

Abstract
Current concepts suggest that stress-induced release of neuromodulators such as corticotropin-releasing factor (CRF) can drive drug-dependent behaviors. Although previous drug exposure can enhance behavioral and neurochemical responses to stress, it is unclear how such drug exposure alters the CRF modulation of excitatory synapses onto ventral tegmental area (VTA) dopamine neurons, a key locus of drug- and stress-induced neuroadaptation. Here, we demonstrate that, after repeated cocaine exposure, the magnitude and duration of the CRF-induced potentiation of NMDA receptor (NMDAR)-mediated neurotransmission was significantly increased compared with naive and saline-treated mice. Furthermore, CRF enhanced AMPA receptor (AMPAR)-mediated transmission only in mice that were exposed to cocaine. Increased frequency of AMPAR-mediated spontaneous miniature EPSCs and the intracellular blockade of CRF potentiation of AMPAR-mediated transmission suggest both presynaptic and postsynaptic effects of CRF. Importantly, pharmacological experiments revealed that CRF receptor 1 and protein kinase A pathways were newly recruited after repeated cocaine for the enhancement of CRF-induced NMDAR potentiation and the appearance of AMPAR potentiation. Thus, enhanced CRF-induced potentiation of excitatory synaptic transmission onto VTA dopamine neurons after cocaine preexposure is likely to produce an abnormal increase in dopamine release during stressful events and could augment activation of addictive behaviors in response to stress.
Introduction
Stress plays an important role in addiction. Both physical and psychological stress can enhance the acquisition of drug self-administration and reinstatement of drug seeking (Erb et al., 1996; Piazza and Le Moal, 1998; Sinha, 2001; Goeders, 2002). Conversely, repeated drug exposure can alter the behavioral responses to stress (Antelman et al., 1980; Hamamura and Fibiger, 1993; Blatchford et al., 2005) and the dopamine release produced by stressors (Kalivas and Stewart, 1991; Hamamura and Fibiger, 1993; Suzuki et al., 2002). Although there is considerable interest in understanding the cellular bases of the interaction of drugs of abuse and stress, the underlying mechanisms remain unclear.
The stress hormone corticotropin-releasing factor (CRF) plays a key role in the neuroendocrine and behavioral responses to stress, primarily via the hypothalamic pathway (Bale and Vale, 2004) but also through interaction with extrahypothalamic structures. Of particular interest to our study is that CRF contributes to stress-related addictive behaviors by acting on the mesolimbic dopamine system (Sarnyai et al., 2001; McFarland et al., 2004; Lodge and Grace, 2005; Wang et al., 2005, 2007) in addition to other subcortical brain regions (Sarnyai et al., 2001; Gallagher et al., 2008; Koob and Le Moal, 2008). The mesolimbic dopamine pathway consists of dopaminergic neurons in the ventral tegmental area (VTA) and their projections to various targets including nucleus accumbens, prefrontal cortex, and amygdala (Fields et al., 2007), and a growing body of evidence indicates that this pathway is critical in modulating addictive behaviors (for review, see Kauer and Malenka, 2007; Bonci and Borgland, 2009).
Several lines of evidence indicate a critical role for CRF and the mesolimbic dopamine system in stress-related addictive behaviors. Neurons containing CRF project to and make synapses within the VTA (Tagliaferro and Morales, 2008). Injection of CRF directly into the VTA increases locomotor activity (Kalivas et al., 1987) and drug taking (Wang et al., 2005), whereas injection of CRF antagonists inhibits stress-induced reinstatement of cocaine seeking (Wang et al., 2005, 2007). Also, repeated drug exposure enhances the locomotor response to central injection of CRF (Erb et al., 2003). Moreover, electrophysiological recordings have shown that CRF regulates excitability of VTA dopamine neurons (Korotkova et al., 2006; Wanat et al., 2008) and synaptic inputs onto these neurons (Ungless et al., 2003).
The glutamatergic synaptic inputs onto VTA dopamine neurons are particularly important, since these excitatory inputs tightly regulate dopamine neuron firing activity and ultimately dopamine release (Overton and Clark, 1997). Furthermore, these glutamatergic synapses have been shown to undergo long-lasting adaptations after exposure to drugs of abuse (Ungless et al., 2001; Borgland et al., 2004; Kauer, 2004; Bellone and Lüscher, 2006; Chen et al., 2008) and to be modulated by stress (Fitzgerald et al., 1996; Saal et al., 2003). Here, we examined whether previous experience with cocaine would modify the effects of CRF on glutamate receptor function in VTA dopamine neurons.
Materials and Methods
Slice preparations for electrophysiology.
All procedures conformed to animal care standards set forth by the National Institutes of Health and the Ernest Gallo Clinic and Research Center. Male C57BL/6 mice (Charles River) were housed four per cage with their mother across the duration of the experiment, with food and water available ad libitum and a 12 h light/dark cycle with the lights on at 7:00 A.M. After 2 d of saline injections to habituate, mice were given intraperitoneal injections of either saline (0.9% NaCl) or cocaine (15 mg/kg dissolved in saline) for 5 d and were decapitated within 24–30 h after the last injection. A block of brain tissue containing VTA from mice aged postnatal day 26 (P26) to P28 was sliced in the horizontal plane (190 μm) in an ice-cold, high-sucrose solution containing (in mm) 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, and 75 sucrose. Slices were transferred to a holding chamber containing artificial CSF (aCSF; in mm: 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 18 NaHCO3, and 10 glucose) and equilibrated at 31–34°C for at least 1 h. Picrotoxin (100 μm) was added to the aCSF during recordings to block GABAA receptor-mediated IPSPs, and aCSF was aerated with 95% O2/5% CO2 and perfused over the slice at ∼2 ml/min. For miniature EPSC (mEPSC) recordings, lidocaine (500 μm) and AP-5 (50 μm) were also added.
