

Abstract
Presynaptic reuptake, mediated by the dopamine (DA) transporter (DAT), terminates DAergic neurotransmission and constrains extracellular DA levels. Addictive and therapeutic psychostimulants inhibit DA reuptake and multiple DAT coding variants have been reported in patients with neuropsychiatric disorders. These findings underscore that DAT is critical for DA neurotransmission and homeostasis. DAT surface availability is regulated acutely by endocytic trafficking, and considerable effort has been directed toward understanding mechanisms that govern DAT's plasma membrane expression and postendocytic fate. Multiple studies have demonstrated DAT endocytic recycling and enhanced surface delivery in response to various stimuli. Paradoxically, imaging studies have not detected DAT targeting to classic recycling endosomes, suggesting that internalized DAT targets to either degradation or an undefined recycling compartment. Here, we leveraged PRIME (PRobe Incorporation Mediated by Enzyme) labeling to couple surface DAT directly to fluorophore, and tracked DAT's postendocytic itinerary in immortalized mesencephalic cells. Following internalization, DAT robustly targeted to retromer-positive endosomes, and DAT/retromer colocalization was observed in male mouse dopaminergic somatodendritic and terminal regions. Short hairpin RNA-mediated Vps35 knockdown revealed that DAT endocytic recycling requires intact retromer. DAT also targeted rab7-positive endosomes with slow, linear kinetics that were unaffected by either accelerating DAT internalization or binding a high-affinity cocaine analog. However, cocaine increased DAT exit from retromer-positive endosomes significantly. Finally, we found that the DAT carboxy-terminal PDZ-binding motif was required for DAT recycling and exit from retromer. These results define the DAT recycling mechanism and provide a unifying explanation for previous, seemingly disparate, DAT endocytic trafficking findings.
SIGNIFICANCE STATEMENT The neuronal dopamine (DA) transporter (DAT) recaptures released DA and modulates DAergic neurotransmission, and a number of DAT coding variants have been reported in several DA-related disorders, including infantile parkinsonism, attention-deficit/hyperactivity disorder and autism spectrum disorder. DAT is also competitively inhibited by psychostimulants with high abuse potential. Therefore, mechanisms that acutely affect DAT availability will likely exert significant impact on both normal and pathological DAergic homeostasis. Here, we explore the cellular mechanisms that acutely control DAT surface expression. Our results reveal the intracellular mechanisms that mediate DAT endocytic recycling following constitutive and regulated internalization. In addition to shedding light on this critical process, these findings resolve conflict among multiple, seemingly disparate, previous reports on DAT's postendocytic fate.
Introduction
Dopamine (DA) neurotransmission is required for vital functions in the CNS, including locomotion, reward, and sleep/arousal (Iversen and Iversen, 2007). Dysregulated DA neurotransmission is linked to multiple neurological and psychiatric disorders such as Parkinson's disease, schizophrenia, attention-deficit/hyperactivity disorder (ADHD), autism spectrum disorder (ASD), and bipolar disorder (Sawa and Snyder, 2002; Chen et al., 2004; Sulzer and Schmitz, 2007; Sharma and Couture, 2014). Following evoked DA release, extracellular DA is cleared rapidly via presynaptic reuptake, mediated by the plasma membrane DA transporter (DAT) (Gether et al., 2006; Torres and Amara, 2007). DAT is potently inhibited by addictive psychostimulants, such as cocaine and amphetamine, as well as by therapeutic drugs, such as methylphenidate (Ritalin) (Torres and Amara, 2007; Kristensen et al., 2011). Multiple DAT coding variants have been reported in ADHD, ASD, and infantile Parkinsonism, and these DAT mutants exhibit anomalous DA efflux, loss of function, or DAT membrane instability due to endocytic dysfunction (Mazei-Robison et al., 2008; Sakrikar et al., 2012; Hamilton et al., 2013; Bowton et al., 2014; Cartier et al., 2015). Therefore, DA homeostasis is exquisitely sensitive to altered DAT activity, and mechanisms that regulate DAT function and availability are predicted to significantly impact DA-dependent processes.
DAT surface expression is dynamically regulated by endocytic trafficking (Melikian, 2004; Eriksen et al., 2010; Bermingham and Blakely, 2016). DAT internalizes constitutively, and either acute PKC activation or amphetamine exposure accelerates DAT endocytosis, thereby decreasing DAT surface expression and function (Daniels and Amara, 1999; Melikian and Buckley, 1999; Saunders et al., 2000). Considerable effort has been directed toward understanding DAT's postendocytic fate. Multiple studies indicate that DAT recycles back to the plasma membrane under basal and regulated conditions (Loder and Melikian, 2003; Boudanova et al., 2008a; Furman et al., 2009b; Chen et al., 2010; Richardson et al., 2016). Paradoxically, DAT postendocytic tracking studies using either antibody feeding or fluorescent cocaine analogs to track DAT reported that DAT targets to prelysosomal and lysosomal pathways, not to classical recycling endosomes (Eriksen et al., 2009; Eriksen et al., 2010; Vuorenpää et al., 2016). In contrast, neuronal fractionation studies (Rao et al., 2011) and tracking studies in heterologous cells and primary cultured neurons suggest differential DAT targeting (Hong and Amara, 2013). Moreover, a recent antibody feeding study performed in acute mouse brain slices reported little surface DAT internalization in either DAergic terminals or somatodendritic compartments (Block et al., 2015). These conflicting reports have raised considerable debate among transporter biologists and have failed to identify the mechanism(s) that governs DAT's postendocytic fate.
The retromer complex mediates endosomal export of cargo destined for either retrograde transport to the trans-Golgi network (TGN), or for rapid recycling to the plasma membrane (Seaman, 2004; Burd and Cullen, 2014). Multiple neuronal proteins recycle via a retromer-dependent mechanism, including β2-adrenergic receptors, ionotropic glutamate receptors, wntless, and Alzheimer-associated sortilin-related receptor 1 (Rogaeva et al., 2007; Zhang et al., 2012; Choy et al., 2014; Varandas et al., 2016). Moreover, retromer disruption is closely linked to multiple neurological disorders, including Alzheimer's and Parkinson's diseases (Muhammad et al., 2008; Tsika et al., 2014; Dhungel et al., 2015; Small and Petsko, 2015).
Previous discrepancies in DAT postendocytic mechanisms prompted us to investigate the DAT postendocytic itinerary in the absence of either bound ligands or antibodies, and to identify the molecular mechanisms governing DAT postendocytic sorting. In the current study, we took advantage of PRIME labeling (Uttamapinant et al., 2010) to covalently couple fluorophore to the DAT surface population and subsequently track DAT's temporal-spatial postendocytic itinerary. Our results reveal a critical role for retromer in DAT endocytic trafficking and implicate the DAT carboxy-terminal PDZ-binding motif in retromer-mediated DAT recycling.
Materials and Methods
Materials.
Picolyl azide (pAz) and Bis[(tertbutyltriazoyl)methyl]-[(2-carboxymethyltriazoyl)methyl]-amine (BTTAA) were synthesized as described previously (Uttamapinant et al., 2012). Rat anti-DAT antibody (MAB369) was from Millipore and mouse anti-actin antibody (sc-56459) was from Santa Cruz Biotechnology. Mouse anti-EEA1 (610456) and mouse anti-rab11 (610656) antibodies were from BD Transduction. Rabbit anti-rab7 antibody (D95F2) was from Cell Signaling Technology and goat anti-Vps35 antibody (NB100–1397) was from Novus Biologicals. Horseradish peroxidase-conjugated secondary antibodies were from Millipore (goat anti-rat), Jackson ImmunoReseach (donkey anti-goat), and Pierce (goat anti-mouse). Alexa Fluor-conjugated anti-rat, anti-mouse, and anti-rabbit secondary antibodies were from Invitrogen and donkey anti-goat Alexa Fluor 488 (minimal cross-reaction to mouse serum proteins) and donkey anti-rat Alexa Fluor 594 (minimal cross-reaction to mouse serum proteins) were from Jackson ImmunoResearch. Alkyne-Alexa Fluor 594 was from Invitrogen. [3H]DA (dihydroxyphenylethylamine 3,4-[ring-2,5,6-3H]) was from PerkinElmer. Sulfo-NHS-SS-biotin, Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and streptavidin agarose were from Thermo Fisher Scientific. Phorbol 12-myristate 13-acetate (PMA), AIM-100, β-CFT (WIN35428), and GBR12909 were from Tocris Bioscience. All other chemicals and reagents were from Thermo Fisher Scientific or Sigma-Aldrich and were of highest grade possible.
cDNA constructs and mutagenesis.
To generate ligase acceptor peptide (LAP)-hDAT-pcDNA3.1(+), hDAT was subcloned into pBS-SKII− as a shuttle vector and degenerate mutations were introduced into hDAT codons corresponding to amino acids 193 and 204, adding BsaBI and HpaI sites (Quikchange mutagenesis kit; Agilent Technologies). Sense and antisense oligonucleotides encoding the LAP-peptide sequence, flanked by linkers (GSSGSSGGFEIDKVWHDFPAGSSGSSG; LAP peptide sequence is underlined), were annealed and ligated into the blunt BsaBI/HpaI site, and the final LAP-DAT cDNA was excised subcloned into pcDNA3.1(+) at HindIII/XbaI sites. LAP-DAT-AAA-pcDNA3.1(+) was generated by mutating the last three amino acids of hDAT into alanines (LKV to AAA) using Quikchange mutagenesis kit (Agilent Technologies). All DNA sequences were determined by the dideoxy chain termination (Genewiz).
