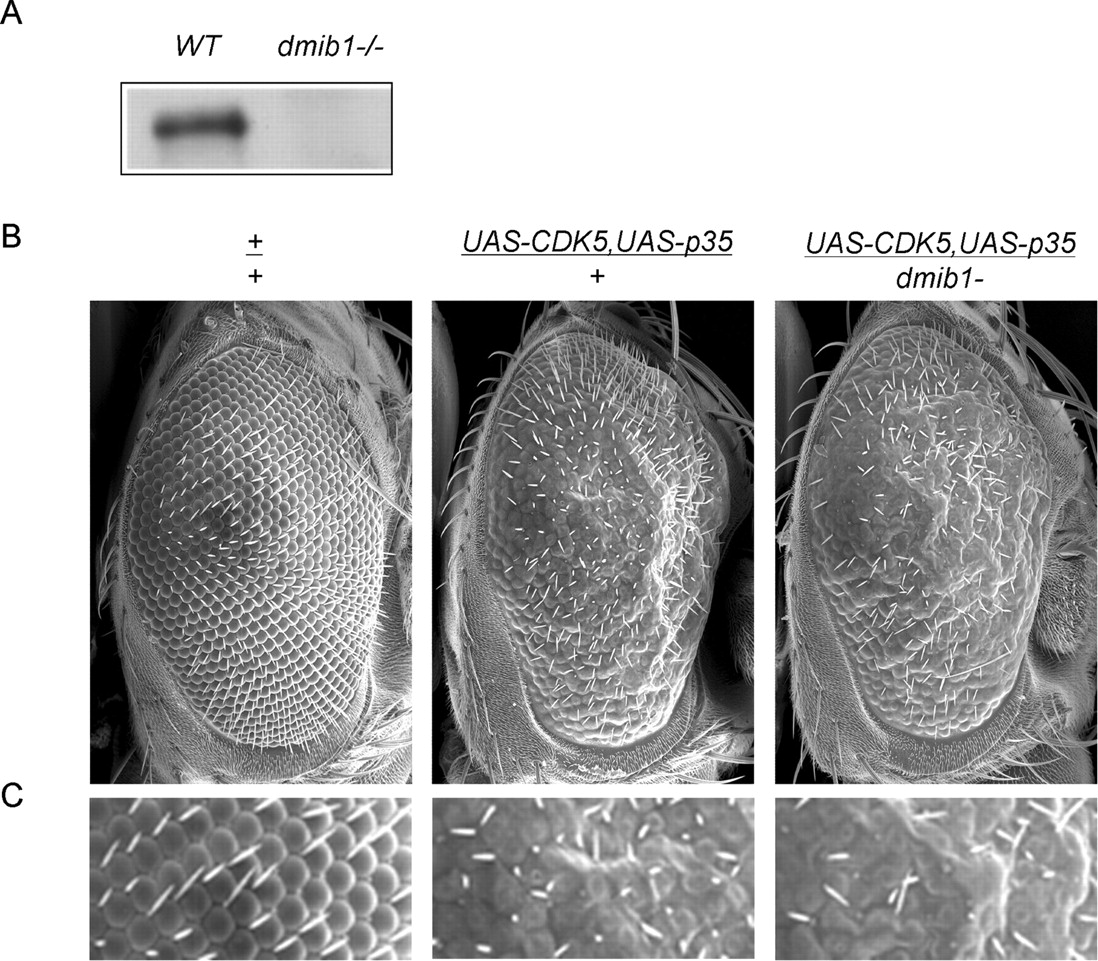
Genetic interaction of dMib1 and p35/CDK5 in Drosophila. A , The absence of Drosophila dMib in the mutant (dmib1−/−) was confirmed by immunoblotting with Mib antibodies. B , The partial loss of dMib1 in heterozygote enhances the rough eye phenotype caused by p35/CDK5 overexpression. The fly genotypes are also shown. C , High-magnification pictures of images in B .