Electrophysiology.
Whole-cell voltage-clamp recordings (Hamill et al., 1981) were made using a Multiclamp 200B amplifier (Molecular Devices). Cells were visualized with an upright microscope using infrared differential interference contrast illumination. Electrodes (3–4 MΩ) contained (in mm) 120 cesium methanesulfonate, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 tetraethylammonium-Cl, 2.5 MgATP, and 0.25 NaGTP (pH 7.2–7.3, 275–285 mOsm). A bipolar stimulating electrode was placed rostral to the recording electrode and was used to stimulate excitatory afferents at 0.1 Hz. Series resistance (20–30 MΩ) and input resistance were monitored on-line with a 4 mV depolarizing step (50 ms) given after every afferent stimulus. Putative VTA dopamine neurons were identified by the presence of an Ih current (Johnson and North, 1992), using a series of 500 ms hyperpolarizing steps (in 10 mV increments) from a holding potential of −70 mV. Ih is present in both dopamine and nondopamine VTA neurons in rats (Margolis et al., 2006); however, previous studies in mice found that 98% of Ih-containing neurons were tyrosine hydroxylase positive, indicative of dopamine neurons, when recordings are focused on neurons just medial to the medial terminal nucleus of the accessory optic tract (Wanat et al., 2008), where nearly all the recordings in the present study were performed. Neurons were voltage clamped at −70 or +40 mV to record AMPA receptor (AMPAR)- or NMDA receptor (NMDAR)-mediated EPSCs, respectively, and EPSCs were filtered at 2 kHz, digitized at 10 kHz, and collected on-line using Igor Pro software (Wavemetrics). mEPSCs were recorded in cells voltage clamped at −70 mV in the presence of lidocaine (500 μm) and AP-5 (50 μm) to suppress spontaneous action potential-driven release and NMDAR mEPSCs. AMPAR mEPSCs were collected with an acquisition rate of 100 kHz and filtered at 2 kHz using Clampex 9.2 (Molecular Devices). The Mini Analysis Program (Synaptosoft) was used to analyze data with filtering during analysis at 1 kHz. Detection parameters in the Mini Analysis Program were set at an amplitude threshold of 7 pA. Briefly, the software calculates the peak amplitude by taking the amplitude at the local maximum minus the average baseline, and then this value is compared with the amplitude threshold. A 10 ms period to search local maximum was used. Time before peak for baseline was 5 ms, and the period to average baseline was 1 ms. The period to search a decay time (the time to decay) was 10 ms, and the fraction of peak to find a decay time was 0.5 ms. Events were eliminated manually if the time to the peak was >1 ms or the time to the decay point was >3 ms. For a given cell, mEPSCs were collected for 4 min, and data were only included if the holding current at −70 mV was <200 pA and the root mean square noise was <2 pA to assure a high signal-to-noise ratio. All data were also visually inspected to prevent noise disturbance of the analysis. For comparisons of probability distributions, a Kolmogorov–Smirnov test was performed with a threshold of p < 0.05. Only one experiment was performed on a single neuron from a given brain slice.
All chemicals were purchased from Sigma, except human/rat CRF (Bachem).
Statistical analysis.
All values are expressed as mean ± SEM. Statistical significance was assessed using a two-tailed Student's t test, a one-way ANOVA, a two-way repeated-measures ANOVA with a Bonferroni's post hoc test for multiple group comparisons, or a Kolmogorov–Smirnov test.
Results
To examine how chronic exposure to cocaine could alter CRF responsiveness at the cellular level in VTA dopamine neurons, C57BL/6 mice received five intraperitoneal injections (one per day) of either 15 mg/kg cocaine or an equivalent volume of saline. Slice preparation and electrophysiological recordings occurred 24–28 h after the final injection. This cocaine treatment regimen was chosen because it induces behavioral sensitization in mice (Thomas et al., 2001) and also facilitates the locomotor response to CRF given 1–2 d after the last cocaine treatment (Erb et al., 2003).