Cell culture and transfections.
The rat mesencephalic cell line 1Rb3AN27 was kindly provided by Dr. Alexander Sorkin (University of Pittsburgh, Pittsburgh, PA) and maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mm glutamine, and 100 units/ml penicillin/streptomycin at 37°C and 5% CO2. Pooled, stable AN27 cell lines expressing wild-type hDAT, LAP-hDAT, or LAP-hDAT-AAA were generated by transfecting 2 × 105 cells/well in 6-well culture plate with 1 μg of plasmid DNA using Lipofectamine 2000, at a lipid: DNA ratio of 2:1 (w/w). Stably transfected cells were selected with 200 μg/ml G418 (Invitrogen) and resistant cells were trypsinized, pooled, and maintained under selective pressure in 80 μg/ml G418. SK-N-MC cells were from the American Type Culture Collection and were maintained in MEM (Sigma-Aldrich M2279) supplemented with 10% fetal bovine serum (Invitrogen), 2 mm l-glutamine, 102 U/ml penicillin/streptomycin at 37°C and 5% CO2. Pooled #14 stable SK-N-MC cell lines expressing hDAT were generated by transfecting 1 × 106 cells/well in 6-well culture plate with 3 μg of plasmid DNA using Lipofectamine 2000 at a lipid: DNA ratio of 2:1 (w/w). Stably transfected cells were selected in 500 μg/ml G418 (Invitrogen) and resistant cells were pooled and maintained under selective pressure in 200 μg/ml G418.
[3H]DA uptake assay.
Cells were seeded onto 96-well plates 1 d before performing assays at 2 × 104 or 7.5 × 104 cells per well for single-point and kinetic studies, respectively. Cells were washed twice with KRH buffer containing the following (in mm): 120 NaCl, 4.7 KCl, 2.2 CaCl2, 1.2 MgSO4, 1.2 KH2PO4,and 10 HEPES, pH 7.4, and preincubated in KRH buffer supplemented with 0.18% glucose at 37°C for 30 min in the presence of either vehicle or the indicated drugs. For single-point assays, uptake was initiated by adding 1 μm [3H]DA in KRH buffer supplemented with 0.18% glucose and 10 μm each pargyline and sodium ascorbate (KRH/g/p/a). For uptake kinetics, 10× mixtures were prepared by diluting a 1/20th volume [3H]DA into 60 μm unlabeled DA KRH/g/p/a. The 60 μm [3H]DA solution was serially diluted into KRH/g/p/a to generate [3H]DA cocktails at 10× the indicated final substrate concentrations. Cells were preincubated (20 min at 37°C) with KRH buffer supplemented with 0.18% glucose (KRH/g) and uptake was initiated by adding a 1/10 volume of 10× concentrated [3H] substrate mixture. Nonspecific uptake at each DA concentration was defined in parallel in the presence of 10 μm GBR12909. Assays proceeded for 10 min at 37°C and were terminated by washing cells rapidly three times with ice-cold KRH buffer. Cells were solubilized in scintillation fluid and accumulated radioactivity was measured by liquid scintillation counting in a Wallac Microbeta plate counter.
Endocytic rate measurements by reversible biotinylation.
Cells were plated at 3 × 105 cells per well in 6-well plate 24 h before conducting experiments. Cells were washed three times with ice-cold PBS, pH 7.4, supplemented with 1 mm MgCl2 and 0.1 mm CaCl2 (PBS2+); surface proteins were biotinylated twice, 15 min at 4°C with 2.5 mg/ml sulfo-NHS-SS-biotin and quenched twice for 15 min at 4°C with 100 mm glycine/PBS2+ (quench solution). Internalization was initiated by washing cells rapidly three times with prewarmed (37°C) PBS2+ supplemented with 0.18% glucose/0.1% IgG/protease-free BSA (PBS2+/g/BSA) containing either vehicle or the indicated drugs, and incubating for 10 min at 37°C. Zero time point and strip controls were kept at 4°C in parallel. Endocytosis was arrested by washing cells rapidly with ice-cold NT buffer (150 mm NaCl, 20 mm Tris, pH 8.6, 1.0 mm EDTA, pH 8.0, 0.2% protease free/IgG free BSA). Residual surface biotin was stripped by reducing twice with 100 mm TCEP in NT buffer for 25 min at 4°C, followed by 3 washes with PBS2+. Cells were lysed in RIPA containing protease inhibitors for 20 min at 4°C with constant shaking and insoluble cellular debris was pelleted at 14,000 × g for 10 min at 4°C. Lysate protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific), compared with BSA standards, and biotinylated proteins were isolated from equivalent amounts of total cellular protein by batch streptavidin–agarose affinity chromatography as described previously (Loder and Melikian, 2003; Holton et al., 2005; Boudanova et al., 2008a; Boudanova et al., 2008b; Gabriel et al., 2013; Wu et al., 2015). Samples were resolved by SDS-PAGE and DAT was detected by immunoblot using a monoclonal rat anti-DAT antibody (MAB369; Millipore). Nonsaturating DAT bands were detected using a VersaDoc gel documentation system and were quantified using Quantity One software (Bio-Rad). Relative internalization rates were calculated as a percentage of biotinylated DAT internalized compared with DAT surface levels at t = 0.
W37VLplA expression and purification.
His6-taggedW37VLplA was expressed and purified as described previously (Uttamapinant et al., 2013). Briefly, pYFJ16-His6-W37VLplA plasmid was transformed into BL21 E. coli and bacteria were incubated at 37°C with shaking until they attained log phase growth. Protein expression was induced with 100 μg/ml isopropyl-β-d-thiogalactopyranoside and proceeded for 8 h at room temperature. Bacteria were lysed and His6-W37VLplA was purified by nickel-affinity chromatography. Eluted protein was dialyzed for 8 h twice against 20 mm Tris base, 1 mm DTT, 10% (v/v) glycerol, pH 7.5, at 4°C and protein concentrations were determined by A280 absorbance (NanoDrop; Thermo Fisher Scientific) using an extinction coefficient of 46250 M−1 cm−1. Ligase aliquots were stored at −80°C.
PRIME labeling and postendocytic tracking studies.
Live AN27 cells stably expressing the indicated LAP-DAT constructs were covalently labeled with alkyne-Alexa Fluor 594 as described previously (Uttamapinant et al., 2010; Uttamapinant et al., 2013). Briefly, cells were seeded onto glass coverslips in 24-well plates at a density of 8 × 104 cell/well 1 d before assaying. Cells incubated with a ligation mixture containing 10 μm W37VLplA, 200 μm pAz, 1.0 mm ATP, and 5.0 mm MgCl2 in PBS/3% BSA for 20 min at room temperature, followed by 3 washes with PBS2+. A low Cu2+ click labeling solution containing 10 mm CuSO4, 50 mm BTTAA, and 100 mm sodium ascorbate and was prepared and incubated at room temperature for 10 min in a closed tube. The labeling solution was diluted 200× with PBS2+ (producing final concentrations of 50 μm CuSO4, 250 μm BTTAA, and 500 μm sodium ascorbate), supplemented with alkyne-Alexa Fluor 594 to a final concentration of 20 μm, and incubated with cells for 10 min at room temperature. Cells were washed three times with room temperature PBS2+ and internalization was initiated by washing cells rapidly and incubating in prewarmed (37°C) PBS2+/g/BSA containing the indicated drugs. Cells were fixed at the indicated postendocytic time points in 4% paraformaldehyde for 10 min at room temperature and subsequently blocked, permeabilized, and stained with indicated primary antibodies and Alexa Fluor 488-conjugated secondary antibodies as described previously (Navaroli et al., 2011). Note that all of the antibodies directed against endosomal markers were carefully vetted for specificity by their ability to recognize native and GFP-tagged proteins via immunoblot and by their ability to label GFP-tagged endosomal markers in situ. Dried coverslips were mounted in ProLong Gold with DAPI to stain nuclei and were cured thoroughly before performing imaging.
Wide-field microscopy.
Cells were visualized with a Zeiss Axiovert 200M microscope using a 63×, 1.4 numerical aperture (NA) oil-immersion objective and 0.2 μm optical sections were captured through the z-axis with a Retiga-1300R or Regita-R1 cooled CCD camera (Qimaging). 3D z-stack images were deconvolved with a constrained iterative algorithm using measured point spread functions for each fluorescent channel using Slidebook version 5.0 software (Intelligent Imaging Innovations). All representative images shown are single, 0.2 μm planes through the center of each cell.
Image processing for colocalization.
Given an unprocessed, multicolor, 3D image stack, the DAT image was used to identify an extracellular background region of interest for background fluorescence estimation and the range of contiguous optical sections (z-axis planes) containing in-focus DAT signal (i.e., the cell). The sample fields imaged were chosen to contain at least 1 extracellular region at least 5 μm across. This background region was determined automatically by first taking the maximum intensity projection (in Z) of the DAT stack. Then, the (x,y) position of the 2D region (x ± radius, y ± radius) having the lowest average intensity within this projection was saved. The radius used was nominally 20 pixels (±2 μm). For each of the color image stacks, at each z plane, the average intensity of this 2D region was subtracted from all pixels of the plane, leaving as positive signal the fluorescence greater than the extracellular background. The outline of the box containing this background region was superimposed on the DAT maximum projection image, as well as the maximum projections of the other color stacks, for visual inspection and verification before proceeding with the analysis.
