

Abstract
We tested a hypothesis that the spinal plasticity induced within a few hours after nerve injury may produce changes in cortical activities and an initial phase of neuropathic pain. Somatosensory cortical responses elicited by vibratory stimulation were visualized by transcranial flavoprotein fluorescence imaging in mice. These responses were reduced immediately after cutting the sensory nerves. However, the remaining cortical responses mediated by nearby nerves were potentiated within a few hours after nerve cutting. Nerve injury induces neuropathic pain. In the present study, mice exhibited tactile allodynia 1–2 weeks after nerve injury. Lesioning of the ipsilateral dorsal column, mediating tactile cortical responses, abolished somatic cortical responses to tactile stimuli. However, nontactile cortical responses appeared in response to the same tactile stimuli within a few hours after nerve injury, indicating that tactile allodynia was acutely initiated. We investigated the trigger mechanisms underlying the cortical changes. Endogenous glial cell line-derived neurotrophic factor (GDNF), found in the Meissner corpuscles, induced basal firing ∼0.1 Hz or less in its Aβ tactile afferents, and disruption of the basal firing triggered the potentiation of nontactile cortical responses. Application of 10 nm LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid], a specific antagonist of group II metabotropic glutamate receptors (mGluRs), on to the surface of the spinal cord also induced the potentiation of nontactile cortical responses. Together, it is suggested that low-frequency afferent firing produced by GDNF in touch-sensitive nerve fibers continuously activated spinal group II mGluRs and that failure of this activation triggered tactile allodynia.
Introduction
Injuries to peripheral sensory nerves induce neuropathic pain (Bennett and Xie, 1988; Kim and Chung, 1992; Decosterd and Woolf, 2000) and somatic cortical plasticity (Merzenich et al., 1983). However, the relationship between these two phenomena has not been well investigated thus far, because the underlying mechanisms are considered to be different. Neuropathic pain is established as a consequence of a complex cascade of inflammation and gene expression (Campbell and Meyer, 2006), whereas cortical plasticity is found within a few hours after peripheral nerve cutting (Calford and Tweedale, 1988; Björkman et al., 2009). The complexity of the mechanisms involved in neuropathic pain prevents us from identifying the primary etiology and developing an effective therapy for neuropathic pain. However, the early cortical plasticity may be intimately related to the initial phase of neuropathic pain, because both are closely correlated in the phantom pain in the chronic phase (Flor et al., 1995). The beginning of neuropathic pain after nerve injury has not yet been investigated, mainly because neuropathic pain is evaluated by behavioral tests performed after recovery from the surgical procedures for inducing neuropathic pain (Bennett and Xie, 1988; Kim and Chung, 1992; Decosterd and Woolf, 2000). In the present study, we investigated the initial phase of neuropathic pain as a sensory modality shift from tactile information, which is mainly mediated by the ipsilateral dorsal column–medial lemniscal system in the spinal cord (Jain et al., 1995), to nociceptive information, which is mainly mediated by the contralateral spinothalamic tract (Peschanski et al., 1985). Based on the anatomical features of the spinal afferent pathways, we found that the sensory modality shift from tactile to nociceptive information was initiated at the spinal cord level within a few hours after nerve injury.
The behavioral tests for investigating neuropathic pain are affected by many factors, and therefore, manipulations or drug applications that affect the results of the behavioral tests may have no direct effect on neuropathic pain itself. However, observation of the sensory modality shift at the spinal cord level might serve as a straightforward method for investigating neuropathic pain. Activity-dependent fluorescence changes derived from mitochondrial flavoproteins have been used in vivo for visualizing brain activities (Shibuki et al., 2003; Reinert et al., 2004). Cortical fluorescence changes can be imaged through the intact skull without surgical damage to the brain in mice (Tohmi et al., 2006; Kubota et al., 2008; Kitaura et al., 2010). Flavoprotein signals are resistant to and spontaneously recover from photobleaching (Kubota et al., 2008) and quantitatively reflect neural activities (Tohmi et al., 2006). Therefore, we used this technique to quantify the cortical changes after peripheral nerve cutting. This approach combined with manipulation of spinal neural circuits enabled us to investigate the initial phase of neuropathic pain. Using this method, we obtained evidence suggesting that neuropathic pain is triggered by conduction blockade of afferent basal firing, which is produced by glial cell line-derived neurotrophic factor (GDNF) (Paratcha and Ledda, 2008) in touch-sensitive nerve fibers, and the consequent failure of sustained activation of group II metabotropic glutamate receptors (mGluRs) in the spinal cord.
Materials and Methods
The Ethics Committee of Niigata University approved the experimental protocols used in this study. Male C57BL/6 mice between 8 and 10 weeks old and purchased from Charles River Japan were used for all experiments.
Surgical procedures.
The surgical procedures and imaging experiments were performed as described previously (Tohmi et al., 2006; Kubota et al., 2008; Kitaura et al., 2010). Mice were anesthetized with urethane (1.65 g/kg, i.p.). During the experiments, body temperature was monitored using a rectal probe and maintained at 38°C using a silicon rubber heater. When the cortical responses to stimulation of the forepaw were investigated, the left brachial plexus was exposed, and the median and ulnar (M/U) nerves or radial and musculocutaneous (R/Mc) nerves were isolated for later nerve cutting. We also investigated the cortical responses to hindpaw stimulation, because it was difficult to analyze the effects of the spinal cord lesioning on the responses to forepaw stimulation. When the cortical responses to stimulation of the hindpaw were investigated, the left tibial nerve was isolated. After subcutaneous injection of bupivacaine (AstraZeneca), disinfected head skin was incised and the skull over the right somatosensory cortex was exposed. The surface of the skull was cleaned with sterile saline, and a small piece of metal was attached to the skull with dental acrylic resin (Super Bond; Sun Medical). The piece of metal was screwed to a manipulator to fix the head position. The surface of the skull was covered with liquid paraffin to prevent drying and to keep the skull transparent. These surgical operations were usually finished within 60 min. Recordings were started at 1 h after the induction of anesthesia. Additional doses of urethane (0.1–0.2 g/kg, s.c.) were administered when necessary. At the end of the recording experiments, the mice were killed by intraperitoneal injection of an overdose (300 mg/kg) of pentobarbital (Kyoritsu Seiyaku). When cortical responses were recorded at 1–2 weeks after M/U nerve cutting, the mice were allowed to recover from the anesthesia after suturing the wound and covering the surface of the skull with dental acrylic resin (Tohmi et al., 2006). Fradiomycin (Mochida Pharmaceutical) and ampicillin (Meiji Seika) were applied to avoid infection.