We first examined whether repeated in vivo cocaine administration altered the effects of CRF on NMDAR- and AMPAR-mediated EPSCs. NMDAR EPSCs were evoked while holding neurons at +40 mV, and amplitudes were measured 20 ms after the stimulation artifact, a time point when EPSCs are purely NMDAR mediated (Ungless et al., 2001), which was confirmed by pharmacological assessment showing that the AMPA/kainate receptor antagonist CNQX (6-cyano-7-nitroquinoxaline-2,3-dione) had no effect on amplitudes of NMDAR currents at this time point (Fig. S1, available at www.jneurosci.org as supplemental material). The effects of CRF were measured at the 10–15 min time period after the CRF application; CRF-induced NMDAR EPSC potentiation reached a plateau at this time period with a greater effect at 10–15 min compared with the 5–10 min time period of the application in all three groups (effects of 5–10 min vs 10–15 min after the CRF application in all three groups; p < 0.05, t test). In agreement with our previous study (Ungless et al., 2003), bath-applied CRF (1 μm) induced a slowly developing potentiation of NMDAR EPSCs in VTA dopamine neurons from naive and saline-treated mice (Fig. 1) (naive: 128 ± 2.7% of baseline, n = 4; saline: 127. 4 ± 1.8% of baseline, n = 5; increase determined as the average from 10–15 min after the initiation of CRF application; both p < 0.001). However, with previous exposure to cocaine, the initial CRF-induced potentiation of NMDAR EPSCs was markedly enhanced (Fig. 1) (152.4 ± 3% of baseline; n = 6; p < 0.001) and significantly greater than those from naive and saline-treated groups (p < 0.001). Since repeated cocaine exposure strengthens excitatory inputs onto VTA dopamine neurons by increasing the ratio of AMPAR-mediated to NMDAR-mediated EPSCs (Borgland et al., 2004), we examined whether CRF might modulate AMPAR EPSCs in neurons held at −70 mV (Borgland et al., 2006). Bath application of CRF (1 μm) had no effect on AMPAR EPSCs in naive (Fig. 2A,D) (103.9 ± 2.7% of baseline; n = 6) and saline-treated (Fig. 2B,D) (103.2 ± 1% of baseline; n = 5) mice but induced a small but significant potentiation in VTA AMPAR EPSCs in cocaine-treated mice (Fig. 2C,D) (cocaine: 122.1 ± 1.4% of baseline; n = 5; p < 0.001). Thus, previous exposure to cocaine enhanced the CRF responsiveness of excitatory synaptic transmission onto VTA dopamine neurons such that CRF potentiates both NMDAR- and AMPAR-mediated EPSCs.

Repeated cocaine exposure enhances potentiation of NMDAR-mediated synaptic transmission by CRF in the VTA. A, Top, Representative traces of NMDAR EPSCs evoked at +40 mV before (baseline) and 15 min after application of 1 μm CRF onto VTA dopamine neurons from naive (left), saline-treated (middle), and cocaine-treated (right) mice. Middle, Examples of the effect of 1 μm CRF on NMDAR EPSCs. Bottom, Summarized responses to 1 μm CRF showing that the CRF increase in NMDAR EPSCs persisted after washout, with a significantly greater magnitude and duration in cocaine-treated mice (right) relative to saline-treated (middle) and naive (left) mice. Black horizontal bars indicate a 10 min application of CRF. B, Summary of data from A with each colored bar representing the mean ± SEM of EPSCs at two different time periods shown in A with matched colors (peach, CRF initial, 10–15 min after application of CRF; pale violet, CRF late, 15–20 min after washout of CRF). Note that potentiation of NMDAR EPSCs after washout following CRF application was much larger in neurons from cocaine-treated mice. ***p < 0.001, CRF change versus baseline (one-way ANOVA with Bonferroni's post test within a group); ###p < 0.001, one-way ANOVA with Bonferroni's post test across groups at two different time points.

CRF potentiates AMPAR-mediated synaptic transmission onto the VTA only in cocaine-treated mice. A, B, CRF (1 μm) did not increase AMPAR EPSCs evoked at −70 mV in neurons from naive (A) and saline-treated (B) mice. C, Potentiation of VTA AMPAR EPSCs during CRF application from cocaine-treated mice. Insets show representative traces of AMPAR EPSCs before (baseline) and 7 min after application of CRF. The stimulation artifact was eliminated from all traces. D, Summary of data from A–C, representing the mean ± SEM averaged from 5–10 min after CRF application. ***p < 0.001, one-way ANOVA with Bonferroni's post test across groups during CRF application.
In addition to a greater initial CRF increase in NMDAR EPSCs after chronic cocaine, CRF also produced a sustained enhancement of NMDAR EPSCs that persisted after washout of CRF. The CRF-induced potentiation of NMDAR EPSCs was diminished in saline-treated and naive mice after a 15–20 min washout (Fig. 1) (naive, 112.3 ± 2.9% of baseline; saline, 109.4 ± 3% of baseline). In contrast, in cocaine-treated mice, the CRF-induced potentiation of NMDAR EPSCs was still present after a 15–20 min washout (Fig. 1) (150.3 ± 4% of baseline; p < 0.001) and was significantly greater than in saline-treated or naive mice at this time point (Fig. 1B) (p < 0.001). Importantly, after repeated cocaine, CRF did not produce enduring effects on AMPAR EPSCs (Fig. 2C), suggesting that the sustained CRF effect after cocaine was selective for postsynaptic NMDARs.