The in-focus DAT data planes were also determined automatically, by first calculating the normalized total energy, Ê, of each Z plane, defined as follows:
 and
and
 where I([x, y], z) is the pixel intensity at position [x, y] in plane z, Ī(z) is the average intensity of plane z, and n(z) is the total number of pixels in plane z. Starting from the bottom of the stack (i.e., the first z plane) and moving up, the first in-focus plane zbot was defined as the z plane where Ê(z) exceeds ÊT = 0.5. Similarly, starting from the top (i.e., the last z plane) and moving down, the last in-focus plane ztop was where Ê(z) exceeds the threshold ÊT. This was generally a conservative threshold, keeping a few out-of-focus planes at the top and the bottom. The planes zbot to ztop were extracted from the background-corrected multicolor stacks for deconvolution. The point-spread function of the microscope system was determined from images of slides of four-color, 175 nm diameter beads (PS-Speck Microscope Point Source Kit; Thermo Fisher Scientific) All images were subjected to regularized, constrained, iterative deconvolution as described previously (Carrington et al., 1995) with the same smoothness parameter (α = 5·10−5) and integrated until the algorithm reached convergence (0.001 level). All image processing was performed using custom software.
where I([x, y], z) is the pixel intensity at position [x, y] in plane z, Ī(z) is the average intensity of plane z, and n(z) is the total number of pixels in plane z. Starting from the bottom of the stack (i.e., the first z plane) and moving up, the first in-focus plane zbot was defined as the z plane where Ê(z) exceeds ÊT = 0.5. Similarly, starting from the top (i.e., the last z plane) and moving down, the last in-focus plane ztop was where Ê(z) exceeds the threshold ÊT. This was generally a conservative threshold, keeping a few out-of-focus planes at the top and the bottom. The planes zbot to ztop were extracted from the background-corrected multicolor stacks for deconvolution. The point-spread function of the microscope system was determined from images of slides of four-color, 175 nm diameter beads (PS-Speck Microscope Point Source Kit; Thermo Fisher Scientific) All images were subjected to regularized, constrained, iterative deconvolution as described previously (Carrington et al., 1995) with the same smoothness parameter (α = 5·10−5) and integrated until the algorithm reached convergence (0.001 level). All image processing was performed using custom software.
Colocalization analysis.
All image analysis was performed using custom software. Restored images were first segmented via a manually set threshold to identify the signal in each independent channel. Each 3D restored image was projected (via maximum intensity projection) to 2D and then displayed to maximize contrast between signal and background. Three independent thresholds were then chosen for each image at low, medium, and high levels. The criteria used to define these thresholds were as follows: “low” allowed for some diffuse background signal within the cells, “medium” removed all the diffuse cellular signal, and “high” eliminated the margins of labeled structures. To avoid biasing the analysis, thresholds were chosen without knowledge of the experimental conditions of the image. All pixels above threshold in the 3D images were retained, whereas those below threshold were set to 0. DAT colocalization with the indicated proteins was calculated by counting the number of positive (>0) pixels in the 3D DAT image that were also positive in the corresponding position of the indicated 3D protein image and dividing by the total number of positive DAT pixels. Colocalization percentages were calculated at three different thresholds for each channel; however, the percentages calculated using the medium threshold were chosen for statistical analysis and data presentation. The low and high thresholds were used to evaluate whether conclusions drawn from the colocalization results at the medium thresholds varied dependent upon the threshold chosen. Comparison among these conditions revealed that relative changes in DAT/protein colocalization across a given time course were not dependent upon the threshold level chosen; rather, only the baseline (t = 0) percent colocalization was affected by a given threshold level.
Brain slice immunohistochemistry and confocal microscopy.
All animals were handled according to University of Massachusetts Medical School Institutional Animal Care and Use Committee Protocol A1506 (H.E.M.). Adult male C57/Blk6 mice were perfused transcardially with 4% paraformaldehyde and brains were removed and postfixed for 1 d at 4°C, followed by dehydration in PBS/30% sucrose at 4°C for 2–3 d. Then, 25 μm coronal sections were taken through the striatum and midbrain using a sliding microtome (Leica) and slices were blocked in PBS with 0.2% Triton X-100, 5% normal donkey serum, and 1% H2O2. For DAT and Vps35 immunofluorescence, sections were coincubated overnight with rat anti-DAT (1:2000) and goat anti-Vps35 (1:500) in PBS with 0.2% Triton X-100, 5% normal donkey serum, and 1% H2O2. Slices were rinsed in PBS and incubated with donkey anti-goat and donkey anti-mouse Alexa Fluor (1:2000 each) for 1 h at room temperature. Unbound secondary antibodies were washed in PBS and slices were mounted onto glass slides, dried, and coverslipped in Prolong Gold mounting medium containing DAPI (Invitrogen). Images were acquired with a Leica TCS SP5 II laser scanning confocal microscope (Cell and Developmental Biology Core, University of Massachusetts Medical School) using either a 20×, 0.7 NA (HCX PL APO CS 20.0 × 0.70 IMM; Leica) or a 63×, 1.4 NA oil-immersion (HCX PL APO CS 63.0 × 1.40 OIL, Leica) objective. Then, 0.4 μm optical sections were captured through the z-axis and 3D z-stack images were imported into ImageJ using the Bio-Format Importer plugin. All images shown are single, representative 0.4 μm planes.
Short hairpin RNA (shRNA), lentiviral production, and transduction.
Human Vps35-targeted shRNAs cloned into the pGIPZ lentiviral vector were from GE Healthcare Dharmacon and were purchased from the University of Massachusetts Medical School RNAi Core.
Full-length hairpin sequences were as follows: nonsilencing (Luciferase 693): TGCTGTTGACAGTGAGCGCTCTAAGAACGTTGTATTTATATAGTGAAGCCACAGATGTATATAAATACAACGTTCTTAGATTGCCTACTGCCTCGGA; hVps35 sh#32: TGCTGTTGACAGTGAGCGACTGACAGATGAGTTTGCTAAATAGTGAAGCCACAGATGTATTTAGCAAACTCATCTGTCAGGTGCCTACTGCCTCGGA.
Initial shRNA efficacies were determined by immunoblotting cell lysates obtained from HEK293T cells transiently transfected (Lipofectamine 2000) with the indicated pGIPZ-shRNA versus control plasmids. Replication incompetent lentiviral particles were produced as described previously (Wu et al., 2015) and titers were determined 48 h after transfection by counting GFP-positive cells in transduced HEK293T cells.
For lentiviral transduction, 2.5 × 105 DAT SK-N-MC cells/well were seeded into 12-well plates 1 d before viral transduction and infected with 3 ml of the indicated crude lentivirus supplemented with 8 μg/ml polybrene. Virus was removed 24 h after infection and replaced with medium supplemented with 1 μg/ml puromycin to select transduced cells. Cells were assayed 96 h after infection.
Endocytic recycling assay.
DAT delivery to the cell surface from endocytic compartments was measured using a continuous biotinylation assay, as described previously (Boudanova et al., 2008a). DAT-SK-N-MC cells were treated with the indicated lentiviral particles for 96 h at 37°C and surface DAT was biotinylated at 4°C as described above. Endocytic trafficking was initiated by washing cells rapidly 3 times in prewarmed PBS2+/g/BSA (37°C) and incubating in PBS2+/g/BSA containing 1.0 mg/ml NHS-SS-biotin at 37°C for the indicated times, to covalently couple biotin to proteins newly delivered to the plasma membrane. To compensate for NHS group hydrolysis, the biotinylation reagent was replenished after the initial 20 min incubation for the 30 and 45 min time point samples. Labeling was terminated by washing 3 times with ice-cold quench solution and residual biotinylation reagent was quenched by incubating twice with quench solution for 15 min at 4°C. Parallel samples were maintained at 4°C to define the baseline biotinylation level at t = 0. Cells were lysed and biotinylated proteins were separated from nonbiotinylated by batch streptavidin affinity chromatography, as described above. Supernatants were concentrated using Amicon Ultra spin concentrators with a 30 kDa molecular weight cutoff (Millipore) and both biotinylated and supernatant DAT fractions were detected by immunoblot. The percentage biotinylated DAT at each time point was calculated as follows:
 Biotinylated DAT values at each time point were normalized to the baseline biotinylated DAT value determined at t = 0 for each independent experiment. Time course data were fitted to a one-phase association kinetic equation as follows:
Biotinylated DAT values at each time point were normalized to the baseline biotinylated DAT value determined at t = 0 for each independent experiment. Time course data were fitted to a one-phase association kinetic equation as follows:
 where K is the rate constant and tau is the time constant defined as 1/k.
where K is the rate constant and tau is the time constant defined as 1/k.
Cell surface biotinylation.