Fluorescence imaging.
Cortical images (128 × 160 pixels or 2.56 × 3.20 mm) of endogenous green fluorescence (λ = 500–550 nm) in blue light (λ = 470–490 nm) were recorded at nine frames per second using a cooled charge coupled device camera (ORCA-R2; Hamamatsu Photonics). The camera was attached to a binocular epifluorescence microscope (M165 FC; Leica Microsystems) with a 75 W xenon light source and a 1× objective lens. Images were taken in recording sessions repeated at 50 s intervals. Brush vibration at 50 Hz for 600 ms was applied to the palm of the forepaw or the sole of the hindpaw using a solenoid mechanical stimulator (DPS-290; Dia Medical). The amplitude of the vibration was ∼0.2 mm. Images elicited by the stimulation were averaged over 24 trials. Spatial averaging in 5 × 5 pixels and temporal averaging in three consecutive frames were used for smoothing and improving the image quality. The images were normalized, pixel by pixel, with respect to a reference image, which was obtained by averaging five images taken immediately before the stimulation. The normalized images are shown in a pseudocolor scale representing the fractional fluorescence changes (ΔF/F0). The response amplitude was evaluated as values of ΔF/F0 in a square window of 20 × 20 pixels or 400 × 400 μm, including the response peak, which was found by visual inspection of serial pseudocolor images at 0.6–1.0 s after the stimulus onset. The location of the window was adjusted to maximize the response amplitude.
Spinal cord lesioning.
When the dorsal column or spinothalamic tract was lesioned, the spinal cord was fixed with a spinal cord clamp (STS-A; Narishige), and the dorsal surface was exposed. The left dorsal column or spinothalamic tract was destroyed using an ultrasonic cutter (NE87; NSK) at the T12 level. The lesioned area produced by the ultrasonic cutter was more restricted than that produced by passing currents (data not shown). When both tracts were destroyed in the same mouse, the spinothalamic tract lesion was made at 1 mm caudal to the dorsal column lesion. To verify the lesioned area, the spinal cord was isolated after the recording experiments and fixed with 10% paraformaldehyde. Serial spinal cord sections of 100 μm thickness were cut using a microslicer (PRO-7; Dosaka), and the translucent images were observed.
Manipulations applied to the M/U nerves.
When tetrodotoxin (TTX) (160 μm) and TTX plus a tarantula venom peptide (ProTx-I) (50 μm) were applied to the M/U nerves, the compounds were mixed with saline containing 100 mg/ml fibrinogen. A fibrin clot of ∼5 μl in volume was formed by mixing this solution and 100 U/ml of thrombin in the presence of 2.2 mm Ca2+. To localize the effects of TTX and ProTx-I, the M/U nerves surrounded by the fibrin clot were isolated from other tissues using a piece of silicon sheet.
When electrical stimulation was applied to the central cut end of the M/U nerves, a bipolar electrode was attached to the cut end. Monophasic current pulses (100 μs, 10 μA) were continuously delivered at 0.1 Hz throughout the recordings.
Electrophysiology.
Unit recordings from peripheral nerve fibers were made at the left brachial plexus. The peripheral cut ends of the M/U nerves were inserted in a recording chamber filled with saline. A fire-polished glass micropipette with a tip diameter of 7.5 μm was filled with saline for use as a recording electrode. One or a few fibers at the cut end were sucked into the micropipette, and unit activities were recorded using an amplifier (MEG-1200; Nihon Kohden) bandpass filtered between 1.5 Hz and 10 kHz. Unit recordings were also made in the central cut end of the sural nerve composed of sensory fibers.
Histology.
Mice deeply anesthetized with chloral hydrate (400 mg/kg, i.p.) were perfused with 4% paraformaldehyde in a 0.1 m phosphate buffer, pH 7.4. The forepaw was immersed in the same fixative at 4°C for 12 h. After decalcification in a 5% solution of disodium ethylene diamine tetraacetate (Dojindo Laboratories) for 3 weeks at 4°C, the tissues were equilibrated in 30% sucrose overnight for cryoprotection. Sections at 20 μm thickness were prepared using a cryostat. After inhibition of endogenous peroxidase activity by incubation with 0.3% hydrogen peroxide in methanol for 30 min, the sections were treated with 2.5% normal goat serum (Vector Laboratories) for 60 min. The sections were primarily incubated with a rabbit polyclonal antiserum against human GDNF (1:250; Santa Cruz Biotechnology) overnight at room temperature. After washing, the sections were sequentially reacted with biotinylated goat anti-rabbit IgG (1:1000; Vector Laboratories) for 2 h and peroxidase-conjugated avidin (ABC kit; Vector Laboratories) for 90 min at room temperature. The final visualization was achieved using 0.04% 3–3′-diaminobenzidine and 0.0125% hydrogen peroxide in 0.05 m Tris-HCl, pH 7.6. The immunostained sections were counterstained with 0.03% methylene blue.
Subcutaneous injection of compounds into the forepaw.