CRF signals through two different G-protein-coupled receptors, CRF receptor type 1 (CRFR1) and CRFR2, and these receptors are linked to multiple signaling pathways (Hauger et al., 2006; Hillhouse and Grammatopoulos, 2006; Gallagher et al., 2008). Therefore, we sought to determine which receptor subtypes and intracellular pathways underlie the enhanced CRF responsiveness of VTA NMDARs after chronic cocaine. In saline-treated mice, the initial NMDAR EPSC potentiation by CRF was reduced by the selective CRFR2 antagonist atressin2B (AST2B) (Fig. 3C,D) (113.1 ± 3.2% of baseline; n = 5; p < 0.01 compared with data without antagonists from Fig. 1). In contrast, the selective CRFR1 antagonist NBI 27914 (NBI) did not affect the initial CRF effects on NMDAR EPSCs in saline-treated mice, even at a relatively high dose (500 nm) (Fig. 3A,B) (121.6 ± 3.2; n = 4; p > 0.05); a role for CRFR2 but not CRFR1 agrees with what was observed in naive mice (Ungless et al., 2003). Importantly, in cocaine-treated mice, the initial CRF-induced potentiation of NMDAR EPSCs was significantly reduced by inhibition of CRFR2 (Fig. 3G,H) (AST2B: 123.8 ± 10% of baseline; n = 6) and also by inhibition of CRFR1 (Fig. 3E,F) (NBI: 126 ± 2% of baseline; n = 4; both p < 0.001 compared with data without antagonists from Fig. 1). Although neither NBI nor AST2B completely blocked the initial CRF effect in cocaine-treated mice, coapplication of the two antagonists nearly prevented the CRF effects (Fig. 3I,J) (96.6 ± 1.3% of baseline; n = 4) as it did in naive and saline-treated mice (Fig. S2, available at www.jneurosci.org as supplemental material) (naive: 103.2 ± 1.3% of baseline, n = 4; saline: 108 ± 1.7% of baseline, n = 4). Interestingly, the initial increase by CRF in NMDAR EPSCs in cocaine-treated mice with the CRFR1 antagonist (Fig. 3F) (126%) was very similar to that of saline-treated mice without antagonists (Fig. 1B) (127%), suggesting that a CRFR1-mediated initial enhancement of NMDAR EPSCs was recruited after repeated cocaine exposure, as well as a CRFR2-mediated increase in NMDAR EPSCs apparent in both control and cocaine-treated mice.

Enhanced CRF-induced potentiation of NMDAR EPSCs in VTA neurons from cocaine-treated mice is mediated through both CRFR1 and CRFR2. Bar graphs represent data from two different time periods, indicated by matched colors (peach, CRF initial, 10–15 min after application of CRF; pale violet, CRF late, 15–20 min after CRF washout). Data without an antagonist are the same as in Figure 1. A, B, The CRFR1 antagonist NBI (500 nm) had no effect on the initial time period of CRF-induced NMDAR EPSC potentiation. C, D, The CRFR2 antagonist AST2B (100 nm) decreased the CRF-induced potentiation of NMDAR EPSCs at the initial time periods in saline-treated mice. E–H, NBI (E, F) and astressin2B (G, H) significantly reduced the CRF-induced NMDAR EPSC potentiation in cocaine-treated mice at both the initial and later time points. I, J, Coapplication of NBI and AST2B fully blocked the enhanced CRF effect after chronic cocaine. **p < 0.01; ***p < 0.001, comparing CRF effect with and without antagonists (two-way ANOVA with Bonferroni's post test).
In addition to recruitment of novel CRF receptor mechanisms after repeated cocaine exposure, the intracellular signaling molecules underlying the initial CRF potentiation of NMDAR EPSCs by CRF were also altered after cocaine exposure. For these experiments, either the protein kinase A (PKA) inhibitor, protein kinase inhibitor (PKI; 20 μm), or the PKC inhibitor bisindolylmaleimide-1 (BIS; 1 μm) was included in the recording pipette, and cells were equilibrated before CRF was tested. In saline-treated mice, the initial CRF-induced potentiation of NMDAR EPSCs was reduced by the PKC inhibitor BIS (Fig. 4C,D) (112.7 ± 1.5% of baseline; n = 5; p < 0.001) but not the PKA inhibitor PKI (Fig. 4A,B) (132.4 ± 3% of baseline; n = 4), as has been reported for naive mice (Ungless et al., 2003). In contrast, in cocaine-treated mice, the initial enhancement of NMDAR EPSCs by CRF was significantly reduced by PKA inhibition (Fig. 4E,F) (122 ± 1.8% of baseline; n = 5) or PKC inhibition (Fig. 4G,H) (111.5 ± 1.1% of baseline; n = 8). Combined PKA and PKC inhibition also greatly reduces the CRF effect (Fig. 4I,J) (112.9 ± 3.5% of baseline; n = 6), as seen in saline-treated mice (Fig. S2, available at www.jneurosci.org as supplemental material) (102 ± 1% of baseline; n = 4; both p < 0.001 compared with data without inhibitors from Fig. 1). Furthermore, as observed for the contribution of CRFR1, the initial CRF-induced enhancement of NMDAR EPSCs in cocaine-treated mice with PKA inhibition (Fig. 4F) (122% of baseline) was very similar to that of saline-treated mice without inhibitors (Fig. 1B) (127% of baseline). Together, these results suggest that a CRFR1- and PKA-mediated initial enhancement of NMDAR EPSCs was recruited after repeated cocaine exposure.

PKA contribution to the initial CRF-induced potentiation of NMDAR EPSCs appears only after chronic cocaine. Data without inhibitors are the same as in Figure 1. A, B, Intracellular application of the PKA inhibitor PKI (20 μm) did not affect CRF-induced NMDAR EPSC potentiation at the initial time period in saline-treated mice. C, D, Intracellular application of the PKC inhibitor BIS (1 μm) reduced the CRF-induced NMDAR EPSC potentiation at the initial time period in saline-treated mice. E–H, Intracellular PKI (E, F) and BIS (G, H) decreased the augmented NMDAR EPSC potentiation by CRF in cocaine-treated mice at both time points. I, J, Coapplication of PKI and BIS completely blocked the enhanced CRF effect after chronic cocaine. ***p < 0.001, comparing CRF effect with and without inhibitors (two-way ANOVA with Bonferroni's post test).