DAT surface levels in SK-N-MC cells were determined by steady-state biotinylation as described previously (Navaroli et al., 2011; Gabriel et al., 2013; Wu et al., 2015). Briefly, after infection with the indicated lentiviral particles, cells were labeled twice for 15 min at 4°C with 1.0 mg/ml sulfo-NHS-SS-biotin in PBS2+ and excess biotinylation reagent was quenched twice for 15 min at 4°C with PBS2+/100 mm glycine. Excess glycine was removed by washing 3 times in ice-cold PBS2+ and cells were lysed in RIPA buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) containing protease inhibitors. Lysates were cleared by centrifugation and protein concentrations were determined with the BCA protein assay kit (Pierce). Biotinylated proteins from an equivalent amount of cellular protein were recovered by batch streptavidin affinity chromatography (overnight at 4°C) and bound proteins were eluted in denaturing SDS-PAGE sample buffer for 30 min at room temperature with rotation. Samples were analyzed by SDS-PAGE and the indicated proteins were detected by immunoblotting with the indicated antibodies. Immunoreactive bands were detected with SuperSignal West Dura (Pierce) and captured using the VersaDoc Imaging station (Bio-Rad). Nonsaturating bands were quantified using Quantity One software (Bio-Rad).
Statistical analysis.
Data are presented as means of results from each independent experiment ± SEM, as indicated in figure legends. For experiments in which two conditions were compared, data were analyzed using an unpaired, two-tailed Student's t test. For experiments in which three or more conditions were evaluated, statistical significance was calculated using either a one-way or two-way ANOVA, followed by post hoc multiple-comparisons tests, as indicated in the figure legends. The initial slope for each time course was determined for data points between t = 0 and the plateau, defined as the point after which no significant differences were detected. Data from independent experiments were analyzed by linear regression and slope values were averaged. All statistical analyses were performed using GraphPad Prism 6.0 software.
Results
DAT expression, function, and trafficking tolerate LAP peptide incorporation into extracellular loop 2
Previous studies investigating DAT postendocytic trafficking relied primarily on either antibody feeding or DAT bound to fluorescent cocaine analogs, either of which have potential to target DAT's postendocytic destination(s) artifactually. We sought to track DAT endocytic trafficking by covalently labeling the DAT cell surface population with small fluorophores, thereby creating a “fluorescent DAT.” To accomplish this, we took advantage of the recently reported PRIME labeling approach (Uttamapinant et al., 2013). This method covalently couples fluorophore to cell surface proteins that encode an extracellular ligase acceptor peptide (LAP), which is a substrate for bacterial lipoic acid ligase (LplA). To generate a DAT construct compatible with extracellular LAP labeling, we replaced hDAT second extracellular loop (EL2) residues 193–204 with a 27 aa peptide that encodes the 13 aa LAP sequence flanked by linkers of 7 aa in length (LAP-DAT). We first tested whether the LAP peptide insertion had any deleterious effect on DAT expression, function, functional downregulation, or regulated endocytosis when stably expressed in the rat mesencephalic cell line 1Rb3AN27 (AN27). LAP-DAT exhibited robust, saturable [3H]DA uptake in these cells (Fig. 1A) and kinetic analyses revealed no significant difference between LAP-DAT and wild-type DAT (WT-DAT) Vmax values (WT-DAT: 99.3 ± 18.2 vs LAP-DAT: 94.4 ± 19.7 pmol/min/mg; p = 0.86, Student's t test, n = 6) or affinity for DA (Km: WT-DAT 2.8 ± 0.4 vs LAP-DAT 4.8 ± 1.3 μm; p = 0.17, Student's t test, n = 6). Immunoblot analysis revealed that LAP-DAT expressed at levels comparable to WT-DAT, with a similar ratio of mature (∼75 kDa) to immature (∼55 kDa) biosynthetic species (Fig. 1B, top). LAP-DAT was also subject to PKC-induced function downregulation comparable to WT-DAT. PKC activation with PMA decreased WT-DAT and LAP-DAT function significantly to 68.7 ± 3.4% and 58.2 ± 7.5% control levels, respectively (Fig. 1B, bottom). We next tested whether LAP-DAT constitutively internalized and was subject to stimulated endocytosis in response to Ack1 inhibition, as we reported previously (Wu et al., 2015). Basal LAP-DAT internalization rates were not significantly different from WT-DAT (p = 0.20), and AIM-100 treatment increased both LAP-DAT (134.5 ± 5.3% control levels) and WT-DAT (162.6 ± 16.8% control levels) internalization significantly (Fig. 1C). Therefore, appending the DAT EL2 domain with the LAP peptide did not affect DAT biosynthesis, function, or regulated endocytosis, which is consistent with previous studies in which epitopes were engineered into EL2 (Sorkina et al., 2006; Navaroli et al., 2011). It should be noted, however, that, although LAP-DAT expression and function were comparable to WT-DAT in AN27 cells, we observed marked LAP-DAT proteolytic cleavage when expressed in either SK-N-MC or SH-SY5Y cells (data not shown), precluding use of these cells for LAP-DAT studies.

DAT expression, function, downregulation, and endocytosis tolerate LAP peptide incorporation into the second extracellular loop. A, DA uptake kinetics. Saturable DA uptake was measured in stable WT-DAT AN27 or LAP-DAT AN27 cells as described in the Materials and Methods. Average data are presented ± SEM; n = 6. B, LAP-DAT expression and PKC-mediated downregulation. Top, Immunoblot analysis Representative immunoblots characterizing WT-DAT and LAP-DAT expression in AN27 cells and actin loading controls. Arrows indicated mature, fully glycosylated (m) and immature, partially glycosylated (i), DAT species. Bottom: [3H] DA uptake assay. Cells were treated ±1 μm PMA for 30 min at 37°C and [3H]DA uptake was measured with 1 μm DA, as described in the Materials and Methods. Average data are expressed as a percentage of vehicle-treated controls ± SEM. **Significant compared with vehicle-treated control, p < 0.01, Student's t test, n = 4. C, Internalization assay. WT-DAT and LAP-DAT internalization rates were measured in AN27 cells ±20 μm AIM-100, as described in the Materials and Methods. Top, Representative immunoblots showing the total surface protein at t = 0 (T), strip controls (S), and internalized protein during either vehicle (V), or AIM-100 (A) treatments. Bottom, Average internalization rates are presented as the percentage of vehicle-treated controls ± SEM. *Significantly greater than vehicle control, p < 0.05, one-tailed Student's t test, n = 6–10. D, PRIME strategy for labeling surface DAT. A LAP was engineered into the DAT second extracellular loop. pAz was covalently bound to surface DAT encoding LAP peptide (LAP-DAT) via LplA-mediated coupling, followed by conjugation with alkyne-Alexa Fluor 594 via low-Cu2+ click chemistry, resulting in a fluorescently labeled surface DAT population. E, LAP-DAT labeling. Stable LAP-DAT AN27 cells were labeled with alkyne-Alexa Fluor 594 (red) as described in Materials and Methods, followed by fixation and staining with anti-DAT antibody (green). Two representative fields are shown and all cells in the field are indicated by DAPI staining (blue).
We next tested whether LAP-DAT could be covalently coupled to fluorophore using the PRIME strategy (see schematic; Fig. 1D) by coupling alkyne-Alexa Fluor 594 to LAP-DAT AN27 cells and subsequently fixing, permeabilizing, and costaining cells with an anti-DAT antibody. Alkyne-Alexa Fluor 594 signal was detected at the surface of cells expressing LAP-DAT, whereas nearby nontransfected cells (identified using DAPI staining) were not labeled (Fig. 1E). Moreover, the alkyne-Alexa Fluor 594 signal overlapped with the anti-DAT antibody signal at the cell surface. These results demonstrate that PRIME labeling is both highly efficacious and specific for LAP-DAT. As a negative control, we performed PRIME labeling on cells expressing WT-DAT and observed no labeling (data not shown). We observed that a linker between the DAT polypeptide backbone and the LAP peptide was absolutely required to achieve high-efficiency labeling and earlier attempts at labeling LAP-DAT constructs, either without linkers or with shorter linkers, failed to label cells efficiently (data not shown).
Tracking DAT temporal-spatial postendocytic trafficking via PRIME labeling
We next used LAP-DAT to temporally and spatially track DAT's postendocytic fate. We labeled the DAT surface population at room temperature (18–22°C; conditions of minimal endocytosis), stimulated internalization by shifting cells to 37°C, and then fixed/stained cells at various postendocytic time points to quantify DAT colocalization with several endosomal markers (see schematic; Fig. 2A). As a proof of concept, we first characterized DAT trafficking from the plasma membrane to early endosomes, given consistent reports that DAT colocalizes to EEA1/rab5-positive vesicles shortly after internalization (Daniels and Amara, 1999; Melikian and Buckley, 1999; Eriksen et al., 2009). At the 0 min time point, 4.0 ± 0.2% of the LAP-DAT signal colocalized with EEA1 (Fig. 2B,C). This minimal colocalization was observed between DAT and each of the endocytic markers investigated throughout our study and we attributed this baseline signal to low-level DAT internalization that occurred during the room temperature labeling procedure. Under basal conditions, DAT/EEA1 colocalization increased rapidly and significantly over the first 10 min of internalization and peaked at 21.3 ± 1.4% colocalization, which translates to a 430.9% enhancement over baseline. DAT/EEA1 colocalization plateaued at subsequent time points, although there was a trend for decreased DAT/EEA1 colocalization at the 60 min time point (Fig. 2C; p = 0.08). We also observed significant, time-dependent DAT/EEA1 colocalization upon stimulating DAT internalization with 20 μm AIM-100 (Fig. 2C). However, we observed a significant decrease in the initial slope of DAT/EEA1 colocalization over time in cells treated with AIM-100 compared with vehicle-treated cells (initial slope = vehicle: 1.7 ± 0.4 vs AIM-100: 0.87 ± 0.1, p = 0.02, Student's t test, n = 30; Fig. 2C). These results demonstrate that the PRIME labeling strategy is effective for tracking DAT from the cell surface to endosomal destinations. Note that, in the interest of space, we have only presented DAT internalization images under control conditions because any AIM-100-induced differences are not discernible by visual inspection alone.

