

Abstract
Oxidative stress influences cell survival and homeostasis, but the mechanisms underlying the biological effects of oxidative stress remain to be elucidated. The protein kinase MST1 (mammalian Ste20-like kinase 1) plays a major role in oxidative stress-induced cell death in primary mammalian neurons. However, the mechanisms that regulate MST1 in oxidative stress responses remain largely unknown. In the present study, we demonstrate that the protein kinase c-Abl phosphorylates MST1 at Y433, which triggers the stabilization and activation of MST1. Inhibition of c-Abl promotes the degradation of MST1 through C terminus of Hsc70-interacting protein (CHIP)-mediated ubiquitination, and thereby attenuates cell death. Oxidative stress induces the c-Abl-dependent tyrosine phosphorylation of MST1 and increases the interaction between MST1 and FOXO3 (Forkhead box O3), thereby activating the MST1-FOXO signaling pathway, leading to cell death in both primary culture neurons and rat hippocampal neurons. The identification of the c-Abl tyrosine kinase as a novel upstream activator of MST1 suggests that the c-Abl-MST1 signaling cascade plays an important role in cellular responses to oxidative stress.
Introduction
In mammalian systems, mammalian Ste20-like kinase (MST) is ubiquitously expressed and is best known for its function in promoting cell death. The MST family of kinases consists of MST1, MST2, MST3, and MST4 (Taylor et al., 1996; Schinkmann and Blenis, 1997; Lin et al., 2001). Each of these proteins harbors an N-terminal kinase domain and a C-terminal regulatory domain, classifying them structurally as members of the Class II germinal center kinase family of Sterile 20-like kinases (Dan et al., 2001). The mechanisms leading to MST activation and cell death vary depending on stimulus specificity and cell type. Exposure of cells to stress and apoptosis-inducing stimuli such as staurosporine, Fas ligase, and oxidative stress activates MST. Once activated, MSTs form dimers via the SARAH (Salvador/Rassf/Hippo, aa 433–480) domain at the extreme C-terminal region of MST and are autophosphorylated on specific threonine residues in the activation loop (Glantschnig et al., 2002; Hwang et al., 2007).
We discovered that oxidative stress activates MST, and that active MST1 phosphorylates and activates Forkhead box O3 (FOXO3) by disrupting its association with the inhibitory adaptor protein 14-3-3; activated MST1 also promotes neuronal cell death through induction of the proapoptotic gene BIM (Lehtinen et al., 2006). We and others also found that MST activity is regulated by Akt-induced phosphorylation (Jang et al., 2007; Yuan et al., 2010). Recently, we showed that c-Jun N-terminal kinase (JNK) acts not only as a MST1 substrate but also as a feedback activator for MST1 signaling during cell death (Bi et al., 2010). However, although progress in elucidating MST signaling pathways has been made, much remains to be done to gain a full understanding of the role of MST signaling in the regulation of cell death; in particular, the specific signaling upstream regulators that mediate a specific stimulus such as oxidative stress remain unclear.
The ubiquitously expressed c-Abl tyrosine kinase is a transducer of a variety of extrinsic and intrinsic cellular signals including those from growth factors, cell adhesion, oxidative stress, and DNA damage (Woodring et al., 2003; Zhu and Wang, 2004). Recently, c-Abl has been linked to oxidative stress-induced neuronal cell death through Cdk5/GSK3β activation and Tau hyperphosphorylation or through p73 upregulation. STI571, a c-Abl kinase inhibitor, decreases Cdk5 activation and Tau phosphorylation as well as neuronal cell death (Cancino et al., 2009).
In this study, we characterize a novel upstream kinase of the MST1 signaling pathway involved in oxidative stress-induced neuronal cell death. We found that c-Abl phosphorylates the Y433 of MST1 kinase at the C terminus, leading to the stabilization and activation of MST1 as well as enhanced interaction with FOXO3. In mammalian neurons, oxidative stress activates c-Abl, which phosphorylates and activates the MST1-FOXO3 signaling cascade and promotes neuron apoptosis. Inhibition of c-Abl by using c-Abl RNAi or STI571 attenuates oxidative stress-induced MST1 activation as well as cell death both in primary cultured neurons and in rat hippocampal neurons. Together, our findings identify a novel upstream kinase of MST1-FOXO3 signaling that regulates the cellular response to oxidative stress.
Materials and Methods
Plasmids and transfection.
The plasmids used were as follows: pCMV-Myc-c-Abl WT (wild type) and KD (kinase dead) were a gift from Dr. Cheng Cao (Beijing Institute of Biotechnology), and pFlag-CMV10 CHIP (C terminus of Hsc70-interacting protein) was acquired from Dr. Jianxin Sun (University of Medicine and Dentistry of New Jersey, Newark, NJ). MST1-Y433F and other mutants were generated by site-directed mutagenesis. All mutations were verified by sequencing. Fragments of GST-MST1 plasmids were cloned by PCR into pGEX4T1 at BamHI and XhoI restriction sites. Fragments of FLAG-MST1 plasmid were cloned into pFLAG-CMV10 at BglII and BamHI sites by PCR. Mammalian RNAi constructs were designed as described previously (Gaudilliere et al., 2002). The hairpin RNA (hpRNA) targeting sequences used include MST1 hpRNA, GGGCACTGTCCGAGTAGCCAGC; c-Abl hpRNA#1, GACCAACCTGTTCAGCGCT; c-Abl hpRNA#2, AAGCAGCTCG ATGGACCTCCA; CHIP hpRNA, GCACGACAAGTACATGGCGGA. Unless stated otherwise, all transfections were performed in complete medium with Lipofectamine 2000 (Invitrogen) or lipoD293 (SignaGen Laboratories) according to the manufacturer's protocols.