Our results also indicate that the signaling mechanism underlying the cocaine-induced sustained enhancement of NMDAR EPSCs that persists after washout of CRF was similar to that mediating the initial CRF enhancement of NMDAR EPSCs. First, the sustained NMDAR EPSC enhancement after CRF washout in cocaine animals was significantly reduced by antagonists of either CRFR1 or CRFR2 (Fig. 3E–H) (15–20 min after washout: NBI, 110.6 ± 2% of baseline; AST2B, 97 ± 3.2% of baseline; all p < 0.001 compared with data without antagonists from Fig. 1). Second, the sustained NMDAR EPSC enhancement after repeated cocaine required both PKC and PKA (Fig. 4E–H) (15–20 min after washout: PKI, 101.2 ± 1.1 of baseline; BIS, 113.9 ± 2.4% of baseline; all p < 0.001 compared with data without inhibitors from Fig. 1). Coapplication of either the two receptor antagonists or the two kinase inhibitors completely blocked the longer-lasting NMDAR EPSC enhancement by CRF (Figs. 3I,J, 4I,J) (15–20 min after washout: NBI plus AST2B, 98 ± 2.3% baseline, n = 5; PKI plus BIS, 102.3 ± 2% of baseline, n = 6; both p < 0.001 compared with data without antagonists from Fig. 1). Thus, the receptor and intracellular pathways underlying the sustained NMDAR EPSC enhancement after repeated cocaine were similar during the initial and later time periods, requiring CRFR1, CRFR2, PKA, and PKC to increase both the magnitude and duration of the NMDAR EPSC potentiation by CRF.
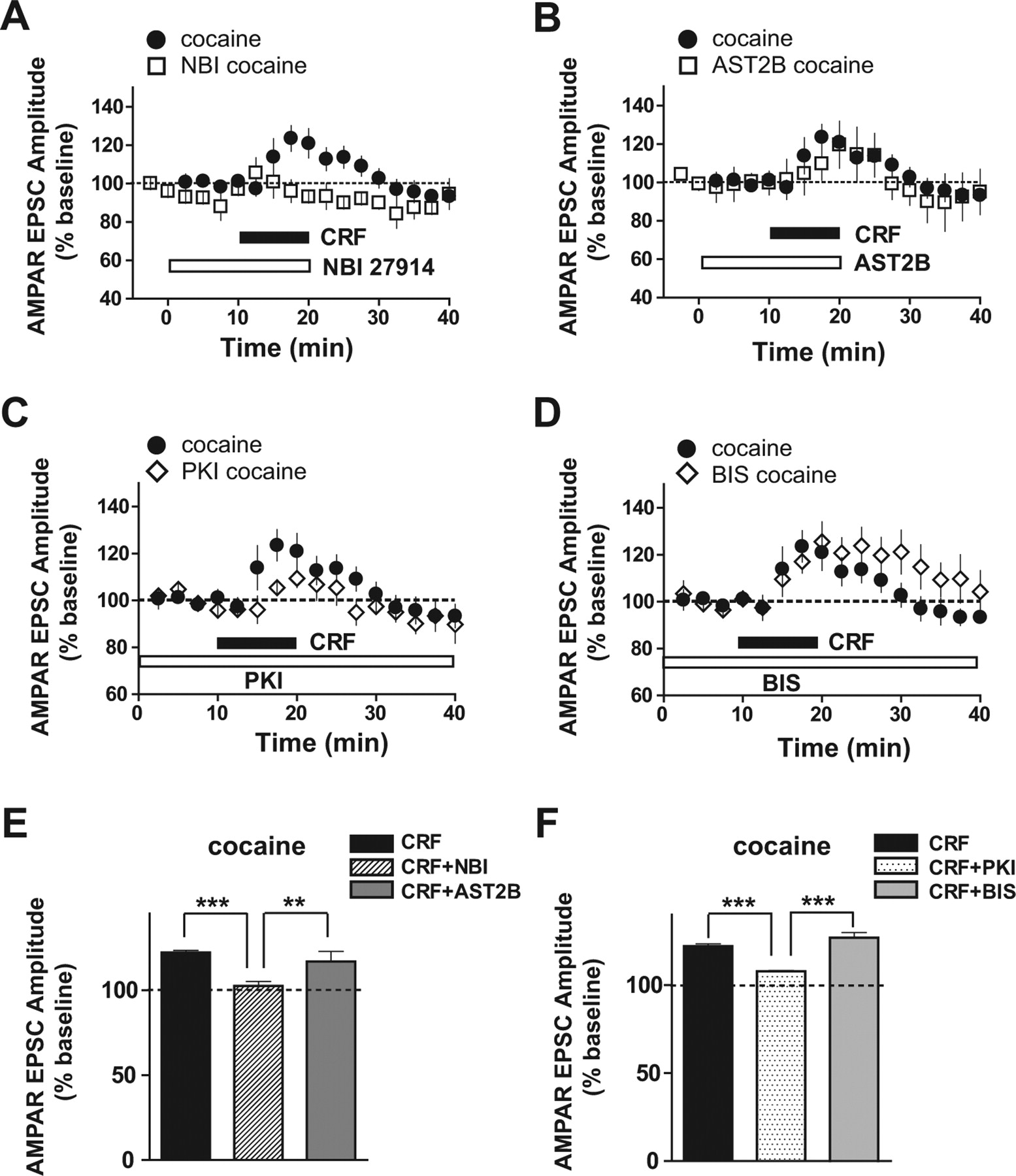
We subsequently examined the receptor and kinase pathways underlying the transient CRF-induced potentiation of AMPAR EPSCs in cocaine-treated mice (Fig. 2) to determine whether CRF-induced potentiation of AMPAR-mediated EPSCs after chronic cocaine occurred through the same signaling pathway observed for NMDAR currents. Interestingly, the AMPAR potentiation by CRF in cocaine-treated mice was still observed in the presence of the CRFR2 antagonist AST2B or PKC inhibitor BIS (Fig. 5B,D,E,F) (CRF: 122.1 ± 1.4, n = 5; CRF plus AST2B: 116.8 ± 6, n = 5; CRF plus BIS: 126.9 ± 3, n = 5). In contrast, the CRF-induced AMPAR EPSC potentiation was significantly decreased by the CRFR1 antagonist NBI or PKA inhibitor PKI (Fig. 5A,C,E,F) (NBI: 102.5 ± 2.6, n = 5; PKI: 107.9 ± 0.5, n = 5; both p < 0.001). Thus, after repeated cocaine exposure, a newly recruited CRFR1- and PKA-dependent mechanism mediated the CRF increase in both AMPAR and NMDAR currents. Furthermore, a single exposure to cocaine failed to potentiate the CRF increase in NMDAR EPSCs but was sufficient to induce CRF potentiation of AMPAR EPSCs (Fig. S3, available at www.jneurosci.org as supplemental material), suggesting that distinct mechanisms likely engage the CRF-induced potentiation of NMDA and AMPA EPSCs. Nonetheless, CRF-induced AMPA EPSC potentiation after cocaine is likely to occur through distinct mechanisms from a cocaine-induced increase in AMPAR/NMDAR current ratio (Ungless et al., 2001) because CRF rapidly increased AMPAR EPSCs only after cocaine preexposure (present study), whereas cocaine exposure previously resulted in a slowly increased AMPAR/NMDAR current ratio (Argilli et al., 2008). Future studies will be necessary to uncover the mechanisms underlying such observations.