LAP-DAT labeling facilitates DAT postendocytic tracking. A, Strategy for tracking DAT postendocytic routes using PRIME labeling. Live LAP-DAT AN27 cells were labeled via PRIME labeling at room temperature, as described in the Materials and Methods. Internalization was induced by shifting cells to 37°C ± 20 μm AIM-100. Cells were then fixed at the indicated time points, permeabilized, and stained for EEA1. B, DAT trafficking to EEA1-positive early endosomes. Representative images of LAP-DAT (red) and the early endosome marker EEA1 (green) are shown at the indicated time points (note: vehicle-treated cells are shown). C, Average data expressed as the percentage of DAT colocalized with EEA1± SEM. Asterisks indicate significant differences, **p < 0.005, ***p < 0.001, two-way ANOVA with Bonferroni's multiple-comparisons test, n = 19–49 cells at each time point imaged across three independent experiments.
After internalization and localization to early endosomes, proteins are targeted to degradative, recycling, or retrograde (i.e., TGN) pathways. Given that DAT recycling and rapid delivery to the plasma membrane has been reported by us and others (Loder and Melikian, 2003; Lee et al., 2007; Gabriel et al., 2013; Richardson et al., 2016), we next investigated whether DAT targets to the classic rab11 recycling endosome (Welz et al., 2014). Under basal conditions, we observed no significant DAT/rab11 colocalization over baseline at either the 5 or 10 min time points (Fig. 3A,B). A small, but statistically significant, increase in DAT/rab11 colocalization (7.4 ± 0.42%) was observed at 20 min after endocytosis, which translated to 46.3% over baseline. During AIM-100 treatment, we observed significant increases in DAT/rab11 colocalization at 45 and 60 min time points and significantly more LAP-DAT colocalized with rab11 during AIM-100 treatment compared with control cells at the 60 min time point (Fig. 3B). These results indicate that although the majority of DAT does not traverse the conventional recycling pathway, a small but significant DAT population enters rab11-positive endosomes and this is increased when DAT endocytosis is stimulated by acute Ack1 inhibition.
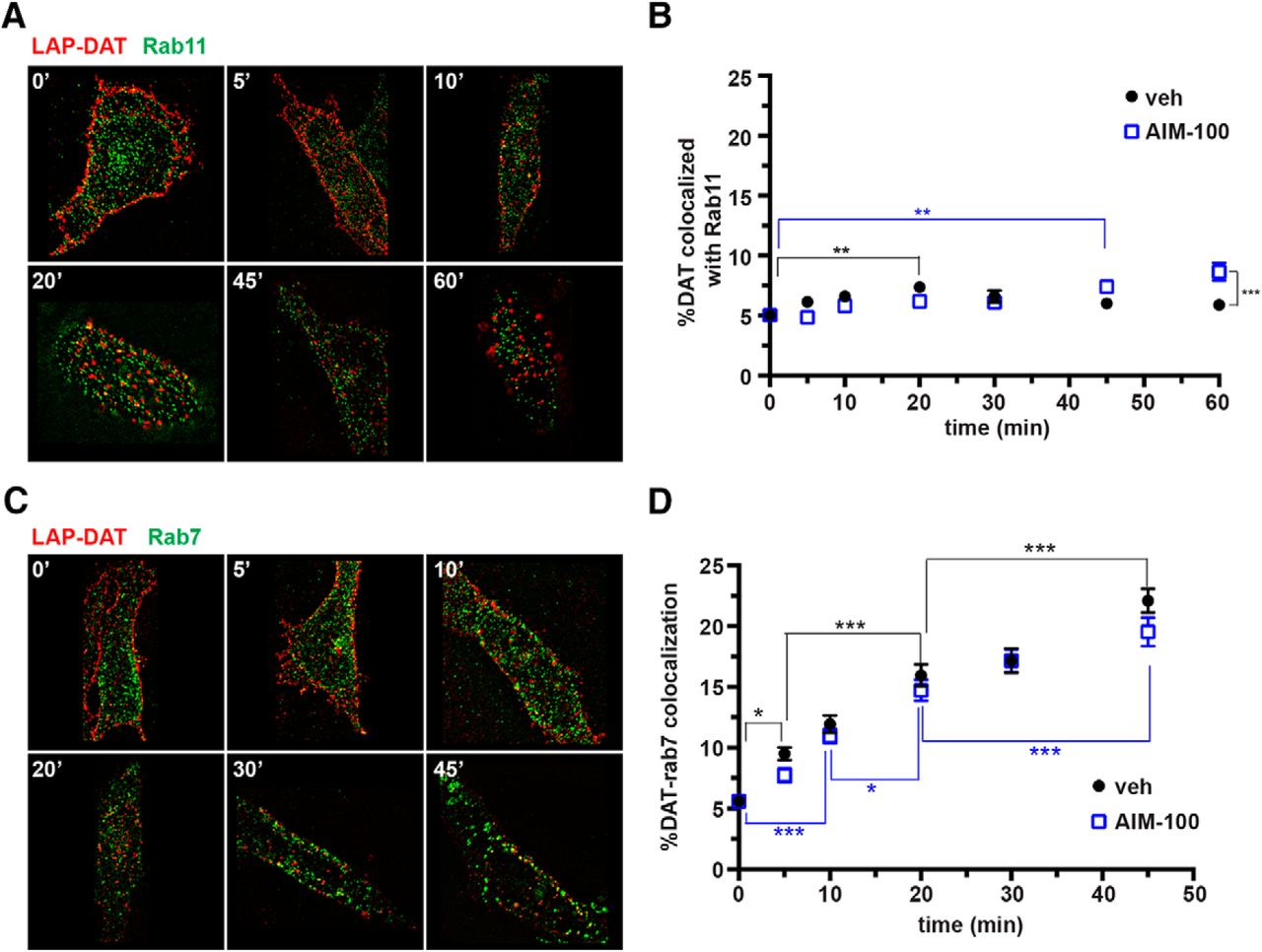
DAT targets to rab11- and rab-7-positive endosomes in PRIME labeling studies. Live LAP-DAT AN27 cells were labeled via PRIME labeling at room temperature, internalized for the indicated times at 37°C, and probed for either rab7 or rab11 as described in the Materials and Methods. A, C, Representative images showing LAP-DAT (red) with recycling endosome marker rab11 (green, A) or late endosome marker rab7 (green, C) at the indicated time points. B, D, Average data expressed as the percentage of DAT colocalized with either rab11 (B) or rab7 (D) ± SEM. Asterisks indicate significant differences, *p < 0.05, **p < 0.005, ***p < 0.001, two-way ANOVA with Bonferroni's multiple-comparisons test, n = 28–30 cells at each time point imaged over three independent experiments.
Given that the majority of internalized DAT did not colocalize with rab11-positive endosomes, we next investigated whether DAT targeted to degradation by staining late endosomes using rab7 as a marker. Under basal conditions, DAT exhibited a slow, linear (r2 = 0.95) increase in rab7 colocalization over time, with 22.1 ± 0.97% DAT/rab7 colocalization observed at the 45 min time point, which translated to a 295.5% elevation over baseline (Fig. 3C,D). AIM-100 treatment had no effect on the rate of DAT/rab7 colocalization (slope = vehicle: 0.34 ± 0.04 vs AIM-100: 0.31 ± 0.04, p = 0.62, Student's t test, n = 30). These results suggest that a fraction of DAT moves slowly into rab7-positive endosomes after internalization, with no kinetic difference in late endosome targeting during basal versus stimulated DAT endocytic conditions.
DAT targets to retromer-positive endosomes
Given that DAT expression is quite stable, we questioned whether DAT postendocytic sorting to a rab7-positive compartment was indicative of immediate postendocytic targeting to the degradative pathway, or to another rab7-positive endosomal population. Recent studies indicate that rab7 is also part of the cargo-selective trimer (Vps35-Vps29-Vps26) that recruits proteins to the retromer complex from the endosomal membrane (Rojas et al., 2008; Seaman et al., 2009). Therefore, we hypothesized that DAT may recycle to the plasma membrane via a retromer-mediated mechanism. To test this possibility, we examined whether internalized DAT entered a retromer-positive endosome, as indicated by colocalization with Vps35, a retromer core protein required for cargo recruitment into retromer (Seaman, 2004; Burd and Cullen, 2014). Under basal conditions, we observed DAT/Vps35 colocalization that increased rapidly and significantly over the first 15 min of internalization, and peaked at 24.0 ± 0.88% colocalization, which translates to 413.0% enhanced colocalization over baseline (Fig. 4A,B). We further observed a significant loss in DAT/Vps35 colocalization at 45 min compared with the 15 min peak, likely reflecting DAT exiting from Vps35-positive endosomes over time. During AIM-100 stimulation, DAT/Vps35 colocalization likewise increased significantly over time (Fig. 4B). However, AIM-100 treatment diminished the rate of increase in DAT/Vps35 colocalization significantly compared with vehicle-treated cells (initial slope = vehicle: 1.2 ± 0.30 vs AIM-100: 0.63 ± 0.10, p = 0.04, Student's t test, n = 24–30) and peaked at 30 min postinternalization (Fig. 4B). We further observed no significant loss of DAT/Vps35 colocalization after the 30 min peak. These results suggested that DAT is targeted to the retromer complex after either basal or stimulated endocytosis and is recruited away from retromer-positive endosomes over time under basal conditions.