Anti-GDNF antibody (40 μg), anti-GFRα1 antibody (20 μg), and control mouse IgG (40 μg) and goat IgG (20 μg) were dissolved in saline. A short interfering RNA against GDNF (GDNF siRNA) (1 nmol) and control RNA (1 nmol) were dissolved into RNAase-free water. Drug solutions (20 μl) were placed in a microsyringe (Gastight #1705; Hamilton) and injected using a fine hypodermic needle with an outer diameter of 200 μm (Nanopass needle; Terumo). The injection was performed under anesthesia with pentobarbital (70 mg/kg, i.p.) or urethane (1.65 g/kg, i.p.). The needle was carefully inserted into the disinfected palm of the forepaw using a manipulator to minimize tissue damage. The drug solutions were administered during a period of 2 h using a motorized injector (IM-1; Narishige).
Behavioral test for tactile allodynia.
The forepaw-withdrawal thresholds to mechanical stimuli were measured using a plantar aesthesiometer (model 37400; Ugo Basile). Mice were anesthetized with pentobarbital (70 mg/kg, i.p.), and underwent cutting of the M/U nerves, a sham operation, or subcutaneous injection of the GDNF siRNA. The thresholds were measured at 1–3 weeks after the operation. Diazepam (3 mg/kg, i.p.) was applied to the mice for stable measurements. The hindpaw-withdrawal thresholds to mechanical stimuli were also measured in mice before and 3 h after pharmacological conduction block of the peroneal nerve. In this experiment, mice were anesthetized with pentobarbital after the hindpaw-withdrawal thresholds were measured, and a bupivacaine solution (3%, 20 μl) was locally injected to the peroneal nerve. The hindpaw-withdrawal thresholds were measured again 3 h after the injection in the same mice recovered from the anesthesia. The bupivacaine was dissolved in saline containing 3% carboxymethylcellulose, which was used to localize the effect of bupivacaine, and 0.5% indigo carmine, which was to confirm the injected site after the thresholds were measured.
Drugs.
TTX, fibrinogen, and thrombin were obtained from Wako Pure Chemicals. GDNF siRNA and control RNA were purchased from Qiagen. Anti-GDNF antibody, control mouse IgG, anti-GFRα1 antibody, and control goat IgG were from R & D Systems. ProTx-I was obtained from Peptide Institute. Bupivacaine (powder) and carboxymethylcellulose was obtained from Sigma. Indigo carmine was from Tokyo Chemical Industry. LY354740 [(+)-2-aminobicyclo[3.1.0]-hexane-2,6-dicarboxylate monohydrate] was from Santa Cruz Biotechnology, and LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid] was from Tocris Bioscience.
Statistical analysis.
The statistical significance of differences among values was analyzed using StatView software (SAS Institute). Unpaired data were evaluated by the Mann–Whitney U test, whereas paired data were evaluated by the Wilcoxon's signed rank test. The χ2 test was used to evaluate the significance of the effects on the number of units or Meissner corpuscles. Only p values <0.05 are shown in the figures.
Results
Somatic cortical potentiation after peripheral nerve cutting
Peripheral nerve cutting produces potentiation of the somatic cortical responses mediated by the nearby nerves (Tinazzi et al., 1997). We quantified this potentiation using transcranial flavoprotein fluorescence imaging. After vibration was applied to the palm of the left forepaw via a small brush of fine horsehair, fluorescence increases appeared in the right somatosensory cortex (Fig. 1A). These responses were immediately diminished after cutting the left M/U nerves innervating the palm of the forepaw (Fig. 1B,C). However, some responses mediated by the R/Mc nerves innervating the back of the forepaw remained after M/U cutting. The remaining responses were potentiated within 3 h after M/U cutting compared with the responses recorded immediately after the nerve cutting. Additional potentiation of the somatic responses was found after 1–2 weeks (Fig. 1B,C), and finally, R/Mc cutting completely abolished the potentiated responses. We observed no apparent changes in the cortical responses in sham-operated mice (Fig. 1C). The size of the response area, in which ΔF/F0 was larger than the half-maximal amplitude, was only slightly increased to 124 ± 12% (mean ± SEM, n = 6) at 3 h after M/U cutting compared with that before the nerve cutting. However, it was significantly increased to 189 ± 20% (n = 7, p < 0.02, Wilcoxon's signed rank test) at 1 week and to 158 ± 20% (n = 7, p < 0.03) at 2 weeks after M/U cutting.

Cortical potentiation after peripheral nerve cutting. A, Cortical responses in the somatosensory cortex elicited by brush vibration applied to the palm of the forepaw. Original and pseudocolor images in ΔF/F0 of the bilateral somatosensory cortices are shown. The time after the stimulus onset is shown in each pseudocolor image. B, Averaged responses before and after M/U cutting in the right somatosensory cortex. The time after the M/U cutting is shown in each panel. The peak responses after the stimulus onset are shown. The last image was obtained after R/Mc cutting. C, Time course of the changes in the response amplitudes after M/U cutting. The data are means and SEM obtained from 10 (or fewer as shown in parentheses) mice. Significant increases in the response amplitudes from the data immediately after M/U cutting were evaluated by the Mann–Whitney U test and are shown with p values. The black dots with bars show the data obtained from seven sham-operated mice.