Tissue culture.
Cerebellar granule neuron cultures were prepared from postnatal day 6 rat pups. At 72 h after initial plating, transfections were performed using a calcium-phosphate method as described previously (Lehtinen et al., 2006). For the dual-luciferase reporter assays, cells were transfected with 1 μg of 3xIRS or BIM-luciferase reporter plasmid and 200 ng of the tk-Renilla luciferase reporter plasmid, the latter serving as an internal control for transfection efficiency. At 24 h after transfection, or 72 h for RNAi experiments, cells were left untreated or were treated with H2O2 for 12 h and subjected to luciferase assays. Means ± SD of normalized firefly/Renilla luciferase values relative to the control neurons are shown.
Primary cultures of rat hippocampus neurons isolated from embryonic day 18 (E18) rat embryos were performed as described previously (Kaech and Banker, 2006). For transfection, cultures of E18 plus 3∼5 day in vitro (DIV) neurons were transiently transfected with the indicated plasmid DNA, together with a plasmid encoding pEGFP at a 3:1 ratio using the Lipofectamine 2000 (Invitrogen) protocol as described previously (Dalby et al., 2004). At 24 h after transfection, or 72 h for RNAi experiments, cultures were left untreated or were treated with H2O2 (60–100 μm) for 24 h, fixed, and subjected to a cell survival assay as described previously (Lehtinen et al., 2006). To measure survival of transfected neurons, GFP-positive neurons were counted in a blind manner as either alive or dead according to the appearance of Hoescht 33258 (Sigma) staining. Approximately 150 cells were counted per experiment.
Immunoblotting, coimmunoprecipitation, and kinase assays.
Immunoblotting and coimmunoprecipitation were performed as described previously (Yuan et al., 2010). The antibodies used were MST1, c-Abl, phospho-MST1 (Thr183)/MST2 (Thr180), and ERK1/2 (Cell Signaling Technology); c-Abl (K12), HA, GST, 14-3-3β and β-actin (Santa Cruz Biotechnology); FLAG-M2 and phospho-c-Abl (Tyr412) (Sigma); phosphotyrosine (p-Tyr) (4G10), NeuN, and FOXO3 (Millipore); neuron-specific βIII tubulin antibody (Tuj 1) and MST1 (Abcam); GFP and phospho-FOXO3 (Ser207) (Invitrogen); and BIM (Stressgen) and Multi Ubiquitin (MBL International). Polyclonal antibody to Y433-phosphorylated MST1 was generated in rabbits (NewEast Biosciences) using a synthetic peptide corresponding to phospho-MST1 (425–439) [p-Y433] [CKIPQDGDpYEFLKSW]. In vitro kinase assays were performed as described previously (Bi et al., 2010). Briefly, recombinant active MST1 kinase (Millipore) was incubated under the following reaction conditions: 100 mm Tris, pH 7.4, 20 mm MgCl2, and 10 μCi ATP, with 1 μg of GST-FOXO3-FD (Forkhead domain) or Histone H2B as substrate. Immunoprecipitated c-Abl was incubated with 1 μg of GST-MST1 or GST-MST1-KD in reaction buffer containing 30 mm Tris, pH 7.4, 20 mm MgCl2, 1 mg/ml BSA, 100 μm ATP. Kinase reactions were separated by SDS-PAGE and analyzed by autoradiography or by immunoblotting using the indicated antibody.
Immunofluorescence and immunohistochemistry.
Freshly fixed neurons were first washed with PBS three times and blocked with 20% goat serum in PBS containing 0.2% Triton X-100 to reduce nonspecific antibody binding. Neurons were then incubated with the primary antibody at 4°C overnight. After washing with PBS three times, Alexa Fluor 488- or 546-conjugated secondary antibody (Invitrogen) was used to detect the signal. The secondary antibody was incubated at room temperature for 1 h, and then nuclear morphology was visualized using the DNA dye Hoechst 33258 (Sigma).
Formalin-fixed, paraffin-embedded brain sections were deparaffinized in xylene three times for 10 min, followed by rehydration in graded alcohol (100%, 95%, 85%, and 70%), and then washed with PBS three times for 5 min. Antigen retrieval was performed by microwaving the slides in 10 mm citrate buffer, pH 6.0, for 10 min, followed by cooling at room temperature for 20 min, and then washing with PBS three times for 5 min. The slides were transferred to a humidified chamber and incubated with 20% normal goat serum for 20 min to reduce nonspecific binding. The slides were incubated with pY433 MST1 antibody (1:50) at 4°C overnight. After washing with PBS three times, Alexa Fluor 488- or 546-conjugated secondary antibody (Invitrogen) was used to detect the signal. Negative control slides omitted the primary antibody or were blocked with pY433 MST1 peptide.
Animals and stereotaxic drug delivery.
Animals were maintained in the Animal Care Facility at our institute, and all experiments involving animals were approved by the Institutional Animal Care and Use Committee at the Institute of Biophysics, Chinese Academy of Sciences. Male Sprague Dawley rats (∼200 g) were anesthetized with chloral hydrate, 400 mg/kg, with unilateral injections performed stereotaxically into the upper leaf of the dentate gyrus in the dorsal hippocampus (−3.5 mm anteroposterior, +2.0 mm mediolateral, and −2.7 mm dorsoventral from the dura, according to bregma) as described previously (Cancino et al., 2008). The animals were injected at a rate of 0.25 μl/min with 4 μl of DMSO or rotenone (2.5 μg/μl) (Sigma), or rotenone plus STI571 (20 mm).