The genetic interaction between p35/CDK5 and Mib1 was tested by monitoring adult eye phenotype. Overexpression of p35/CDK5 caused a rough eye phenotype as described previously (Connell-Crowley et al., 2000) (Fig. 10 B), whereas the heterozygotic line of the dMib1 mutant (dmib1−/+) had a smooth eye phenotype (data not shown). Crossing the p35/CDK5 transgenic line with the dMib1 mutant line showed that the partial loss of dMib1 in the heterozygote enhanced the rough eye phenotype induced by p35/CDK5 (Fig. 10 B,C). This result also supports the negative regulation between p35/CDK5 and dMib1 in neuronal development.
Discussion
Using large-scale proteomic analysis of the PSD samples, we identified Mib1 as a novel ligase in the PSD fraction. The localization of Mib1 in the PSD was validated by immunohistochemical analysis and electron microscopy. Neuronal culture experiments suggested that Mib1 inhibits neuronal outgrowth through its ligase activity. Moreover, more than 20 Mib-interacting proteins were revealed by affinity purification coupled with mass spectrometry, including the CDK5 complex, a positive regulator of neuronal morphogenesis. Interestingly, the negative interplay between Mib1 and CDK5 was suggested by the observation that CDK5 overexpression led to the degradation of Mib1, which was dependent on their association, the CDK5 kinase activity, and the Mib1 ligase activity. Their reciprocal inhibition was also supported by several functional assays in neuronal cultures and in the fly.
Apart from previous analyses of Mib1 function in Notch signaling during neurogenesis, our experiments demonstrated that Mib1 plays an important role in inhibiting neurite outgrowth in postmitotic neurons. This phenomenon could be, at least to some degree, explained by the activation of Notch signaling, because several reports suggested that the Notch pathway negatively modulates neurite morphology. The fly carrying mutations in Notch or Delta displayed intersegmental nerve with abnormal axon guidance without changing its neuronal identity (Giniger et al., 1993). Defects in neuron morphogenesis were also found in loss-of-function mutants of other Notch pathway genes, such as Numb (Huang et al., 2005), Kuzbanian, a Drosophila protease required for the cleavage in Notch activation (Fambrough et al., 1996), and presenilin (Wittenburg et al., 2000). The involvement of Notch signaling in neurite development was further observed using cultured N2a cells (Franklin et al., 1999), cortical neurons (Sestan et al., 1999; Redmond et al., 2000), or hippocampal neurons (Berezovska et al., 1999). We also obtained the similar results by overexpressing the constitutively active Notch intracellular domain in neurons (data not shown). However, the downstream targets of Notch signaling that control neurite morphology remain to be determined.
However, Mib1 may function through pathways independent of Notch signaling. Indeed, our proteomic survey of the Mib1 interactome discovered many proteins that may or may not be relevant to Notch activation. For example, FAM, an orthologue of Drosophila fat facets (faf), was identified to interact with Mib1 (Fig. 3 D). FAM is a deubiquitinating enzyme that removes ubiquitin tag from substrates. Neuronal overexpression of fat facets (faf) causes a dramatic increase of synaptic boutons in neuromuscular junctions (DiAntonio et al., 2001). FAM/faf has been also suggested to control vesicle trafficking (Chen et al., 2003), synaptic plasticity (Chen et al., 2002), and Notch signaling (Overstreet et al., 2004). The association of ligases and deubiquitinating enzymes appears to be a common theme to modulate the ubiquitination of protein targets (Nijman et al., 2005). Moreover, δ-catenin is another interesting protein isolated by Mib1 affinity purification. δ-catenin is associated with severe mental retardation in cri-du-chat syndrome and its gene knockout in mice causes severe impairments in synaptic function (Israely et al., 2004). Some of the catenins including α-, β-, and δ-catenin family members are critical for neuronal morphogenesis (Yu and Malenka, 2003; Abe et al., 2004; Elia et al., 2006).