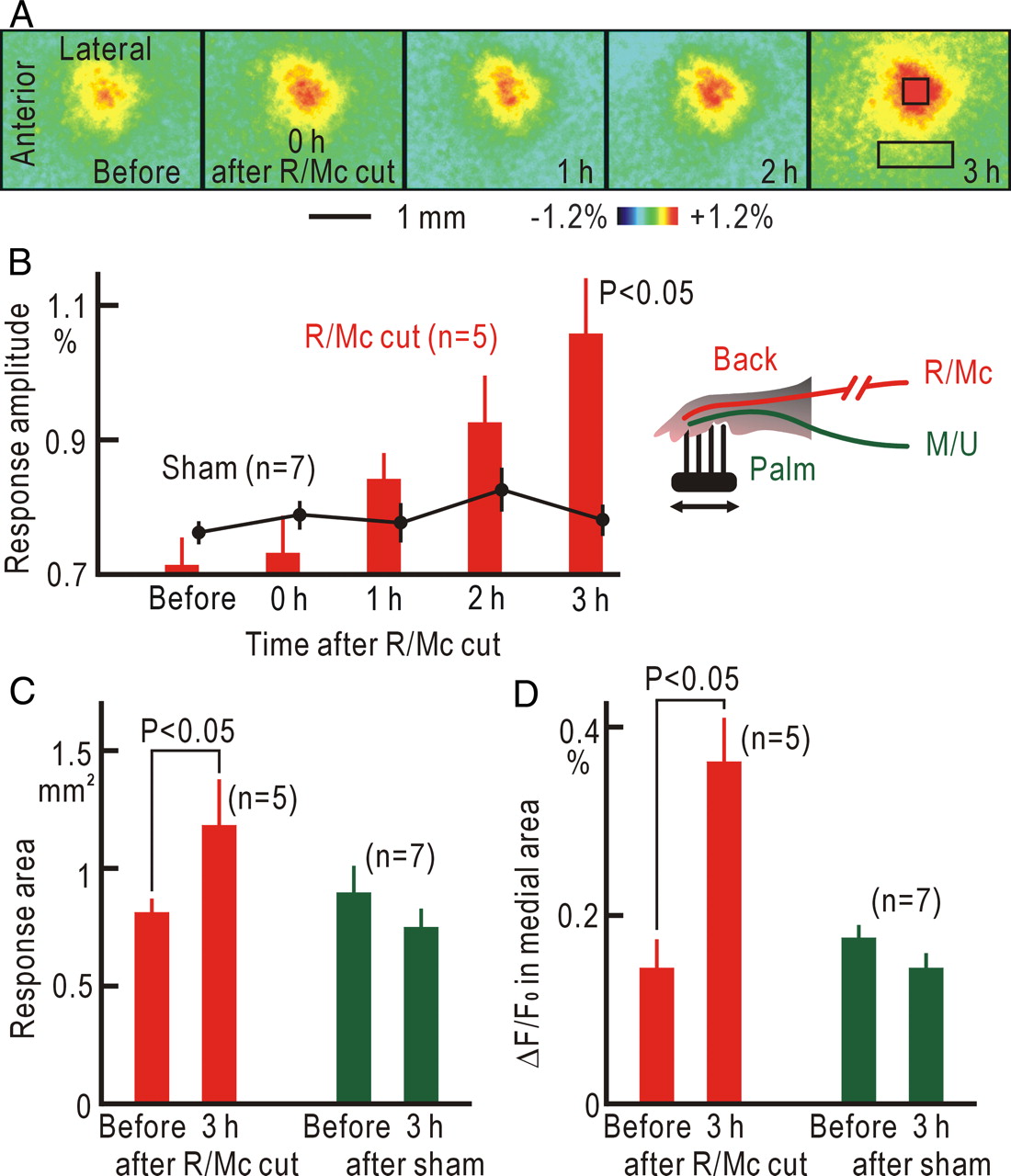
When the R/Mc nerves were cut first, the somatic cortical responses did not show any immediate changes (Fig. 2A,B). However, the responses exhibited slow and significant potentiation within 3 h after R/Mc cutting (p < 0.05). The size of the response area was also significantly increased (p < 0.05) at 3 h after R/Mc cutting, whereas it was slightly decreased in sham-operated mice (Fig. 2C). It has been reported that peripheral nerve injury triggers long-term potentiation in the anterior cingulate cortex (Xu et al., 2008). Therefore, we investigated the fluorescence responses in the cortical areas medial to the response center. The ΔF/F0 values measured in a rectangular window of 400 × 1200 μm locating 1 mm medial to the response center (Fig. 2A) were significantly increased (p < 0.05) at 3 h after R/Mc cutting compared with that before the nerve cutting, whereas they were slightly decreased in sham-operated mice (Fig. 2D).

Cortical potentiation after R/Mc cutting. A, Cortical responses before and after R/Mc cutting. The square window in the last panel shows the site of the response center, and the rectangular window shows the medial cortical area in which ΔF/F0 was measured in D. B, Time course of the cortical potentiation after R/Mc cutting. The black dots with bars show the data obtained from seven sham-operated mice. C, Size of response area before and 3 h after R/Mc cutting or sham operation. D, ΔF/F0 in a rectangular window locating 1 mm medial to the response center before and 3 h after R/Mc cutting or sham operation. Statistical significance was evaluated by the Wilcoxon's signed rank test in B–D.