Statistical analysis.
Statistical analysis of the data was performed with a two-tailed Student's t test, or one-way ANOVA followed by Fisher's PLSD post hoc test using Origin software (Version 8). Data are presented as the mean ± SEM except for analyses of luciferase assays, where mean ± SD is shown, and the number of experiments are indicated in each figure. *p < 0.05 or **p < 0.01 denotes statistical significance.
Results
MST1 mediates oxidative stress-induced cell death in a c-Abl kinase-dependent manner
As previously reported, protein kinase c-Abl mediates oxidative stress-induced neuronal cell death (Alvarez et al., 2004; Lee et al., 2008). We found that STI571, a specific Abl kinase inhibitor, significantly abrogated the cell death response to rotenone, a mitochondrial complex I inhibitor, which was stereotaxically delivered to rat brain to induce oxidative stress (Fig. 1A). To determine whether MST1 might mediate oxidative stress-induced cell death via c-Abl, STI571 was used to treat primary neurons transfected with MST1 or a control vector. As expected, MST1 overexpression induced cell death in response to hydrogen peroxide, according to the appearance of Hoescht staining (Fig. 1B). In contrast, cell death induced by hydrogen peroxide and/or MST1 overexpression was mitigated by STI571, which effectively abrogates H2O2-induced c-Abl autophosphorylation at Y412 (Fig. 1B, inset). We next used c-Abl RNAi in primary culture of neurons to knock down endogenous c-Abl protein, and found that c-Abl knockdown protects neurons from either hydrogen peroxide or MST1 overexpression-induced cell death (Fig. 1C). Together, these data suggest that c-Abl plays an important role in oxidative stress-induced neuronal cell death and likely functions upstream of MST1 in response to hydrogen peroxide.

MST1 mediates oxidative stress-induced cell death in a c-Abl kinase-dependent manner. A, The rat hippocampus was injected unilaterally with DMSO, rotenone, or rotenone plus STI571 using a stereotaxic procedure. Animals were killed 24 h after injection and perfused with fixing solution. Brains were sectioned and analyzed for apoptosis by TUNEL. Representative images of TUNEL staining of brain DG are shown in the left panel (scale bar, 100 μm). Neuronal survival quantified by TUNEL staining is shown in the right panel. STI571 protected neurons from rotenone-induced neuronal cell death. (ANOVA; p < 0.01, n = 3). B, Hippocampal neuron cultures [E18 + 3 DIV) were transfected with pEGFP, alone or together with a FLAG-MST1 expression plasmid, at a ratio of 1:3. At 24 h after transfection, neurons were left untreated or were treated with H2O2 for 20 h, with or without 5 μm STI571. Transfected neurons were subjected to immunofluorescence using a mouse monoclonal antibody to NeuN together with the DNA dye Hoechst 33258 to reveal neuronal nuclei. Representative images of neurons are shown in the left panel (scale bar, 20 μm) with white arrowheads indicating healthy transfected neurons and red arrowheads indicating transfected neurons that are undergoing apoptosis. The percentage of cell death of GFP-positive neurons is represented as the mean ± SEM. The inset panel shows that STI571 inhibits c-Abl activation. MST1 overexpression-induced cell death and inhibition of c-Abl kinase activity by STI571 significantly protected neurons from hydrogen peroxide (ANOVA; p < 0.01, n = 3). C, Hippocampal neurons (E18 + 3 DIV) were transfected with pEGFP, alone or together with the FLAG-MST1 expression plasmid, and c-Abl RNAi or control vector as indicated. At 72 h after transfection, neurons were left untreated or were treated with H2O2 and analyzed as in B. Percentage of cell death is represented as the mean ± SEM. Cell death was significantly increased upon H2O2 treatment in U6-transfected neurons (ANOVA; p < 0.01, n = 3) but not in c-Abl knockdown neurons. Exposure of MST1-transfected neurons to hydrogen peroxide reliably induced cell death (ANOVA; p < 0.01, n = 3), and this neuronal cell death was dramatically reduced by c-Abl knockdown (ANOVA; p < 0.01, n = 3).
c-Abl phosphorylates MST1 at Y433 in vitro and in vivo
Since c-Abl is a non-receptor tyrosine kinase, we next tested the possibility that MST1 is a direct substrate of c-Abl. We found that recombinant GST-fused MST1 protein was directly phosphorylated by c-Abl by using an in vitro kinase assay followed by immunoblotting with an anti-pan-tyrosine antibody (Fig. 2A). We also found that WT, but not KD, c-Abl phosphorylates MST1 upon expression in HEK 293T cells (Fig. 2B). We further narrowed down the phosphorylation site(s) to the C terminus (aa 326–487) of MST1 (Fig. 2C). Using tandem mass spectrometry (MS/MS) analysis, we observed phosphorylation of only one phospho-tyrosine residue (Y433) in the C-terminal domain of MST1 (Fig. 2D). This tyrosine residue is highly conserved among MST1 proteins across species including vertebrates and Xenopus laevis, and the YxxL (x means any amino acid) motif in MST1 serves as a conserved sequence for phosphorylation by c-Abl (Fig. 2E) (Barilá et al., 2000; Srinivasan et al., 2009; Zuckerman et al., 2009). c-Abl phosphorylates MST1-WT in vivo as demonstrated using an anti-pan-tyrosine antibody, but it failed to phosphorylate MST1-Y433F, in which Y433 was replaced with phenylalanine (Fig. 2F).