CRF-induced potentiation of AMPAR EPSCs after repeated cocaine exposure is mediated via CRFR1 and PKA. Data without antagonists or inhibitors are the same as in Figure 2. A, B, CRF-induced AMPAR EPSC enhancement after chronic cocaine was blocked by NBI (A) but not AST2B (B). C, D, PKI decreased the CRF-induced AMPAR EPSC potentiation after chronic cocaine (C) but not BIS (D). E, Averaged data from A and B from 5–10 min after CRF application. F, Averaged data from C and D. **p < 0.01; ***p < 0.001, one-way ANOVA with Bonferroni's post test.
Intra-VTA injection of CRF increases glutamate levels in cocaine-experienced rats (Wang et al., 2005); therefore, we investigated whether chronic cocaine might alter CRF regulation of presynaptic glutamatergic function in the VTA. AMPAR mEPSCs were recorded at a holding potential of −70 mV in the presence of lidocaine and AP-5. We analyzed AMPAR mEPSCs obtained during the last 4 min of the 10 min CRF application since the effect of CRF on AMPAR currents in cocaine-treated mice peaked ∼8–10 min during the 10 min application (Fig. 2C). The frequency of AMPAR mEPSCs, an indicator of presynaptic action, was significantly increased by CRF selectively in cocaine- but not saline-treated mice (Fig. 6A,C) (saline: baseline, 0.8 ± 0.4 Hz; CRF, 1.2 ± 0.6 Hz; cocaine: baseline:,1.1 ± 0.2 Hz; CRF, 2 ± 0.3 Hz; p < 0.05, t test; n = 7 each for saline and cocaine groups). Accordingly, probability distributions of frequency showed a significant difference after CRF only in cocaine-treated animals (saline, p > 0.05; cocaine, p < 0.01; Kolmogorov–Smirnov test), suggesting that CRF can increase presynaptic glutamate release or the number of synaptic contacts (Malenka and Nicoll, 1999) in cocaine-treated mice. In contrast, although cocaine exposure significantly increased AMPAR mEPSC amplitude relative to saline (p < 0.05), as shown previously (Ungless et al., 2003), CRF did not alter AMPAR mEPSC amplitude in saline-treated (baseline, 11 ± 0.8 pA; CRF, 11 ± 0.5 pA; n = 7; both p > 0.05 both with t test and Kolmogorov–Smirnov test) or cocaine-treated (baseline, 14 ± 1 pA; CRF, 13 ± 0.8 pA; n = 7; p > 0.05 both with t test and Kolmogorov–Smirnov test) mice (Fig. 6A,B). These data suggest that CRF increases the probability of glutamate release via a presynaptic mechanism after cocaine exposure. These experiments, together with the fact that the CRF-induced increase of AMPAR EPSCs in cocaine-treated mice was prevented by intracellular application of a PKA inhibitor (Fig. 5C,F), suggest that repeated cocaine exposure enables CRF to increase VTA glutamatergic function both presynaptically and postsynaptically.