Sensory modality shift as a part of somatic cortical potentiation
Lesioning of the ipsilateral dorsal column abolishes most of the tactile responses in the contralateral somatosensory cortex (Jain et al., 1995), whereas lesioning of the contralateral spinothalamic tract eliminates thalamic nociceptive responses (Peschanski et al., 1985). We used these anatomical features of the spinal afferent pathways to investigate the properties of the cortical responses that appeared after peripheral nerve cutting. The cortical responses elicited by brush vibration applied to the sole of the hindpaw were immediately diminished after cutting the tibial nerve at the level of the lower leg and potentiated within 3 h (Fig. 3A,D), as observed after M/U cutting. Lesioning of the left dorsal column at T12 of the spinal cord blocked the right cortical responses elicited by brush vibration applied to the sole, and no recovery was observed within 3 h (Fig. 3B,D). When lesioning of the dorsal column was followed by tibial nerve cutting, the cortical responses paradoxically appeared again within 3 h after the nerve cutting (Fig. 3C,D). The right cortical responses elicited by brush vibration were only slightly reduced in amplitude by lesioning of the right spinothalamic tract but leaving peripheral nerves intact to 94 ± 5% (n = 5) (Fig. 3E). However, the right cortical responses that appeared after dorsal column lesioning and tibial nerve cutting were markedly reduced to 2 ± 5% (n = 5) (Fig. 3F). The effects of lesioning of the spinothalamic tract were significantly different between the two experiments (p < 0.01, Mann–Whitney U test). In our preliminary experiments, lesioning of the left dorsal column blocked most of cortical field potentials elicited in the right somatosensory cortex by brush vibration applied to the left sole (data not shown). However, the potentials appeared again in the lesioned mice within 3 h after tibial nerve cutting. These results indicate that tactile information, which had been mediated by the ipsilateral dorsal column to the somatosensory cortex of naive mice, was connected to and mediated by the contralateral spinothalamic tract within 3 h after peripheral nerve cutting. The activities in the primary somatosensory cortex driven via the spinothalamic tract reflect the presence of nociceptive activities at the spinal cord level, although the activities might not be directly correlated with pain perception.

Sensory modality shift after peripheral nerve cutting. A, Cortical responses before, immediately after, and 3 h after tibial nerve cutting. B, Cortical responses before, immediately after, and 3 h after dorsal column (DC) lesioning. The lesioned area in the spinal cord is also shown. C, Cortical responses before and immediately after dorsal column lesioning, and immediately after and 3 h after tibial nerve cutting. The images were obtained in this order from the same mouse. The lesioned area in the spinal cord is also shown. D, Changes in the response amplitudes (ΔF/F0) in each group of mice. Statistical significance was evaluated by the Mann–Whitney U test. E, Cortical responses before and after spinothalamic tract (ST) lesioning. The lesioned area in the spinal cord is also shown. F, Cortical responses 3 h after dorsal column lesioning and tibial nerve cutting (Before) and after additional lesioning of the spinothalamic tract (ST lesion). The lesioned area in the spinal cord is also shown. Although the lesioned area in E was smaller than that in F, most of the spinothalamic tract was destroyed in both experiments.
Trigger mechanisms for cortical potentiation with a sensory modality shift
One of the immediate results elicited by nerve injury is nerve conduction blockade. It has been hypothesized that removal of the surround inhibition driven by tonic nerve firing may induce rapid cortical plasticity (Calford, 2002), although no tonic firing that is sufficiently frequent to drive the surround inhibition has been found in sensory nerves (Michaelis et al., 2000). To scrutinize the possible roles of basal afferent firing, we locally applied TTX to the M/U nerves. We found that the cortical responses were potentiated within 3 h after the conduction blockade (Fig. 4A,C). In some experiments, we also applied ProTx-I, a TTX-resistant sodium channel blocker (Middleton et al., 2002), in addition to TTX, because TTX-resistant sodium channels in the peripheral nerves have roles in hyperalgesia (Lai et al., 2004). However, ProTx-I had no additional effects on the results. When the R/Mc nerves were cut first, the conduction blockade of the M/U nerves did not produce any potentiation (Fig. 4C). Continuous electrical stimulation at 0.1 or 0.02 Hz was applied to the central cut end of the M/U nerves to mimic possible basal firing in intact nerves. The stimulation at these frequencies prevented the induction of the cortical potentiation after M/U cutting (Fig. 4B,C), suggesting the presence of low-frequency basal firing in the M/U nerve fibers.

Cortical potentiation induced by nerve conduction block. A, Cortical responses immediately after and 3 h after conduction block of the M/U nerves with 160 μm TTX. B, Cortical responses immediately after and 3 h after M/U cutting with continuous electrical stimulation (100 μs, 10 μA, 0.1 Hz) to the central cut end. C, Changes in the response amplitudes (ΔF/F0) during 3 h after M/U cutting, a sham operation, TTX (160 μm) application to the M/U nerves, TTX application with R/Mc cutting, or M/U cutting with stimulation of the central cut end at 0.1 or 0.02 Hz. The number of mice is shown for each column. Data obtained with TTX alone or TTX plus ProTx-I (50 μm) were mixed because there were no apparent differences between the treatments. Statistical significance was evaluated by the Mann–Whitney U test.
To confirm the presence of low-frequency basal firing directly, we recorded unit activities from the peripheral cut end of the M/U nerves (Fig. 5A). The cut end of the M/U nerves was separated into nerve fibers in saline, and a single or a few fibers were sucked into a glass micropipette to record fiber activities extracellularly (Fig. 5B). Approximately half of the touch-sensitive units (Fig. 5C) had basal firing at very low frequencies of ∼0.1 Hz or less (Fig. 5D,E). Measurement of the nerve conduction velocity (Table 1) revealed that basal firing was found only in touch-sensitive Aβ fibers (Cain et al., 2001). We also recorded unit activities in the central cut end of the sural nerve composed of sensory fibers, and no basal firing was observed in all of the six Aβ fibers recorded.

Roles of GDNF in basal afferent firing. A, Experimental setup for unit recordings from M/U nerves and a typical waveform of a spike in an Aβ fiber. B, Traces with or without a spike in a C fiber (arrow). Stimulus pulses (intensity, 0.4 mA; duration, 100 μs) were applied to the site 18 mm apart from the tip of the recording pipette. C, Mechanoreceptive activities elicited by brush vibration. D, Basal firing in the same unit shown in C. E, Distribution of the basal firing rates determined by observation of 43 touch-sensitive nerve fibers for 10 min. F, Percentages of units with basal firing (left) and mean firing rates (right) in touch-sensitive fibers recorded in naive mice and groups of mice injected with GDNF siRNA (1 nmol), control RNA (1 nmol), GDNF siRNA plus GDNF (2 pg), anti-GDNF antibody (40 μg), control mouse IgG (40 μg), anti-GFRα1 antibody (20 μg), or control goat IgG (20 μg). The number of units is shown for each column. The number of mechanoreceptive spikes during brush vibration at 50 Hz for 600 ms were counted in the same units and are shown with filled circles in the right. Statistical significance in the left was evaluated by the χ2 test, and that in the right was by the Mann–Whitney U test.