c-Abl phosphorylates MST1 at Y433 both in vitro and in vivo. A, Lysates of HEK 293T cells transfected with Myc-tagged c-Abl or the control vector were immunoprecipitated with Myc antibody and subjected to an in vitro kinase assay using full-length GST-MST1 as substrate. Phosphorylation reactions were analyzed by immunoblotting with a tyrosine-specific phosphorylation antibody. MST1 was tyrosine phosphorylated by c-Abl kinase in vitro. B, Lysates of HEK 293T cells transfected with FLAG-tagged MST1, alone or together with Myc-tagged c-Abl-WT or c-Abl-KD expression plasmid, were immunoprecipitated with FLAG antibody and analyzed by immunoblotting against tyrosine-specific phosphorylation antibody. MST1 was tyrosine phosphorylated by c-Abl kinase in vivo. C, Lysates of HEK 293T cells transfected with FLAG-tagged MST1 (aa 1–487), NT (N terminus, aa 1–326), or CT (C terminus, aa 326–487), alone or together with Myc-tagged c-Abl WT expression plasmid, were immunoprecipitated and analyzed as in B. c-Abl-phosphorylated MST1 at the C terminus. D, Phosphorylation reactions from A were subjected to SDS-PAGE followed by Coomassie Blue staining. The band corresponding to MST1 was excised from the gel and digested with trypsin. Phosphorylation sites were mapped by microcapillary liquid chromatography-MS/MS, resulting in 80% coverage of the MST1 amino acid sequence. A phospho-peptide consistent with phosphorylation at Y433 was identified. E, Alignment of sequences in the C terminus of mammalian MST1 and Xenopus laevis MST1 with other putative c-Abl phosphorylation substrates. Y433 of MST1 at the YxxL motif is evolutionarily conserved. F, Lysates of HEK 293T cells transfected with Myc-tagged c-Abl, alone or together with FLAG-tagged MST1-WT, or Y433F expression plasmid were immunoprecipitated and analyzed as in C. Y433 is the phosphorylation site of MST1 by c-Abl.
To further validate the phosphorylation of MST1 at Y433 in vivo, we raised an antibody to specifically recognize MST1 proteins when phosphorylated at the Y433 site (Fig. 3A). We found that the antibody recognized MST1-WT, but not MST1-Y433F, when coexpressed with c-Abl-WT, and c-Abl-KD failed to induce MST1 phosphorylation at Y433 (Fig. 3B). Together, these results support the conclusion that c-Abl kinase phosphorylates MST1 at Y433 in vitro and in vivo.

Oxidative stress induced phosphorylation of MST1 at Y433 both in vitro and in vivo. A, c-Abl kinase was subjected to an in vitro kinase assay using the recombinant full-length GST-MST1 WT or GST-Y433F as a substrate. Phosphorylation reactions were analyzed by immunoblotting with Y433-specific phosphorylation antibody (p-Y433-MST1). c-Abl phosphorylated MST1 at Y433 in vitro. B, Lysates of HEK 293T cells transfected with the FLAG-tagged MST1- WT, or Y433F mutant alone or together with the Myc-tagged c-Abl-WT, or c-Abl-KD expression plasmid were analyzed by immunoblotting with a p-Y433-MST1 antibody. c-Abl phosphorylated MST1 at Y433 in vivo. C, SH-SY5Y cells were left untreated or treated with 200 μm H2O2 for 1 h with or without 10 μm STI571. Lysates of cells were immunoprecipitated with p-Y433-MST1 antibody followed by immunoblotting with a mouse monoclonal antibody to MST1 (Abcam). D, HT-22 cells were transfected with c-Abl RNAi alone or together with the FLAG-tagged MST1 expression plasmid. At 72 h after transfection, lysates of cells treated with or without 200 μm H2O2 were immunoprecipitated with p-Y433-MST1 antibody and then immunoblotted with FLAG antibody. E, Hippocampal neurons (E18 + 3 DIV) left untreated or treated with H2O2 for 1 h, with or without 10 μm STI571, were subjected to immunocytochemical analysis with the p-Y433-MST1 antibody and a mouse monoclonal antibody to neuron-specific βIII tubulin (Tuj 1), together with the DNA dye Hoechst 33258. H2O2 induced endogenous MST1 Y433 phosphorylation. F, The rat hippocampus was injected, and the rat killed and perfused as in Figure 1A. Brains were then sectioned for anti-p-Y433-MST1 staining. Representative images of the ipsilateral side of brain DG are shown (scale bar, 100 μm). At 24 h after surgery, rotenone induced MST1 Y433 phosphorylation, which was blocked by STI571 in vivo.
c-Abl mediates oxidative stress-induced phosphorylation of MST1
Oxidative stress is known to activate c-Abl kinase activity (Sun et al., 2000). Our next objective was to determine whether oxidative stress induces p-Y433 MST1. c-Abl-mediated Y433 phosphorylation of MST1 was increased in neuroblastoma cells (SH-SY5Y) and mouse hippocampal cells (HT-22) upon treatment with H2O2 (Fig. 3C,D). Furthermore, this phosphorylation was attenuated either by STI571 treatment or by c-Abl knockdown (Fig. 3C,D). The subcellular distribution of p-Y433 MST1 was further investigated by immunocytochemical analysis in neurons. We detected hydrogen peroxide-stimulated p-Y433 MST1 signal accumulated in cytoplasm of neurons, which was specifically blocked by the phospho-peptide containing the pY433 (Fig. 3E). Correspondingly, STI571 treatment impaired phosphorylation signal induced by hydrogen peroxide or rotenone in hippocampal dentate gyrus neurons (Fig. 3E,F). These data support the conclusion that c-Abl phosphorylates MST1 at Y433 in neurons in response to oxidative stress.