Repeated cocaine exposure alters the presynaptic and postsynaptic effects of CRF in VTA neurons. A, Example saline and cocaine AMPAR mEPSC recordings at −70 mV at baseline and after CRF from the same VTA neuron, respectively. B, C, Averaged AMPAR mEPSCs from 4 min baseline before CRF and from the last 4 min of the 10 min CRF exposure, showing that CRF did not alter the mEPSC amplitude in saline- or cocaine-treated mice (B) but enhanced the mEPSC frequency significantly only in cocaine-treated mice (C). D, E, Representative cumulative probability plots of mEPSC amplitude (D) (saline baseline, 200 mEPSCs; saline CRF, 280 mEPSCs; cocaine baseline, 240 mEPSCs; cocaine CRF, 450 mEPSCs) and frequency (E) (saline baseline, 360 mEPSCs; saline CRF, 420 mEPSCs; cocaine baseline, 280 mEPSCs; cocaine CRF, 500 mEPSCs) from individual cells from each group. Note the leftward shift and increased slope with CRF application after chronic cocaine in E. *p < 0.05, CRF change versus baseline (t test).
Discussion
Here, we show that enhancement of glutamatergic transmission in VTA dopamine neurons by CRF was significantly potentiated by previous exposure to cocaine; these results provide evidence of a novel cellular mechanism through which drug exposure can facilitate responsiveness to stress, given the critical role of stress in relapse in humans (Sinha, 2001). In addition to the greater magnitude and duration of the CRF-induced NMDAR EPSC potentiation after chronic cocaine, CRF potentiated AMPAR EPSCs only in cocaine-treated mice, uncovering an additional capacity for potentiation of AMPAR transmission after cocaine exposure in addition to a cocaine-induced increase in AMPAR/NMDAR current ratio (Ungless et al., 2003). Furthermore, analyses of AMPAR mEPSCs and intracellular pharmacological experiments revealed that CRF could modulate presynaptic as well as postsynaptic glutamatergic function. Interestingly, the CRF-induced AMPAR EPSC potentiation and the enhanced CRF potentiation of NMDARs after repeated cocaine were both mediated by a newly recruited CRFR1- and PKA-dependent mechanism. We speculate that this longer-lasting potentiation of VTA NMDAR currents after chronic cocaine could enhance VTA burst firing (Overton and Clark, 1997) or synaptic plasticity (Kauer and Malenka, 2007) after exposure to stress and could also contribute to the long-lasting behavioral effects of brief stressful events (see below).
CRF signaling can occur through activation of two subtypes of receptors: CRFR1 and CRFR2. Although CRF receptors are often reported to couple to the cAMP/PKA signaling pathway, a growing body of evidence suggests that CRF receptors can activate multiple types of G-proteins linking to a variety of second messengers (Hauger et al., 2006). For example, studies in the VTA have found that the PKC pathway can be activated by CRFR1 or CRFR2 depending on the molecular target (Ungless et al., 2003; Wanat et al., 2008) and also that CRFR2 can act through PKA to increase intracellular Ca2+ in dopamine neurons (Riegel and Williams, 2008). Our observation that repeated cocaine exposure recruited a CRFR1- and PKA-dependent enhancement of NMDAR and AMPAR EPSCs during CRF exposure is in agreement with other studies demonstrating CRFR1-dependent signaling after chronic cocaine or ethanol treatment but not under control conditions (Pollandt et al., 2006; Funk et al., 2007), with one study showing a CRFR1- and PKA-dependent regulation of amygdala glutamatergic signaling only after repeated cocaine exposure (Pollandt et al., 2006). Increased CRFR1-dependent signaling after cocaine could reflect increased expression and/or the functional sensitivity of CRFR1 (Hauger et al., 2006; Gallagher et al., 2008). Alternately, several studies have shown increased basal activity of PKA after repeated exposure to cocaine or other drugs of abuse (Terwilliger et al., 1991; Ortiz et al., 1996; Hope et al., 2007), which could amplify CRF receptor signaling. Also, PKA and PKC could act directly on glutamate receptors or through regulatory proteins to increase receptor currents (Newpher and Ehlers, 2008).
Importantly, we found that the CRF enhancement of NMDAR EPSCs was significantly larger and longer-lasting after repeated cocaine exposure. Interestingly, this enhancement of potentiation of NMDAR EPSCs exhibited a novel pharmacology in cocaine-treated mice, with a requirement for both CRFR1 and CRFR2 and for both PKA and PKC. Many physiological actions of CRF exhibit antagonistic interactions of CRFR1 and CRFR2 (Hillhouse and Grammatopoulos, 2006); however, CRFR1 and CRFR2 can also exhibit additive or more complex interactions (Rebaudo et al., 2001; Hauger et al., 2006; Gao et al., 2008). In the amygdala, CRFR2/PKC mediates a potentiation of glutamate receptor currents, with an additional enhancement after cocaine through the CRFR1/PKA pathway recruited (Pollandt et al., 2006). This is similar to the initial CRF effect on VTA NMDAR EPSCs observed here, where enhancement of NMDAR EPSCs through a CRFR2/PKC pathway was present in control and cocaine neurons, and VTA dopamine neurons from cocaine-treated mice had an additional CRFR1/PKA-dependent pathway that further enhanced NMDAR currents. More unusual are cases in which CRFR1 and CRFR2 are both necessary for a physiological action of CRF; some behavioral studies suggest this to be the case (Mousa et al., 2003; Hauger et al., 2006) but do not directly demonstrate that CRFR1 and CRFR2 are acting through the same cell. Together, our results suggest a novel mechanism through which activation of both CRFR1 and CRFR2 resulted in enhanced potentiation of NMDAR function after cocaine.