Classification of M/U nerve fibers
Because the basal firing was found in touch-sensitive or rapidly adapting (RA) mechanoreceptive fibers, it may be produced by a mechanism specific to RA mechanoreceptive fibers. RA mechanoreceptive neurons in the dorsal root ganglia or trigeminal ganglia are characterized by the expression of the receptor tyrosine kinase RET during a certain developmental period (Bourane et al., 2009; Luo et al., 2009). RET is a component of the GDNF family receptors. The excitability of trigeminal ganglion neurons is facilitated by GDNF and GFRα1, another component of the GDNF family receptors (Takeda et al., 2010). We found that GDNF and GFRα1 were present in Meissner corpuscles in the palm of the mouse forepaw (Fig. 6A,D), suggesting that the basal firing in touch-sensitive fibers may be dependent on GDNF and GFRα1. To test this possibility, a GDNF siRNA (1 nmol) was subcutaneously injected into the palm of the forepaw. At 4–5 d after the injection, the number of units with basal firing and the mean firing rate in touch-sensitive nerves were significantly reduced compared with those of mice injected with a control RNA (Fig. 5F). At the same time, the number of GDNF-positive Meissner corpuscles was significantly reduced (Fig. 6B,C). The reduction in the basal firing induced by the GDNF siRNA was immediately rescued by subcutaneous application of 2 pg of GDNF (Fig. 5F). Application of an anti-GDNF mouse antibody (40 μg) or anti-GFRα1 goat antibody (20 μg) also immediately suppressed the basal firing, whereas application of control mouse IgG or control goat IgG did not (Fig. 5F). In these experiments, the compounds were slowly injected into the palm of anesthetized mice during a period of 2 h through a fine hypodermic needle with an outer diameter of 200 μm. Application of these compounds had no effect on the mechanoreceptive activities in touch-sensitive nerves (Fig. 5F), suggesting that almost no tissue damage was produced by the treatment. Together, these results indicate the presence of the basal afferent firing produced by endogenous GDNF in the touch-sensitive nerve fibers.

GDNF and GFRα1 in Meissner corpuscles. A, Darkly stained GDNF in a Meissner corpuscle (red arrow) of the forepaw after injection of the control RNA. B, Loss of GDNF in a Meissner corpuscle (blue arrow) after injection of the GDNF siRNA. C, Percentages of GDNF-positive Meissner corpuscles, identified by blinded tests, after injection of the GDNF siRNA or control RNA. The numbers in parentheses represent the total numbers of Meissner corpuscles. Statistical significance was evaluated by the χ2 test. D, GFRα1 in a Meissner corpuscle (red arrow) of a naive mouse.
Tactile allodynia as a counterpart of the sensory modality shift
The effects of the sensory modality shift at the spinal cord level within 3 h after peripheral nerve cutting should be detected later by behavioral tests, because the potentiation of the somatic cortical responses after peripheral nerve cutting continued for >2 weeks after M/U cutting (Fig. 1B,C). This possibility was investigated by paw-withdrawal tests. The threshold of the forepaw-withdrawal responses to mechanical stimuli was significantly reduced after M/U cutting (Fig. 7A), whereas a sham operation had no clear effect. The mice with left M/U nerve cutting showed the characteristic behaviors of tactile allodynia: they only occasionally stepped on the ground with the left forepaw, and licking of the left forepaw was subsequently observed. The threshold of the paw-withdrawal responses can be measured in mice with peripheral nerve cutting only after recovery from the surgical operation. Therefore, conduction of the peroneal nerve was pharmacologically blocked by local application of bupivacaine to the nerves. The threshold of the hindpaw-withdrawal responses was significantly reduced at 3 h after the application compared with that in mice injected with a vehicle solution (Fig. 7B). These results were compatible with the acute tactile allodynia observed after partial nerve ligation (Seltzer et al., 1990; Brüggemann et al., 2001).

Tactile allodynia. A, Forepaw-withdrawal thresholds to mechanical stimuli before and 1–2 weeks after M/U cutting or a sham operation. Statistical significance was evaluated by the Mann–Whitney U test. B, Hindpaw-withdrawal thresholds to mechanical stimuli before and 3 h after injection with a bupivacaine (3%, 20 μl) or vehicle solution to the peroneal nerve. C, Forepaw-withdrawal to thresholds to mechanical stimuli before and 1–3 weeks after subcutaneous injection of 1 nmol of GDNF siRNA or control RNA.
The mechanical allodynia after peripheral nerve cutting might be produced by tissue injury and inflammation (Marchand et al., 2005) rather than disruption of the basal afferent firing. To rule out this possibility, sustained reduction of the basal firing in touch-sensitive Aβ fibers with minimal tissue damage was produced by subcutaneous application of the GDNF siRNA, as shown in Figure 5E. Mechanical allodynia was found at 1 week after application of the GDNF siRNA (Fig. 7C), whereas injection of a control RNA had no effect. The allodynia induced by the GDNF siRNA disappeared within 3 weeks, suggesting that it only persisted while the effects of the GDNF siRNA were maintained. Together, these results indicate that tactile allodynia is a counterpart of the sensory modality shift at the spinal cord level produced by reduction of the basal firing in touch-sensitive nerve fibers and that tissue injury or inflammation was not required to produce the mechanical allodynia.