c-Abl-mediated phosphorylation of MST1 stabilizes its protein level
Interestingly, we observed that the MST1 protein level was increased when coexpressed with c-Abl-WT, but not when coexpressed with c-Abl-KD (Fig. 2B), and c-Abl knockdown resulted in a reduction in the MST1 protein level (Fig. 4A), leading us to further characterize the effect of the phosphorylation event on MST1 protein stability. As expected, we found that c-Abl-WT, but not c-Abl-KD, increased the MST1 protein level in a dose-dependent manner, and consistent with this, c-Abl kinase failed to increase the MST1-Y433F protein level (Fig. 4B). We next determined the expression of MST1-WT or MST1-Y433F when coexpressed with c-Abl after protein synthesis inhibition by treating cells with cycloheximide (CHX). We found that MST1-WT, but not MST1-Y433F, was stabilized by c-Abl (Fig. 4C), which suggests that c-Abl phosphorylation of the MST1 protein increases its stability. In other experiments, we found that c-Abl knockdown reduced the protein stability of endogenous MST1 (Fig. 4D).

c-Abl-mediated phosphorylation stabilizes MST1 protein. A, Neuro2A cells were transfected with c-Abl RNAi or control vector. At 72 h after transfection, lysates were subjected to immunoblotting with c-Abl, MST1 (Cell Signaling Technology), and actin antibody. B, Lysates of HEK 293T cells transfected with FLAG-tagged MST1-WT or MST1–Y433F mutant, alone or together with increasing amounts of Myc-tagged c-Abl-WT or c-Abl-KD expression plasmid, were analyzed by immunoblotting with the FLAG, Myc, or ERK1/2 antibody. C, Neuro2A cells were transfected with FLAG-tagged MST1-WT or MST1–Y433F mutant, together with Myc-tagged c-Abl expression plasmid. At 24 h after transfection, cells were treated with 50 μg/ml CHX for different time periods. Equal amounts of total protein lysates were subjected to immunoblotting. c-Abl stabilized the protein level of WT-MST1 but not that of Y433F-MST1. D, Neuro2A cells were transfected with c-Abl RNAi or control vector. At 72 h after transfection, cells were treated with 50 μg/ml CHX for the times indicated, and the lysates were subjected to immunoblotting with MST1 (Cell Signaling Technology), c-Abl, or ERK1/2 antibody.
c-Abl phosphorylation of MST1 protects it from ubiquitination
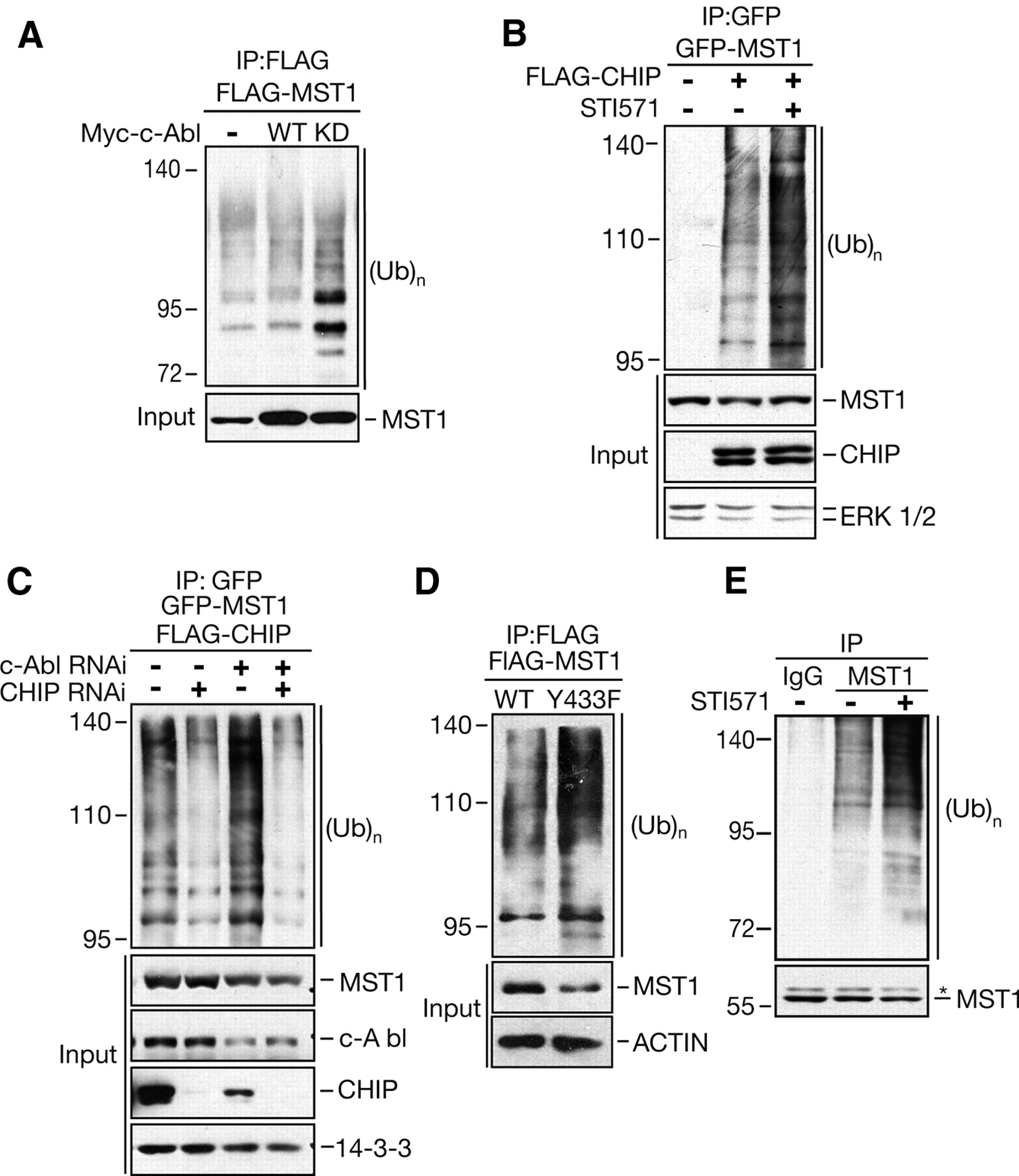
A previous report indicated that MST1 can be destroyed by the proteasomal degradation pathway (Ren et al., 2008). To understand the mechanism, we performed an in vivo ubiquitination assay to investigate whether c-Abl regulates MST1 ubiquitination. As predicted, MST1 cotransfected with c-Abl-KD but not WT was heavily ubiquitinated, with a characteristic ladder indicative of polyubiquitination (Fig. 5A). These data suggest that c-Abl protects MST1 from proteasome-dependent degradation in a kinase-dependent manner.