DAT targets to retromer-positive endosomes. A, B, PRIME labeling studies. Surface DAT was coupled to Alexa594 via PRIME labeling, as described in the Materials and Methods, and DAT/Vps35 colocalization was determined at the indicated postendocytic time points. A, Representative images showing LAP-DAT (red) and retromer marker Vps35 (green) at the indicated time points. B, Average data expressed as the percentage of DAT-positive objects that colocalized with Vps35 signal ± SEM. All later time points were significant different from baseline; Asterisks indicate significant differences, *p < 0.05, **p < 0.001, two-way ANOVA with Bonferroni's multiple-comparisons test, n = 24–30 cells imaged at each time point imaged over three independent experiments. C, D, DAT/Vps35 colocalization in mouse DAergic neurons. Coronal sections were prepared from mouse brains and were costained for DAT and Vps35 and imaged as described in the Materials and Methods. C, Representative single-plane confocal images showing DAT (red) and Vps35 (green) expression in somatodendritic region of the substantial nigra. Scale bar, 50 μm (top) and 1 μm (bottom). D, Representative confocal images showing DAT (red) and Vps35 (green) staining in dorsal striatum. Scale bar, 5 μm. White arrowheads indicate DAT/Vps35 colocalized puncta.
We next investigated whether native DAT colocalized with Vps35 in bona fide DAergic neurons. Immunocytochemistry performed on coronal mouse brain slices revealed DAT/Vps35 colocalization in perinuclear regions of substantia nigra DAergic neurons (Fig. 4C). We also observed discrete DAT/Vps35-positive colocalized puncta in a subset of DAergic terminals in the dorsal striatum (Fig. 4D), consistent with DAT targeting to retromer complex. We also attempted to use PRIME labeling in ex vivo mouse striatal slices to track DAT postendocytic trafficking in native DAergic terminals. We engineered an AAV construct encoding a DIO-HA-tagged LAP-DAT and packaged this construct into AAV2 particles. These were injected stereotaxically into DATIRES-Cre mouse midbrain to facilitate HA-LAP-DAT expression exclusively in dopaminergic neurons. HA-LAP-DAT expressed and was targeted robustly to DAergic terminals in the dorsal striatum (data not shown). However, we were unable to label LAP-DAT efficiently in ex vivo striatal slices prepared from injected mice. Together with our cellular LAP-DAT studies and mouse striatal immunocytochemistry, these results indicate that DAT targets to retromer in both AN27 cells and intact DA neurons, consistent with the premise that internalized DAT is sorted and recycled via a retromer-dependent mechanism.
Retromer complex is required for DAT recycling
We next investigated whether retromer activity was required to recycle DAT back to the plasma membrane. We reasoned that if DAT recycling were retromer dependent, then retromer disruption would decrease the DAT recycling rate and potentially decrease DAT surface levels and target DAT to degradation in lieu of recycling. To test these hypotheses, we used shRNA to knockdown Vps35, an approach previously reported to disrupt retromer function (Choy et al., 2014; Varandas et al., 2016). For these studies, we turned to stably transfected DAT-SK-N-MC cells, in which DAT trafficking is indistinguishable from that observed in ex vivo mouse striatal slices (Gabriel et al., 2013; Wu et al., 2015). Moreover, this system enabled us to study WT-DAT (rather than LAP-labeled) and to make use of commercially available human-directed shRNA constructs targeting hVps35, whereas rat-directed shRNAs for use in AN27 cells are not commercially available. We screened several human Vps35-targeted shRNA constructs and found that shRNA#32 reduced Vps35 expression significantly to 35.2 ± 4.8% control Vps35 levels in DAT-SK-N-MC cells (Fig. 5A). Vps35 knockdown decreased both absolute DAT surface levels (normalized to actin, 54.9 ± 6.4% control; Fig. 5B) and total DAT levels (54.4 ± 4.7% control; Fig. 5C) significantly, whereas nonsilencing lentiviral particles had no effect on DAT expression. To test whether DAT loss in response to retromer disruption was due to impaired endocytic recycling, we used a continuous biotinylation assay to measure DAT recycling rates. We observed robust DAT recycling in cells infected with control lentiviral particles (Fig. 5D,E). In contrast, Vps35 knockdown attenuated DAT recycling significantly, with a significant increase in the recycling tau value (vector: 10.1 ± 1.9 vs shRNA#32: 15.7 ± 2.4, p < 0.05, one-tailed Student's t test, n = 7–8; Fig. 5E). Interestingly, although total DAT levels were diminished, the ratio of surface: intracellular DAT was significantly higher after Vps35 knockdown (Fig. 5E), suggesting that retromer disruption caused total DAT loss via enhanced postendocytic degradation. Indeed, we observed a low-molecular-weight DAT fragment in intracellular fractions (Fig. 5D) that was significantly enhanced after Vps35 knockdown, as normalized to mature intracellular DAT at the 0 min time point (vector: 0.11 ± 0.05; shRNA#32: 1.41 ± 0.82, p < 0.04, Student's t test, n = 3–5). Together, these results demonstrate that retromer is required to maintain DAT surface levels and supports that retromer is a major DAT recycling mechanism.
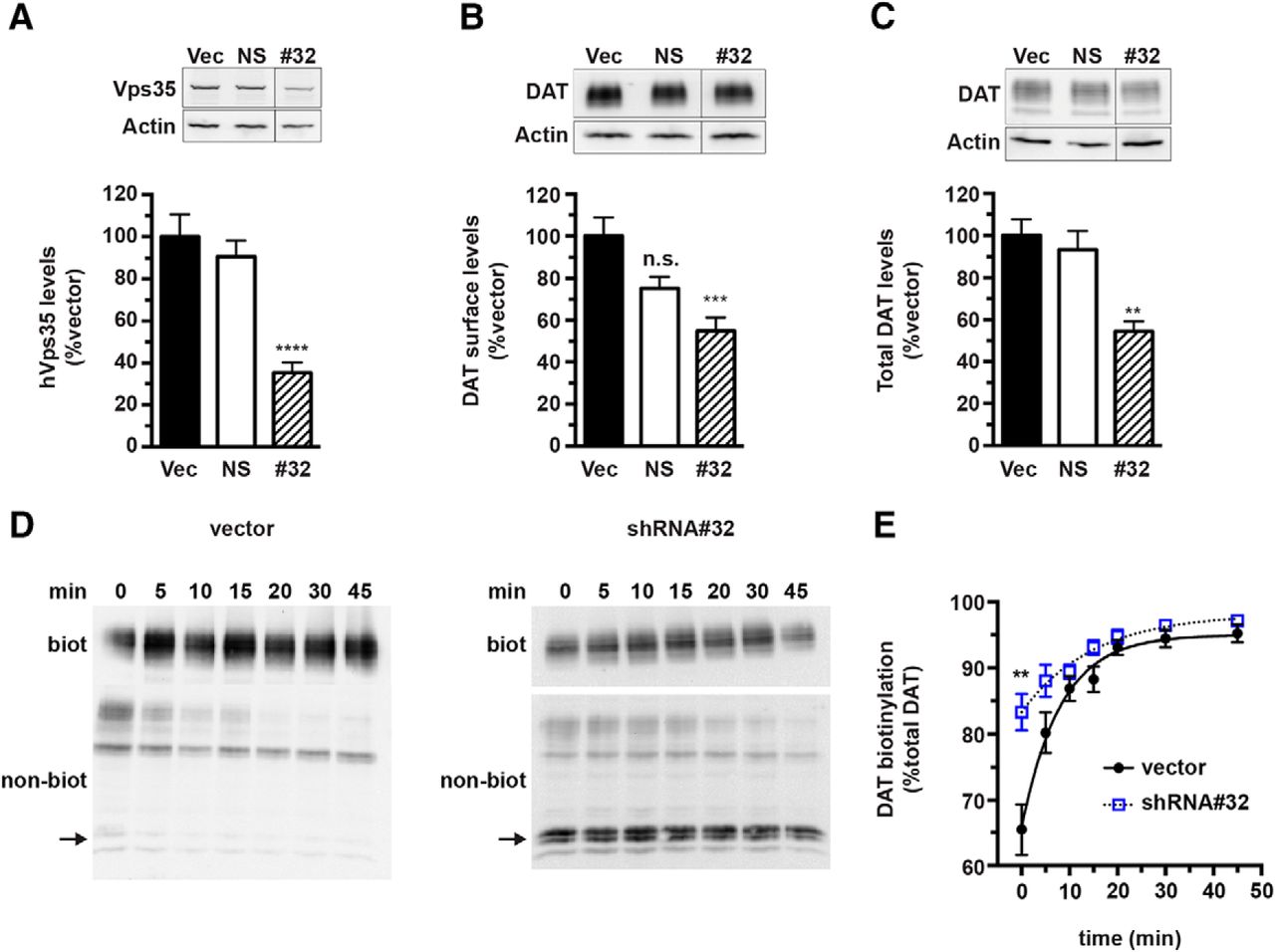
Retromer complex is required for DAT recycling and to maintain DAT surface expression. Lentiviral-mediated hVps35 knockdown in SK-N-MC cells. DAT SK-N-MC cells were transduced with the indicated lentiviral particles, as described in the Materials and Methods and assays were performed 96 h after transduction. A, Vps35 knockdown efficiency. Top, Representative immunoblots of lysates from DAT-SK-N-MC cells transduced with lentiviral particles expressing pGIPZ vector (vec), nonsilencing shRNA (NS), or hVps35 #32 shRNA (#32), probed for Vps35 and actin. Bottom, Average hVps35 protein levels expressed as the percentage of vector-transduced hVps35 levels ± SEM. ****Significantly different from vector control, p < 0.001, one-way ANOVA with Dunnett's post hoc analysis, n = 7. n.s., Not significant. B, DAT surface levels. DAT-SK-N-MC cells were transduced with the indicated lentiviral particles and DAT surface levels were measured by surface biotinylation, as described in the Materials and Methods. Top, Representative immunoblots showing the biotinylated DAT surface fraction and actin loading controls. Bottom, Average DAT surface levels (normalized to actin) expressed as the percentage of vector controls ± SEM. ***Significantly different from vector control, p < 0.005, one-way ANOVA with Bonferroni's multiple-comparisons test, n = 6. C, Total DAT expression. Total DAT levels were quantified by immunoblotting, normalizing to actin loading controls. Top, Representative immunoblots. Bottom, Average total DAT levels (normalized to actin) expressed as the percentage of vector levels ± SEM. **Significantly different from vector control, p < 0.01, one-way ANOVA with Bonferroni's multiple-comparisons test, n = 5. D, E, Endocytic recycling assay. DAT endocytic recycling rates were determined by continuous biotinylation, as described in the Materials and Methods. D, Representative DAT immunoblots of the biotinylated (biot) and nonbiotinylated (nonbiot) DAT fractions at the indicated time points for vector-treated and Vps35 shRNA#32-treated cells. Arrow indicates a low-molecular-weight DAT fragment. E, Average recycling data. Values are expressed as the percentage of total DAT biotinylated over time ± SEM. Two-way ANOVA indicates significant effect of shRNA, p < 0.0001, **Significantly greater than vector control, p < 0.0001, Sidak's multiple-comparisons test, n = 7–8.