Testing the roles of group II mGluRs in the initial phase of neuropathic pain
Observation of the sensory modality shift from tactile to nociceptive information at the spinal cord level serve as a straightforward method to investigate the initial phase of neuropathic pain. We tested the validity of this method by investigating the effects of LY354740, a specific agonist of group II mGluRs, and LY341495, a specific antagonist of group II mGluRs (Simmons et al., 2002). We selected these compounds because the role of the basal firing of touch-sensitive nerve fibers in preventing the induction of tactile allodynia may be attributed to sustained activation of group II mGluRs (Simmons et al., 2002; Jones et al., 2005). When a piece of cotton containing LY354740 (10 nm) was placed on the dorsal surface of the spinal cord around the L3–S1 level, tibial nerve cutting failed to induce cortical potentiation within 3 h (Fig. 8A,B). The amplitudes of the cortical potentiation within 3 h after tibial nerve cutting were significantly reduced in the presence of LY354740 (p < 0.01, Mann–Whitney U test) (Fig. 8E). Application of LY354740 alone had no apparent effect on the cortical responses (n = 5; data not shown). Lesioning of the left dorsal column at the T12 level abolished the cortical tactile responses, and no recovery was observed within 3 h (Fig. 8C). However, when the lesioning was followed by application of LY341495 (100 nm) onto the surface of the spinal cord around the L3–S1 level, cortical responses appeared within 3 h (Fig. 8D). The changes in the amplitudes of the cortical responses within 3 h after dorsal column lesioning followed by LY341495 application were significantly increased compared with those in the absence of LY341495 (p < 0.01) (Fig. 8E). These results confirmed the hypothesis that low-frequency basal firing of touch-sensitive nerve fibers prevented the induction of tactile allodynia by sustained activation of group II mGluRs in the spinal cord.

Roles of group II mGluRs in the sensory modality shift at the spinal cord level within 3 h after tibial nerve cutting. A, Cortical responses before, immediately after, and 3 h after tibial nerve cutting. B, Cortical responses before, immediately after, and 3 h after tibial nerve cutting in the presence of 10 nm LY354740 applied to the spinal cord around the L3–S1 level. C, Cortical responses before, immediately after, and 3 h after dorsal column lesioning. D, Cortical responses before, immediately after dorsal column lesioning, and 3 h after the application of 100 nm LY341495 to the spinal cord around the L3–S1 level. E, Changes in the response amplitudes (ΔF/F0) during 3 h after tibial nerve cutting or the application of LY341495 in each group of mice. Statistical significance was evaluated by the Mann–Whitney U test.
Discussion
Basal firing in sensory nerves
In the present study, basal firing at low frequencies of ∼0.1 Hz or less was found in touch-sensitive Aβ fibers. The basal firing was unlikely produced by injury potentials, because it was observed only in touch-sensitive Aβ fibers and reduced after application of GDNF siRNA. The firing rate of the basal firing was very low, and therefore, its presence and functional roles might have been overlooked in previous studies (Michaelis et al., 2000). However, the disruption of the basal firing triggered acute cortical potentiation and tactile allodynia in the present study. These results suggest that the basal firing in touch-sensitive nerves may work as a safekeeping signal. Physical insults to the skin may fail to elicit nociceptive withdrawal reflexes when sensory nerves originating from the skin area have been injured. Reorganization of cortical somatotopic maps (Calford and Tweedale, 1988; Björkman et al., 2009) and potentiation of cortical responses in response to stimulation of nearby nerves (Tinazzi et al., 1997) are quickly induced within a few hours after nerve injury, possibly for facilitating nociceptive withdrawal reflexes. Our results suggest that the low-frequency basal firing in touch-sensitive nerves has a role to inform the absence of disconnection in peripheral sensory nerves to the spinal cord. Because the firing rate is ∼0.1 Hz or less, the basal firing is unlikely to interfere with the sensory information mediated by the touch-sensitive nerves. When this safekeeping signal is disrupted by nerve injury, cortical potentiation and tactile allodynia can be quickly induced to avoid physical insults to the deafferented skin area.
Basal firing in peripheral sensory nerves plays essential roles in the development of young animals. In the visual system, spontaneous waves of action potentials in the retina are required for the development of cortical circuits (Katz and Shatz, 1996). The spontaneous periodic activity in the spiral ganglion neurons is required for the refinement and maintenance of tonotopic maps in the auditory system (Tritsch et al., 2007). The absence of afferent activities results in sustained elevation of the intracellular calcium concentration, which causes cell death in some of the cochlear nucleus neurons (Lachica et al., 1995; Zirpel et al., 1998). Basal firing in peripheral sensory nerves is also important in the maintenance of sensory functions of adult animals. Cortical map plasticity is induced by deafferentation in not only the somatosensory but also the visual and auditory systems (Merzenich et al., 1983; Calford, 2002). Photophobia is observed in patients with optic nerve injury (Kawasaki and Purvin, 2002), and damage to the inner hair cells produces tinnitus (Saunders, 2007). These sensory abnormalities may be comparable with neuropathic pain in the somatosensory system.
We found that endogenous GDNF induced basal firing at low frequencies ∼0.1 Hz or less in touch-sensitive or RA mechanoreceptive fibers, which are characterized by the expression of RET during a developmental period (Bourane et al., 2009; Luo et al., 2009). We confirmed that GDNF and GFRα1 were present in Meissner corpuscles. GDNF and GDNF family ligands play crucial roles in the development and normal function of the nervous system (Paratcha and Ledda, 2008). In adult animals, GDNF has alleviating effects on neuropathic pain (Boucher et al., 2000; Wang et al., 2003). The present results indicate that GDNF has another role to produce basal firing in touch-sensitive nerves at low frequencies. The reduction in basal firing induced by the GDNF siRNA was rescued by subcutaneous application of only 2 pg of GDNF, and injection of GDNF at a higher dose did not show any additional effect on the basal firing rate in touch-sensitive nerve fibers (data not shown). Therefore, the mechanisms for GDNF-induced basal firing obviously differ from those for the direct excitation produced by brain-derived neurotrophic factor (Kafitz et al., 1999). It has been reported that excitability of the trigeminal ganglion neurons (Takeda et al., 2010) or the midbrain dopaminergic neurons (Yang et al., 2001) is increased by GDNF by closing K+ channels. A similar mechanism may also be responsible for producing the basal firing in touch-sensitive nerve fibers.