c-Abl-mediated phosphorylation affects MST1 ubiquitination. A, HEK 293T cells were transfected with FLAG-tagged MST1 and HA-ubiquitin, alone or together with Myc-tagged c-Abl-WT, or c-Abl-KD expression plasmid. At 36 h after transfection, lysates of cells were immunoprecipitated with FLAG antibody, followed by immunoblotting with HA antibody to detect ubiquitinated MST1. B, HEK 293T cells were transfected with GFP-tagged MST1 and HA-tagged ubiquitin, alone or together with a FLAG-tagged CHIP expression plasmid. At 36 h after transfection, the indicated cells were treated with 10 μm STI571 for 16 h. Cell lysates were immunoprecipitated with GFP antibody and analyzed as in A. C, HEK 293T cells were transfected as in B, together with c-Abl RNAi, CHIP RNAi, or control vector. At 72 h after transfection, lysates of cells were immunoprecipitated and analyzed as in B. D, HEK 293T cells were transfected with FLAG-tagged MST1-WT or MST1–Y433F mutant, together with HA-tagged ubiquitin expression plasmid. At 36 h after transfection, cell lysates were immunoprecipitated and analyzed as in A. E, HT-22 cells were treated with MG132, alone or together with 10 μm STI571 for 16 h. Cell lysates were immunoprecipitated with MST1 antibody (Cell Signaling Technology), followed by immunoblotting with ubiquitin antibody. Total MST1 is shown in the bottom by using the MST1 antibody (Cell Signaling Technology). The asterisk indicates nonspecific bands.
In agreement with previous reports (Ren et al., 2008), we could detect increased ubiquitination of MST1 in the presence of CHIP (Fig. 5B), which is an E3 ubiquitin ligase of the U-box protein family that is involved in regulation of MST1 protein levels through proteasomal degradation. In support of a role for c-Abl in MST1 polyubiquitination, we further observed that c-Abl inhibition resulted in a substantial increase in polyubiquitinated MST1 (Fig. 5B). In agreement with this, c-Abl knockdown in HEK 293T cells resulted in an increase in MST1 polyubiquitination, and this effect was dramatically abolished by CHIP knockdown (Fig. 5C). Moreover, MST1-Y433F alone confers more ubiquitination than MST1-WT in the presence of CHIP (Fig. 5D), and STI571 led to increased endogenous MST1 polyubiquitination in the present of MG132 (Fig. 5E). Together, our findings indicate that Y433 phosphorylation stabilizes the MST1 protein through blocking ubiquitin-mediated proteasomal degradation, likely through reducing MST1 binding to CHIP.
Oxidative stress-induced tyrosine phosphorylation of MST1 activates the MST-FOXO3 signaling pathway
c-Abl-mediated MST1 phosphorylation and stabilization led us to determine whether MST1 is activated by this phosphorylation per se. MST1 kinase activity was monitored by an in vitro kinase assay using Histone H2B or the FD of FOXO3 as the substrate. We demostrate that c-Abl is capable of enhancing kinase activity of WT-MST1 but not MST1-Y433F (Fig. 6A,B). Similarly, inhibition of c-Abl kinase activity by STI571 resulted in a decreased phosphorylation of FOXO3 at Ser 207, corresponding to the reduced phosphorylation of MST1 at Y433 (Fig. 6C). We previously reported that FOXO3 binds to MST1 through the Forkhead domain of FOXO3 (Lehtinen et al., 2006), and here we demonstrate further that the C terminus of MST1 is essential for this association, even though there is a stronger interaction between FOXO3 and full-length MST1 (data not shown). Since we found that both STI571 treatment of neurons and knockdown of endogenous c-Abl in Neuro2A cells greatly reduced hydrogen peroxide-induced interaction between MST1 and FOXO3 (Fig. 6D,E), we next asked whether Y433 phosphorylation in the C-terminal region of MST1 enhanced the interaction between MST1 and FOXO3. As expected, we found that c-Abl increased MST1-WT, but not MST1-Y433F, binding to FOXO3 (Fig. 6F). These results strongly suggest that MST1 phosphorylation by c-Abl in response to hydrogen peroxide enhances the interaction of MST1 and FOXO3.