Effect of cocaine and cocaine analogs on DAT targeting and recycling
Given that previous reports found that cocaine increases DAT surface expression acutely (Daws et al., 2002; Little et al., 2002) and also that previous studies used high-affinity, fluorescent cocaine analogs to track DAT trafficking (Eriksen et al., 2009; Eriksen et al., 2010), we next tested whether either cocaine or high-affinity tropane analogs influence DAT trafficking to either prelysosomal or retromer-positive endosomes. As seen in Figure 6A, incubation with saturating concentrations of the high-affinity cocaine analog WIN35429 had no significant effect on the slope of DAT targeting to rab7-positive endosomes (vehicle: 0.19 ± 0.05; WIN35429: 0.20 ± 0.06, p = 0.75, Student's t test, n = 30 cells sampled over 3 independent experiments), suggesting that high-affinity tropanes do not enhance DAT targeting to degradation. We next tested whether cocaine binding to DAT influenced either targeting to or recruitment from retromer-positive endosomes. Under vehicle-treated conditions, and consistent with our initial findings (Fig. 4), DAT-Vps35 colocalization increased significantly over time, from 0 to 20 min postendocytosis, and then decreased significantly from 20–45 min as DAT exited retromer-positive endosomes (Fig. 6B). In the presence of 100 μm cocaine, DAT-Vps35 colocalization increased significantly from 0–10 min after endocytosis, with no significant difference in the initial rate of DAT entry into Vps35-positive endosomes compared with vehicle (slopes measured from 0–10 min: vehicle = 1.7 ± 0.2; 100 μm cocaine = 1.9 ± 0.4, p = 0.55, Student's t test, n = 3 independent assays with 28–30 total cells; Fig. 6B). However, during cocaine treatment, DAT/Vps35 colocalization plateaued by 20 min and then decreased significantly by 45 min (Fig. 6B). Moreover, cocaine treatment significantly decreased DAT/Vps35 colocalization at the 20 and 45 min postendocytic time points, as compared to vehicle-treated controls (Fig. 6B). Given that cocaine did not alter targeting to degradative endosomes, these results suggest that cocaine may increase DAT recruitment out of retromer-positive endosomes and possibly increase DAT recycling, consistent with previous results demonstrating increased DAT surface expression after cocaine exposure (Daws et al., 2002; Little et al., 2002).

Cocaine accelerates DAT exit from retromer-positive endosomes in PRIME labeling studies. The DAT surface population was coupled to Alexa Fluor 594 via PRIME labeling, as described in the Materials and Methods, and DAT colocalization with either rab7 (A) or Vps35 (B) was measured at the indicated postendocytic time points, ±100 μm WIN35428 (A) or ± 100 μm cocaine (B). Average data are presented. Two-way ANOVA revealed an interaction of both time (p < 0.0001) and drug (p = 0.0018). Asterisks denote significant differences between the indicated samples, Tukey's multiple-comparisons test, ***p < 0.0001, *p < 0.03, n = 28–30 cells imaged over 3 independent experiments.
DAT exit from retromer is dependent upon its C-terminal PDZ-binding motif
We next investigated whether a specific domain either targeted DAT to and/or recruited DAT from retromer. Many cargo proteins are recruited to the retromer complex via PDZ-binding motifs (Lauffer et al., 2010; Clairfeuille et al., 2016; McGarvey et al., 2016). Interestingly, DAT encodes a C-terminal PDZ-binding motif (–LKV), and multiple reports indicate that DAT surface stability is dependent upon this motif (Torres et al., 2001; Bjerggaard et al., 2004; Rickhag et al., 2013). To test whether the DAT PDZ-binding motif is required for retromer-dependent recycling, we mutagenized the DAT C terminus within the LAP-DAT background, converting the –LKV motif to alanine residues (DAT-AAA), and tested whether DAT targeting to retromer-positive endosomes was affected. As seen in Figure 7, both WT-DAT and DAT-AAA targeted robustly to Vps35-positive endosomes and no significant differences were observed in their colocalization with Vps35 at either the t = 0 or t = 20 min time points (Fig. 7A,B). However, we observed significantly less WT-DAT/Vps35 colocalization compared with DAT-AAA at the 45 min time point, consistent with exit of WT-DAT, but not the DAT-AAA mutant, from retromer-positive endosomes. These data suggest that the DAT –LKV sequence may contribute to DAT exit from retromer-positive endosomes. To further test this possibility, we compared WT-DAT versus DAT-AAA recycling in AN27 cells (Fig. 7C,D). Interestingly, we observed a significant difference in recycling between WT-DAT and DAT-AAA and, in several experiments, DAT-AAA recycling failed to fit one-phase association kinetics, consistent with disrupted endocytic recycling.

DAT exit from retromer and recycling require a C-terminal PDZ-binding motif. A, B, PRIME labeling studies. WT-DAT and DAT-AAA surface populations were coupled to alkyne-Alexa Fluor 594 via PRIME labeling, as described in the Materials and Methods, and DAT/Vps35 colocalization was determined at the indicated postendocytic time points. A, Representative images showing WT-LAP-DAT or LAP-DAT-AAA (red) and retromer marker Vps35 (green) at the indicated time points. B, Average data expressed as the percentage of DAT-positive objects that colocalized with Vps35 signals ± SEM. Asterisks indicate significant differences, *p < 0.04, **p < 0.001, two-way ANOVA with Bonferroni's multiple-comparisons test, n = 29–30 cells imaged at each time point over three independent experiments. C, D, Endocytic recycling assay. DAT endocytic delivery to the plasma membrane was measured by continuous biotinylation in AN27 cells stably expressing either WT-LAP-DAT or LAP-DAT-AAA, as described in the Materials and Methods. C, Representative DAT immunoblots of the biotinylated (biot) and nonbiotinylated (nonbiot) DAT fractions at the indicated time points for WT-DAT (top) vs DAT-AAA (bottom). D, Average recycling data. Values are expressed as the percentage of total DAT biotinylated over time ± SEM. Two-way ANOVA indicates a significant effect of DAT construct, p < 0.0003, n = 7.
Discussion
After biosynthesis in midbrain dopaminergic soma, DAT is forward trafficked to distant presynaptic boutons. Given the considerable energy expenditure that neurons invest to generate DAergic terminals, it is evolutionarily advantageous to maintain these complex structures. Endocytic recycling rapidly modulates synaptic function, bypassing the need for de novo biosynthesis. Multiple studies report that DAT undergoes constitutive and regulated internalization (Melikian, 2004; Bermingham and Blakely, 2016). However, DAT's postendocytic fate has long been debated, with numerous reports demonstrating functional DAT recycling (Loder and Melikian, 2003; Boudanova et al., 2008a; Chen et al., 2013; Richardson et al., 2016), whereas tracking studies observe DAT targeting to degradative vesicles (Miranda et al., 2005; Eriksen et al., 2010). These disparate findings may arise from methodological differences among the studies, which use either antibodies (Hong and Amara, 2013; Block et al., 2015) or fluorescent ligands (Eriksen et al., 2009) to interrogate DAT targeting. Although our approach using PRIME labeling is not likely to either perturb the DAT substrate-binding pocket or impose large steric hindrance on DAT, there is still the possibility that appending DAT with a small fluorophore may influence its postendocytic fate.