The interaction between Mib1 and CDK5 may be important for signaling integration during development, as CDK5 was shown to downregulate the protein level of Mib1 by its kinase activity, and genetic experiments in the fly also confirmed this interaction. The Mib1-CDK5 interaction could be transient because, in the steady state, the majority of Mib1 alone and p35/CDK5 itself are not localized in the same cellular compartment. Mib1 is located in the intracellular vesicle-like structures that were shown to be partially colocalized with endosomal marker Rab9 (Itoh et al., 2003) and EEA1. In contrast, p35 may be attached to plasma membrane through myristoylation (Dhavan and Tsai, 2001), whereas CDK5 itself without a regulatory subunit was mostly shown as a diffuse pattern. Importantly, overexpressed Mib1 caused the redistribution of p35/CDK5 in cellular compartments, maybe because of enhanced protein sorting. Although the underlying mechanism needs additional investigation, we hypothesize that p35/CDK5 is ubiquitinated by Mib1 and the modification facilitates the trafficking of the complex without affecting its degradation.
How does the CDK5 activity stimulate the degradation of Mib1? The ligase activity of Mib1 may be directly upregulated by CDK5 phosphorylation and therefore upregulates its self-ubiquitination and degradation. The idea is supported by several lines of evidence: (1) Mib1 degradation was suggested to rely on the interaction with p35/CDK5 and its kinase activity; (2) the protein level of p35 is reversely correlated with that of Mib1 during the development of cultured neurons; (3) Mib1 is a substrate of p35/CDK5 in vitro; and (4) Neither the Mib1 N-terminal domain nor the Mib1 ligase knock-out mutant is downregulated by p35 expression, despite their physical interaction, suggesting the requirement of full-length Mib1 and its ligase activity. The activity of many ubiquitin ligases can be modulated by phosphorylation, as exemplified by E3s such as APC (anaphase-promoting complex) (Tang et al., 2004) and Itch (Gao et al., 2004). It is possible that Mib1 can be regulated by the similar mechanism. However, more biochemical analysis is required to illustrate the detailed molecular mechanisms involved.
In conclusion, we identified the novel function of Mib1 in regulating neurite morphogenesis and demonstrated its physical and functional interaction with p35/CDK5. The interplay between the two proteins links the Notch signaling with the p35/CDK5 pathway for the first time and provides the molecular basis for integrating multiple pathways. Because Mib1 and p35 have paralogs termed Mib2 (Koo et al., 2005a) and p39 (Dhavan and Tsai, 2001), respectively, it is possible that those paralogs function in a similar manner, and that these interactions could function at different stages during neuronal development. Mib2 is only abundantly expressed in adult tissue, which restrains it from functioning in early development, whereas Mib1 is highly expressed in both embryos and adults (Koo et al., 2005a). The expression of p39 occurs postnatally much later than that of p35 (Dhavan and Tsai, 2001). In addition, because Mib1 and CDK5 were found to play important roles in non-neuronal cells (Dhavan and Tsai, 2001), the scenario disclosed in neurons may be applied to those cell types as well.
Footnotes
-
This work was supported in part by National Institutes of Health Grants DA019937 and AG025688 (J.P.). We are grateful to Dr. Baubak Mansour for initial characterization of Mib1, Dr. Holger Patzke for the p35-GFP construct, Dr. Mingsum Lee for the Mib1 construct, Dr. Morgan Sheng for PSD-95 and GluR2 antibodies, Dr. Richard Kahn for EEA1 antibodies, Dr. Daniel Finley for proteasome inhibitor PS341, Dr. Zixu Mao for the CDK5 inhibitor roscovitine, and Dr. Edward Giniger for the fly lines. We thank Drs. Albert Davis, Edbauer Dieter, Kenneth Moberg, James Lah, and Allan Levey for technical advice and thoughtful discussions. We also thank Dr. Hong Yi for the electron microscopy analysis. L.T. is an Investigator of the Howard Hughes Medical Institute.
- Correspondence should be addressed to Junmin Peng at the above address. jpeng{at}genetics.emory.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}