c-Abl regulates oxidative stress-induced activation of MST1. A, Lysates of HEK 293T cells transfected with FLAG-tagged MST1, alone or together with Myc-tagged c-Abl expression plasmid, were immunoprecipitated with FLAG antibody and subjected to an in vitro kinase assay using Histone H2B as the substrate in the presence of [32P]ATP. Phosphorylation reactions were analyzed by autoradiography. B, Lysates of HEK 293T cells transfected with Myc-c-Abl, alone or together with FLAG-tagged MST1-WT or MST1–Y433F expression plasmid, were immunoprecipitated with FLAG antibody and subjected to an in vitro kinase assay using GST-FOXO3-FD as the substrate in the presence of [32P]ATP. Phosphorylation reactions were analyzed by autoradiography. C, HEK 293T cells transfected with Myc-tagged c-Abl, FLAG-tagged MST1, and FOXO3 expression plasmids were left untreated or treated with 10 μm STI571. Lysates of cells were immunoblotted with p-S207 FOXO3 and p-Y433-MST1 antibody. D, Hippocampal neurons were left untreated or were treated with 400 μm H2O2 for 1 h with or without 10 μm STI571. Lysates of neurons were immunoprecipitated with MST1 antibody (Cell Signaling Technology), followed by immunoblotting with FOXO3 antibody. E, HT-22 cells transfected with the c-Abl RNAi or control U6 vector were left untreated or were treated with 400 μm H2O2 for 1 h. Lysates of cells were immunoprecipitated and analyzed as in D. F, HEK 293T cells were transfected with the indicated expression plasmids. Lysates of cells were immunoprecipitated with FLAG antibody, followed by immunoblotting with GFP or p-Y433-MST1 antibody.
c-Abl-MST1 signaling mediates oxidative stress-induced transcriptional activation of FOXO3 and neuronal cell death
To further study the effect of tyrosine-phosphorylated MST1 on the transcription activation of FOXO3, we used 3xIRS or BIM promoter-luciferase in primary granule neurons as a reporter of FOXO3 transactivation (Lehtinen et al., 2006; Yuan et al., 2009). Both MST1 RNAi and dominant-negative (K59R) MST1 significantly decreased FOXO3 transactivation in the presence of c-Abl (to 62.5 ± 9.7% and 42.4 ± 6.3% of control sample, p < 0.05 and p < 0.01, respectively). Similarly, MST1 knockdown led to a decrease in BIM promoter activity (to 83.7 ± 5.7% of control sample, p < 0.01). Moreover, FOXO3 transcription activity was remarkably reduced when cotransfected with MST1-Y433F (to 55.8 ± 15.6% of cells transfected with MST1-WT, p < 0.01), suggesting that MST1-Y433F might function as a dominant-negative form for FOXO3 transcriptional activation. As expected, c-Abl knockdown resulted in a reduction of FOXO3 transcriptional activity on the 3xIRS and BIM promoter-luciferase (to 47.6 ± 7.3% and 28.3 ± 15.3% of control sample, respectively, p < 0.01) in the presence of hydrogen peroxide. Consistently, in contrast to hydrogen peroxide-induced expression of BIM protein, c-Abl knockdown impaired BIM protein level (Fig. 7A). Together, these results indicate that phosphorylation of MST1 at Y433 by c-Abl kinase activates the transcriptional activation of FOXO3 and directly activates the cell death machinery likely through promoting BIM expression.

c-Abl-MST1 signaling mediates oxidative stress-induced transcription and neuronal death. A, Lysates of HT-22 cells transfected with c-Abl RNAi or control vector were left untreated or treated with increasing concentrations of H2O2 (0–100 μm) for 12 h and then subjected to immunoblotting. B, Top, Hippocampal neurons transfected with pEGFP and MST1 RNAi or control vector, alone or together with MST1, MST1R, and MST1R-Y433F expression plasmids, were treated with H2O2 and analyzed as in Figure 1B. MST1R but not Y433F mutants of MST1R blocked the ability of MST1 knockdown to protect neurons from H2O2-induced death (ANOVA; p < 0.01, n = 3). Data are presented as means ± SEM. Lower right, Lysates of 293T cells transfected with FLAG-MST1 or FLAG-MST1R expression plasmids, together with MST1 RNAi or control vector, were immunoblotted with the FLAG and ERK1/2 antibodies. Lower left, Lysates of 293T cells transfected with MST1, MST1R, MST1R-Y433F, or control vector were immunoblotted with the FLAG and ERK1/2 antibodies. C, Model of the c-Abl-MST1 signaling pathway.
We next tested the significance of c-Abl-mediated phosphorylation of MST1 during oxidative stress-induced neuronal cell death. Expression of an RNAi-resistant form of MST1 (MST1R), but not MST1-WT, reversed the ability of MST RNAi to protect neurons from hydrogen peroxide-induced cell death (Fig. 7B) (Lehtinen et al., 2006). In contrast to MST1R, MST1R mutants in which Y433 was replaced with phenylalanine (MST1R-Y433F) failed to mediate neuronal cell death in a MST1 RNAi background (Fig. 7B). In control experiments, mutation of Y433 had no effect on the amount of MST1R expression (Fig. 7B). These results indicate that phosphorylation of MST1 at Y433 is required for its ability to mediate hydrogen peroxide-induced neuronal cell death.
Discussion
In this study, we discovered a novel signaling mechanism underlying oxidative stress-induced neuronal cell death. Our findings indicate that oxidative stress in neurons induces the c-Abl-mediated phosphorylation of MST1 at Y433, leading to increased MST1 protein stability and kinase activity through inhibiting MST1 proteasomal ubiquitination (Fig. 7C). We also defined a novel regulatory mechanism of MST1-FOXO3 signaling by identifying that the interaction between MST1 and FOXO3 can be enhanced via c-Abl-mediated tyrosine phosphorylation. Together, the results show that that c-Abl-MST1 signaling pathway plays a key role in cellular responses to oxidative stress in the mammalian CNS.