Spinal mechanism underlying the initial phase of neuropathic pain
We found that the initial phase of neuropathic pain comprised the sensory modality shift from tactile to nociceptive information at the spinal cord level. The changes in the spinal neural circuits were triggered by disruption of the afferent basal firing, and this effect was mimicked by the application of a group II mGluR antagonist, while the agonists are known to produce alleviating effects on neuropathic pain (Simmons et al., 2002; Jones et al., 2005). Activation of group II mGluRs reduces presynaptic transmitter release (Goudet et al., 2009), hyperpolarizes postsynaptic neurons by opening G-protein-coupled inward rectifier K+ channels (Irie et al., 2006; Lee and Sherman, 2009), and modulates spontaneous Ca2+ spikes (Koga et al., 2010). Stimulation of the M/U and R/Mc nerves produced fluorescence increases in the same cortical area (data not shown). Therefore, the peripheral information mediated by the both nerves could be terminated on the same spinal neurons. If the two postsynaptic sites are sufficiently distant, the reduced glutamate level at the postsynaptic site after injury of M/U or R/Mc nerves might not be rescued by the activities in the remaining nerves. Therefore, nerve injury may produce failure of group II mGluR activation and increases in the excitability and intracellular Ca2+ concentration around the postsynaptic sites.
According to the gate control theory (Melzack and Wall, 1965), nociceptive spinothalamic tract neurons are dynamically regulated by inhibition from various sources, including innocuous afferent inputs, which drive interneurons in the superficial dorsal horn (Daniele and MacDermott, 2009). In animals with injured nerves, spinothalamic tract neurons show increased responsiveness to innocuous mechanical stimuli (Palecek et al., 1992). These changes after nerve injury are explained by a reduced Cl− gradient in the neurons (Coull et al., 2003; Price et al., 2009), because inhibition to nociceptive spinothalamic tract neurons from innocuous afferent inputs is reduced by the change. The reduced Cl− gradient is attributed to downregulation of the neuron-specific KCl cotransporter (KCC2) in the dorsal horn neurons (Coull et al., 2003). To explain the present result with the reduced Cl− gradient in spinothalamic tract neurons, it must be initiated within a few hours after failure of group II mGluR activation. It has been reported that elevation of the intracellular Ca2+ concentration elicits dephosphorylation of KCC2 and the resulting reduction in KCC2 activity within 1 h (Wake et al., 2007). Therefore, functional downregulation of KCC2 or a reduced Cl− gradient could be produced within 3 h after failure of group II mGluR activation or nerve injury. Anyway, the mechanism underlying the initial phase of neuropathic pain may be sufficiently simple to be verified in near future, because it is induced before involvement of inflammation and gene expression.
The initial phase of neuropathic pain as a potential therapeutic target
Although numerous people suffer from neuropathic pain, we have not been able to develop an effective therapy thus far for two main reasons. First, neuropathic pain is triggered by nerve injury and established by a complex cascade of inflammation and gene expression, and the complex mechanisms involved prevent us from identifying the primary etiology (Campbell and Meyer, 2006). However, the initial phase of neuropathic pain induced before inflammation and gene expression may have a relatively simple mechanism. Second, researchers have had to depend on behavioral tests to evaluate new drug candidates for neuropathic pain, although the results of such behavioral tests are affected by many factors. Analysis of the initial phase of neuropathic pain, which can be quantitatively measured as cortical responses within a few hours after nerve injury, may serve as a straightforward test to evaluate effective drugs for neuropathic pain.
An essential question is whether a recovery from the initial phase of neuropathic pain can alleviate chronic neuropathic pain or not. The initial phase of neuropathic pain is followed by chronic phases. Patients with chronic neuropathic pain experience widespread pain that is not otherwise explainable, sensory deficit, burning pain, pain during light stroking of the skin, and attacks of pain without apparent provocation (Campbell and Meyer, 2006). Therefore, the initial phase of neuropathic pain or tactile allodynia is only a part of the syndrome of chronic neuropathic pain. It has been reported that microglia and inflammation have important roles in chronic pain (Tsuda et al., 2003; Marchand et al., 2005). Brain structures outside the spinal cord are deeply involved in chronic pain (Flor et al., 1995; Neugebauer et al., 2009). However, agonists of group II mGluRs have alleviating effects on chronic neuropathic pain (Simmons et al., 2002; Jones et al., 2005). Surgical reconstruction of the injured afferent connection also has alleviating effects on chronic neuropathic pain (Inada et al., 2005), suggesting that restoration of the basal firing via the reconstructed afferent pathways may cure chronic neuropathic pain. These results may imply that the initial phase of neuropathic pain can be a potential therapeutic target of chronic neuropathic pain, probably because the mechanisms underlying the early phase are still important in the later phases.
Footnotes
This work was supported by grants from the Japanese Government. We thank S. Maruyama and A. Matsushima for their technical assistance.
- Correspondence should be addressed to Katsuei Shibuki, Department of Neurophysiology, Brain Research Institute, Niigata University, 1-757 Asahi-machi, Chuo-ku, Niigata 951-8585, Japan. shibuki{at}bri.niigata-u.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}