To further characterize the mechanism underlying the enhanced CRF modulation of VTA glutamatergic function, we examined the effect of CRF on AMPAR mEPSCs, which can help delineate whether altered glutamate signaling reflects presynaptic or postsynaptic alterations. We found that CRF enhanced AMPAR mEPSC frequency only in cocaine mice, suggesting CRF modulation of glutamate release in the VTA after cocaine exposure. Alternately, enhanced AMPAR mEPSC frequency can also result from postsynaptic modification such as an increased number of synaptic contacts (Malenka and Nicoll, 1999), although such changes are less likely to contribute during the short time period of CRF exposure (10 min). However, intracellular application of kinase inhibitors significantly reduced the CRF enhancement of NMDAR and AMPAR EPSCs, strongly suggesting a postsynaptic site of action for CRF. Also, presynaptic modulation of glutamate release should alter both AMPAR and NMDAR EPSCs, and the observation that CRF exposure greatly increased NMDAR EPSCs but not AMPAR EPSCs during the period after CRF washout indicates a postsynaptic site of action for the sustained CRF modulation of NMDAR EPSCs. Thus, repeated cocaine exposure enables CRF to regulate VTA glutamatergic function both presynaptically and postsynaptically, with a predominant role for postsynaptic action of CRF.
Studies of CRF receptor localization have suggested that these receptors can be localized presynaptically or postsynaptically across different brain regions. CRFR2 s have been shown to be localized both postsynaptically (Hauger et al., 2006) and presynaptically (Lawrence et al., 2002; Swinny et al., 2003). In addition, CRFR1 and CRFR2 functions have been reported at both presynaptic and postsynaptic sites (Fu and Neugebauer, 2008; Gallagher et al., 2008; Nie et al., 2009). Within the VTA, CRFR2 has been suggested to be presynaptic (Wang et al., 2007) and postsynaptic (Ungless et al., 2003), and the CRFR1 has been reported to be postsynaptic (Van Pett et al., 2000; Wanat et al., 2008). However, the precise location of the different CRF receptors has not been fully demonstrated, partly because of the poor performance of antibodies.
The cocaine treatment regimen used here was chosen because it induces behavioral sensitization in mice (Thomas et al., 2001) and also facilitates the locomotor response to CRF given 1–2 d after the last cocaine treatment (Erb et al., 2003). Interestingly, we show here that after repeated cocaine exposure, CRF leads to a sustained enhancement of CRF-induced NMDAR EPSC potentiation that persists after washout of the CRF, and which exhibits a different receptor and kinase pharmacology from the initial enhancement of NMDAR EPSCs. This cocaine-induced potentiation of CRF responsivity on VTA NMDAR function would allow stressor-related CRF increases to produce a sustained enhancement in VTA burst firing activity and dopamine release in VTA target regions (Overton and Clark, 1997) and/or to facilitate induction of synaptic plasticity in the VTA (Kauer and Malenka, 2007). Interestingly, persistent changes of glutamatergic transmission in VTA dopamine neurons have been implicated in neural adaptation induced by drugs of abuse (Ungless et al., 2001; Bellone and Lüscher, 2006; Chen et al., 2008), and this mechanism is also thought to be the cellular basis of drug-induced behavioral sensitization (Everitt and Wolf, 2002), a model for development of addiction (Robinson and Berridge, 2001). In addition, repeated cocaine exposure has been shown to enhance locomotor response to stressors (Antelman et al., 1980; Hamamura and Fibiger, 1993; Blatchford et al., 2005). Brief exposure to stressors can have long-lasting behavioral consequences (Kalivas and Stewart, 1991; Miczek et al., 2004), which have been shown to be independent of the corticosterone activation during the stress response (Covington and Miczek, 2005). Furthermore, VTA NMDARs may represent a critical site for stress regulation of drug seeking (Covington et al., 2008) and for regulation of VTA plasticity after chronic drug exposure (Engblom et al., 2008). Thus, we speculate that CRF facilitation of VTA NMDAR and AMPAR function after repeated cocaine could augment the ability of brief stressful events to control behavior.
In summary, our study suggests that the enhanced ability of CRF with chronic cocaine to regulate glutamatergic inputs onto VTA dopamine neurons represents a plausible cellular mechanism where drugs of abuse and stress could interact. After repeated cocaine exposure, a CRFR1/PKA-dependent mechanism was recruited, which allowed CRF to enhance AMPAR currents and to greatly elevate NMDAR currents relative to control mice. Importantly, we found that chronic cocaine significantly enhanced the magnitude and duration of this CRF-induced NMDAR EPSC potentiation, that this potentiation persisted after CRF washout, and that this enhanced NMDAR potentiation showed a novel CRF receptor pharmacology. It has been proposed that repeated exposure to drugs of abuse may produce long-term neuroadaptations that alter stress responsivity in a way that enhances drug seeking (Wang et al., 2007) and CRF increases VTA glutamatergic synapse function, which could facilitate VTA burst firing or induction of synaptic plasticity. Therefore, in this way, previous experience with drugs of abuse may facilitate the ability of stress to drive drug seeking.
Footnotes
-
This work was supported by National Institute on Drug Abuse Grant RO1 A016782 (A.B.) and by funding from the state of California for medical research on alcohol and substance abuse through the University of California, San Francisco (A.B.).
- Correspondence should be addressed to Antonello Bonci, Ernest Gallo Clinic and Research Center, 5858 Horton Street, Suite 200, Emeryville, CA 94608. antonello.bonci{at}ucsf.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}