This elucidation of the c-Abl-induced phosphorylation of MST1 proteins provides the first molecular basis for how MST is activated by phosphorylation in the mammalian system. The molecular link between intracellular signaling and MST activation has long been a major enigma in the Hpo/MST1 research field. Proteolytic cleavage by caspase-3 remains the best known and only mechanistically understood stimulus for MST to date (Radu and Chernoff, 2009). Initially, it was proposed that MST is negatively regulated by phosphorylation, because epidermal growth factor (EGF) stimulation results in MST1 phosphorylation and decreased kinase activity, which can be restored by protein phosphatase 2A (PP2A) (Creasy and Chernoff, 1995). Subsequent studies showed that Akt-mediated phosphorylation of MST1 at T387 inhibits its cleavage and kinase activity (Jang et al., 2007; Yuan et al., 2010), and MST1 together with Akt and PH-domain leucine-rich repeat protein phosphate (PHLPP) constitute an inhibitory triangle that controls the fine balance of apoptosis and proliferation (Qiao et al., 2010). It is evident that phosphorylation and cleavage, alone or together, play an important role in regulating MST activity. Our finding that MST1 can be phosphorylated and activated by c-Abl kinase in response to oxidative stress in neurons provides the first evidence of MST1 regulation through a stress-responsive non-receptor tyrosine kinase during neuronal cell death. We also observed that MST1 p-Y433 phosphorylation in primary neurons, which appeared 1 h after H2O2 treatment, was sustained at 2 h, declined afterward, and was very low at 24 h (data not shown) preceding cell death, is transient, suggesting that Y433 phosphorylation is dynamic and probably regulated by tyrosine dephosphatate(s).
Recent evidence suggests that the C-terminal domain of MST1 is versatile and can exhibit stimulatory activity, depending on the specific substrate (Anand et al., 2008). The SARAH domain has been shown to be critical for the interaction of MST1 and its interacting proteins, such as the WW45 and RASSF family proteins (Zeng and Hong, 2008). Phosphorylation of Y433 may affect the interaction between MST1 and its interacting proteins, which needs to be further investigated. We have shown that hydrogen peroxide stimulates the interaction between MST1 and FOXO3 (Lehtinen et al., 2006). Here, we found that, on the one hand, c-Abl-mediated tyrosine phosphorylation enhances the interaction of MST1 and FOXO3, while on the other hand, this interaction is dramatically reduced by either treatment with STI571 or c-Abl knockdown, suggesting that MST1 tyrosine phosphorylation by c-Abl positively regulates MST1-FOXO3 signaling cascade and leads to neuronal apoptosis upon oxidative stress. In light of these observations, our findings uncover a novel regulatory mechanism underlying the interaction of MST1 and its substrate.
In this study, we focused on the role of the c-Abl-MST1 signaling pathway in response to oxidative stress in mammalian cells, especially neurons. Interestingly, Y433 is highly conserved among MST1 family members across species, but is absent in MST2 or Hippo, indicating that MST1 regulation may have become diversified during evolution. It will be interesting to investigate whether c-Abl regulates the other MST family proteins.
Numerous studies support a role for the co-chaperone and ubiquitin ligase CHIP in countering neurodegenerative diseases through regulating the degradation of phosphorylated tau (Dickey et al., 2006, 2007), modulating the toxicity of several polyglutamine (polyQ) disease proteins (Jana et al., 2005; Miller et al., 2005; Choi et al., 2007), and promoting toxic α-synuclein oligomers (Shin et al., 2005) and leucine-rich repeat kinase-2 (LRRK2) (Ding and Goldberg, 2009; Ko et al., 2009) for degradation. Although the neuroprotective potential of MST1 regulation by CHIP still needs further confirmation in animals, it will be interesting to investigate the relationship between loss of function of CHIP and MST1 activation under oxidative stress conditions.
In summary, the present study defines a critical oxidative stress pathway in neurons mediated by the c-Abl-MST1 complex. Identification of the signaling link between c-Abl and MST1 kinases fills the gap between oxidative stress and MST1 activation-dependent neuronal cell death, and also points to new biological roles of both kinases. Since MST1 mediates oxidative stimuli-triggered neural apoptosis, our findings here also raise the possibility that c-Abl-MST1 signaling might contribute to pathological states including neurodegenerative diseases. Elucidation of the c-Abl-MST1 signaling pathway as a key mediator of oxidative stress-induced neuronal cell death raises an important question of whether inhibition of this signaling pathway might provide a new therapeutic avenue for degenerative neurological diseases.
Footnotes
-
This work was supported by the National Science Foundation of China (Grant No. 30870792 and 81030025), and the National Basic Research Program of China (973 Program, Grant No. 2009CB918704). We thank Dr. S. Perrett, Dr. J. Fleming, and members of the Yuan laboratory for critical reading of this manuscript and helpful discussion. We thank C. Cao for the pCMV-Myc c-Abl WT and KD plasmid, J. X. Sun for the pFLAG-CMV10-CHIP plasmid, J. Y. Wu for HT-22 cells, and B. L. Zhao for the SH-SY5Y cells and stereotaxic drug delivery instruments.
-
The authors declares no competing financial interests.
- Correspondence should be addressed to either of the following: Dr. Yan Wang, Department of Orthopaedics, Chinese PLA General Hospital, Beijing 100853, China, yanwang301{at}yahoo.com; or Dr. Zengqiang Yuan, State Key Laboratory of Brain and Cognitive Sciences, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China, E-mail: zqyuan{at}ibp.ac.cn





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}