Consistent with previous results, we observed robust, rapid DAT entry into EEA1-positive endosomes within 5–10 min after endocytosis. Rather than observing biphasic entry into and exit from these vesicles, we observed enhanced DAT/EEA1 colocalization that plateaued. This suggests that the DAT cell surface population did not internalize en masse. Rather, discrete DAT populations may internalize differentially over time, eventually redistributing labeled DAT throughout the endocytic pathway (Fig. 2C). Alternatively, DAT internalization may be a stochastic process; however, single-particle tracking studies would be required to test this possibility. Surprisingly, we observed a significantly slower initial rate of DAT/EEA1 colocalization during AIM-100-stimulated DAT internalization (Fig. 2C). Basal DAT internalization is clathrin and dynamin independent, whereas PKC- and AIM-100-stimulated DAT internalization are clathrin and dynamin dependent (Gabriel et al., 2013; Wu et al., 2015). Therefore, a DAT subpopulation may target to EEA1-negative early endosomes (Hayakawa et al., 2006; Lakadamyali et al., 2006; Kalaidzidis et al., 2015) after stimulated endocytosis. Alternatively, stimulating DAT internalization may saturate the early endosomal machinery and stall entry kinetics into EEA1-positive early endosomes.
Although rab11-dependent DAT delivery to the plasma membrane has been reported (Loder and Melikian, 2003; Furman et al., 2009a; Sakrikar et al., 2012; Richardson et al., 2016), previous DAT tracking studies have not observed robust DAT entry into classic rab11-positive recycling endosomes (Eriksen et al., 2010; Hong and Amara, 2013). We observed a small, but significant, enhancement in DAT/rab11 colocalization over time under basal conditions and significantly increased DAT targeting to rab11-positive endosomes after stimulated endocytosis (Fig. 3A,B). However, DAT/rab11 colocalization was relatively modest over time (∼46%), with kinetics that differed from DAT entry into early, EEA1-positive endosomes. In contrast, we observed robust and significant DAT colocalization in Vps35-positive vesicles, which peaked at 15–20 min after internalization (Fig. 4A,B), with 513% enhancement over baseline, comparable to that observed for DAT entry into early endosomes (∼500% over baseline). These kinetic similarities suggest that the majority of internalized DAT targets to the retromer-positive endosomes. Retromer and rab11 dependence are not necessarily mutually exclusive because several studies report that rab11 may function in consort with retromer to facilitate recycling (van Weering et al., 2012; Hsiao et al., 2015). Likewise, several reports indicate that rab7 is required for retromer recruitment both in yeast and mammalian cells and acts in consort with rab5 (Rojas et al., 2008; Seaman et al., 2009). Thus DAT trafficking through rab7-positive endosomes is also consistent with a retromer-mediated mechanism. However, DAT movement into rab7-positive versus retromer-positive endosomes exhibited distinctly slower and linear kinetics, suggesting that a DAT subset consistently targets to a degradative pathway. Indeed, we observed a decrease in total and surface DAT in response to retromer disruption, consistent with targeted degradation. However, unlike previous reports (Miranda et al., 2005; Hong and Amara, 2013), we did not observe enhanced DAT targeting to rab7-positive endosomes after stimulated DAT internalization (Fig. 3C,D).
It was not feasible to use PRIME labeling to track DAT's endocytic fate in ex vivo striatal slices. One possible explanation for this obstacle is that the 38 kDa LplA may not be able to efficaciously penetrate a 300 μm acute brain slice. A recent study similarly used antibody feeding to characterize DAT endocytic trafficking in a DAT knock-in mouse expressing an extracellular HA epitope, but found little antibody localization with endosomal markers in dopaminergic terminal regions (Block et al., 2015). It is possible that this result is due to challenges in permeating a relatively thick brain slice (1.0 mm) with a large (150 kDa), globular antibody, similar to our results with PRIME labeling. Moreover, it is unclear whether DAT bound to large Ig would reflect native DAT endocytic targeting accurately, particularly in light of recent studies demonstrating that antibody-bound membrane proteins target to degradation regardless of their endogenous endocytic routes (St Pierre et al., 2011; Bien-Ly et al., 2014). However, a recent antibody feeding study reported differential DAT targeting to degradation or recycling in response to PKC activation or AMPH exposure, respectively (Hong and Amara, 2013).
Although it was not technically feasible for us to track DAT trafficking in native preparations, robust perinuclear DAT/Vps35 colocalization was readily apparent in DAergic soma. This could indicate potential DAT recycling in somatodendritic compartments. Alternatively, this may simply reflect close opposition between retrograde-targeted proteins and TGN-localized DAT undergoing biosynthesis. Although DAT expression is robust in DAergic terminal regions, we only detected DAT/Vps35 colocalization in a small subset of DAT puncta. Interestingly, elegant recent work from Sulzer and colleagues revealed that only a small fraction of depolarized DAergic terminals actually release DA (Pereira et al., 2016). Therefore, DAT recycling may only occur in the small subset of actively releasing terminals. Consistent with this premise, we observe somewhat modest, but significant, DAT surface expression loss after PKC activation in ex vivo mouse striatal slices (Gabriel et al., 2013), which might reflect larger losses in individual active boutons averaged across a more static DAT population in inactive DA terminals. Future studies should more clearly illuminate this possibility.
Although retromer complex was originally characterized as the machinery that recruits cargo destined for retrograde trafficking to the TGN (Seaman et al., 1997; Seaman et al., 1998), recent studies demonstrate that retromer plays a key role in endosomal recycling (Seaman, 2012; Choy et al., 2014). Importantly, retromer disruption via shRNA-mediated Vps35 knockdown resulted in marked DAT depletion from the plasma membrane and a significant decrease in the DAT recycling rate (Fig. 5). However, retromer disruption did not deplete DAT surface expression completely over a 96 h knockdown window and did not block recycling completely. Given that we only achieved a partial Vps35 knockdown, there was likely still some active retromer complex available. Moreover, given that some DAT targets to rab11-positive endosomes, there may have remained some additional, compensatory retromer-independent recycling via a rab11-mediated mechanism.
Retromer-mediated cargo recruitment frequently requires interaction of sorting nexin 27 with a PDZ-binding motif on cargo proteins (Lauffer et al., 2010). DAT encodes a distal C-terminal PDZ binding motif (–LKV) and previous studies implicate this motif in stabilizing mature DAT in DAergic terminals (Torres et al., 2001; Bjerggaard et al., 2004; Rickhag et al., 2013). We found that DAT required the –LKV signal to exit from Vps35-positive endosomes and that mutagenizing this domain altered DAT recycling significantly, consistent with a role for this domain in retromer recruitment. Retromer-mediated endosomal fission in yeast requires Vps1, the yeast dynamin homolog (Chi et al., 2014; Arlt et al., 2015). Interestingly, we reported previously that DAT recycling relies upon a dynamin-dependent mechanism (Gabriel et al., 2013), also consistent with a retromer-mediated mechanism. Moreover, mutating the DAT carboxy-terminal PDZ binding motif reduced DAT surface expression in a mouse knock-in model and the dynamin K44A dominant-negative mutant partially rescued the surface loss of this mutant (Rickhag et al., 2013), suggesting that DAT-AAA is degraded postendocytically due to its inability to be recruited by retromer for recycling. Consistent with this possibility, GluT1 encodes a PDZ-binding domain required for recycling and retromer disruption drives GluT1 to degradation (Steinberg et al., 2013). Moreover, a recent interactome screen identified the DAT homolog GLYT2 (SLC6A9) as a retromer-associated protein (McMillan et al., 2016) and GLYT2 also encodes a carboxy-terminal PDZ binding motif that is required for stable plasma membrane expression (Armsen et al., 2007). In contrast, serotonin transporter expression and stability are insensitive to C-terminal perturbations (Schmid et al., 2001; El-Kasaby et al., 2010). It is also possible that other nearby residues affect DAT recruitment by retromer. Of particular potential interest is R615, particularly since a coding variant at this residue (R615C) was recently identified in an ADHD proband (Sakrikar et al., 2012). When characterized in cell lines, the R615C exhibits marked endocytic dysfunction, which can be rescued by coexpressing constitutively active Ack1 (Wu et al., 2015). Future studies evaluating potential R615C recruitment by retromer should shed significant light onto whether retromer-dependent DAT recycling may contribute to neuropsychiatric disorders.
Footnotes
This work was supported by the National Institutes of Health (Grants DA15169 and DA035224 to H.E.M).
The authors declare no competing financial interests.
- Correspondence should be addressed to Haley E. Melikian, Ph.D., Brudnick Neuropsychiatric Research Institute, Department of Psychiatry, University of Massachusetts Medical School, 303 Belmont Street, Worcester, MA 01604. haley.melikian{at}umassmed.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}